
The Beginning and Development of the Theranostic Approach in Nuclear Medicine, as Exemplified by the Radionuclide Pair 86Y and 90Y

Abstract
:1. Introduction and Historical Background
2. Nuclear Data
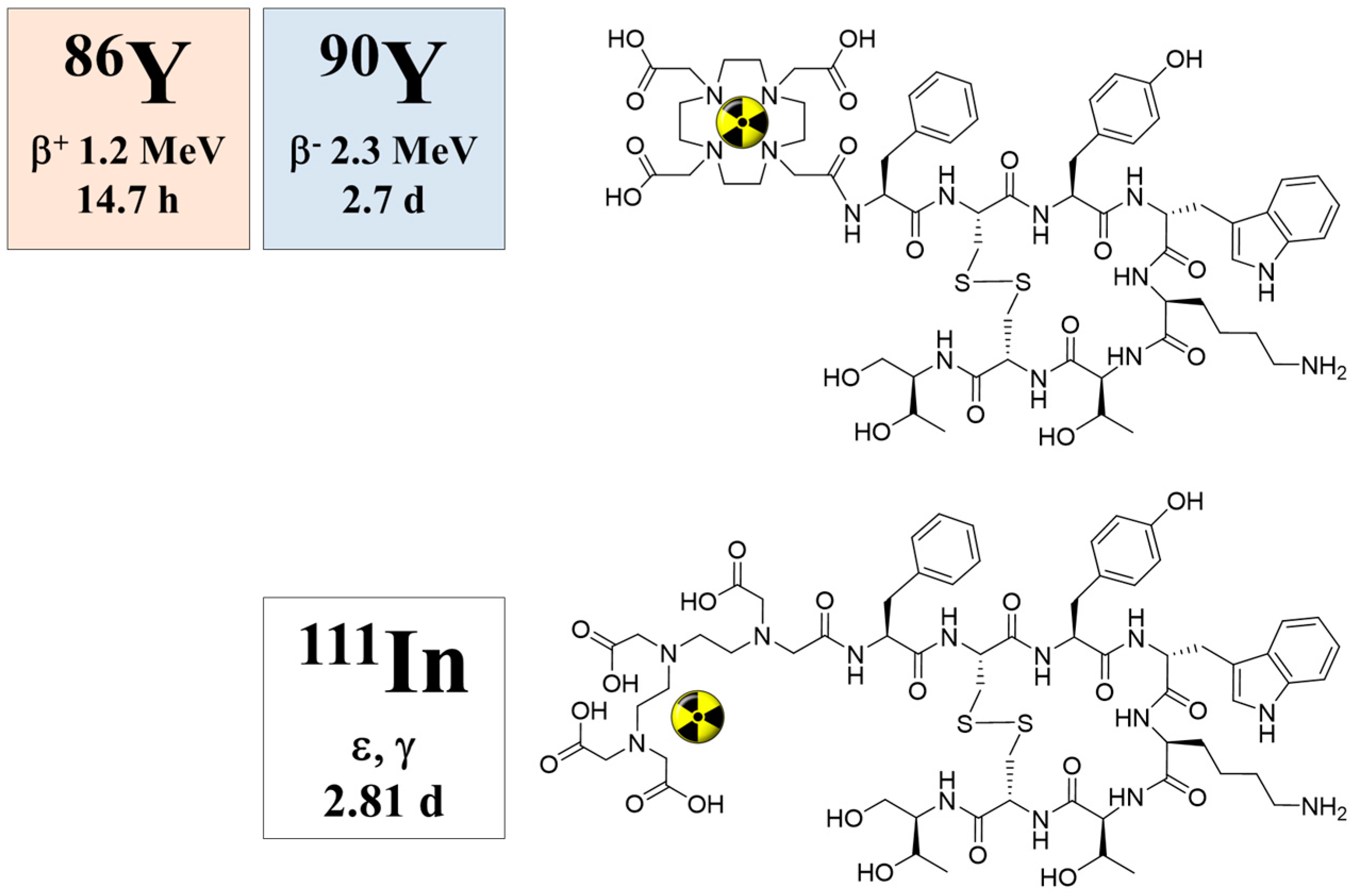
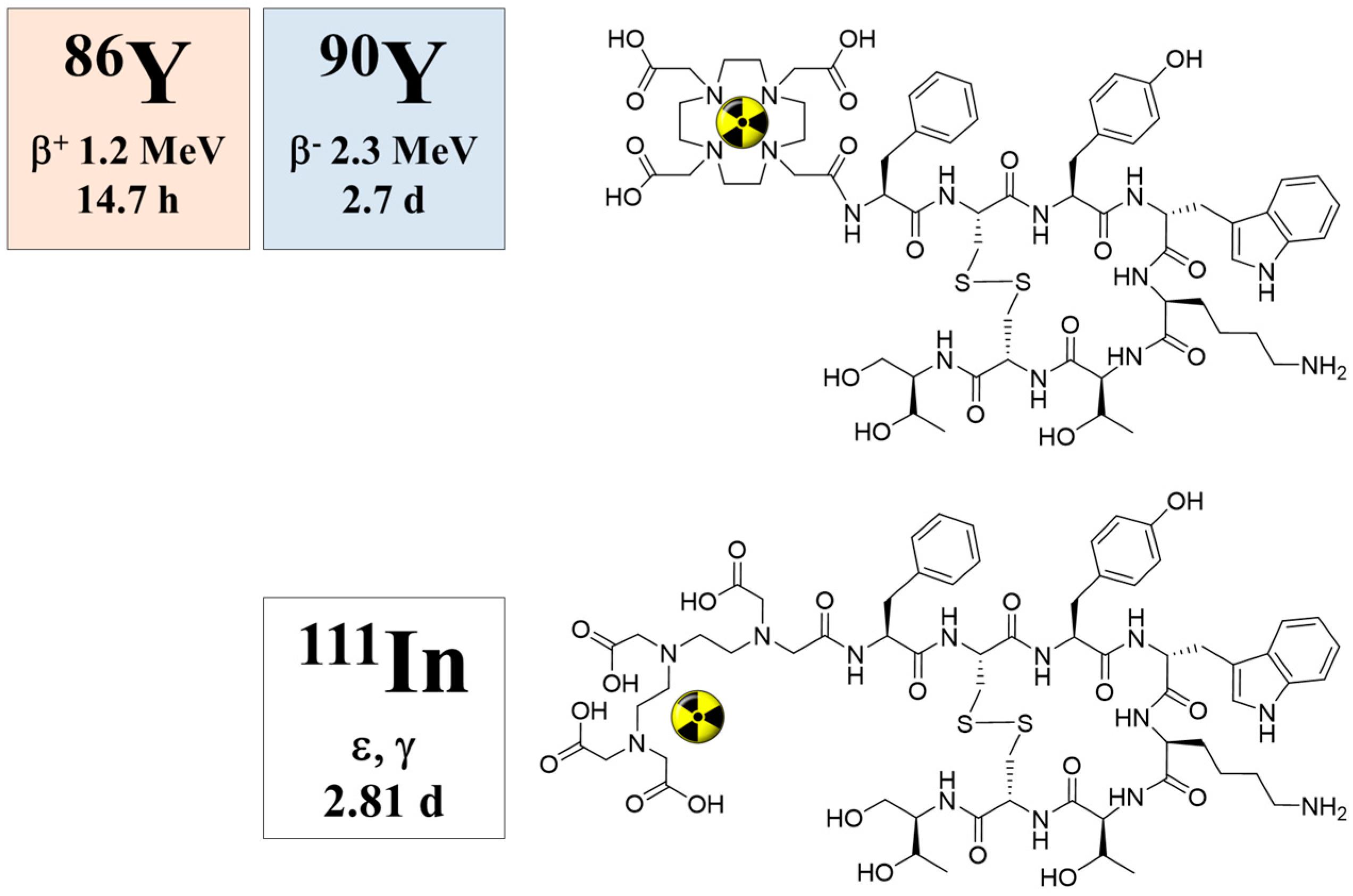
2.1. Decay Data
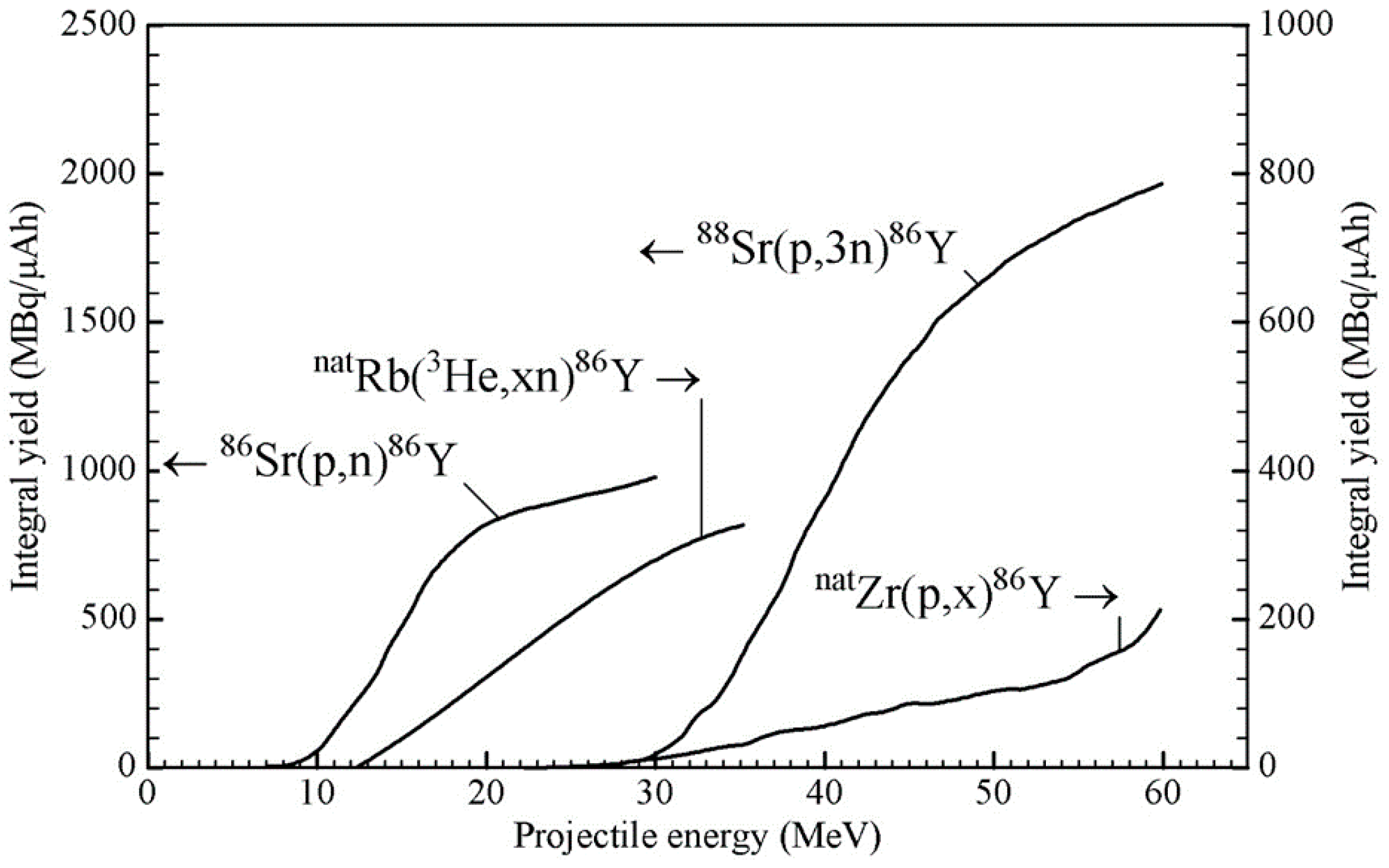
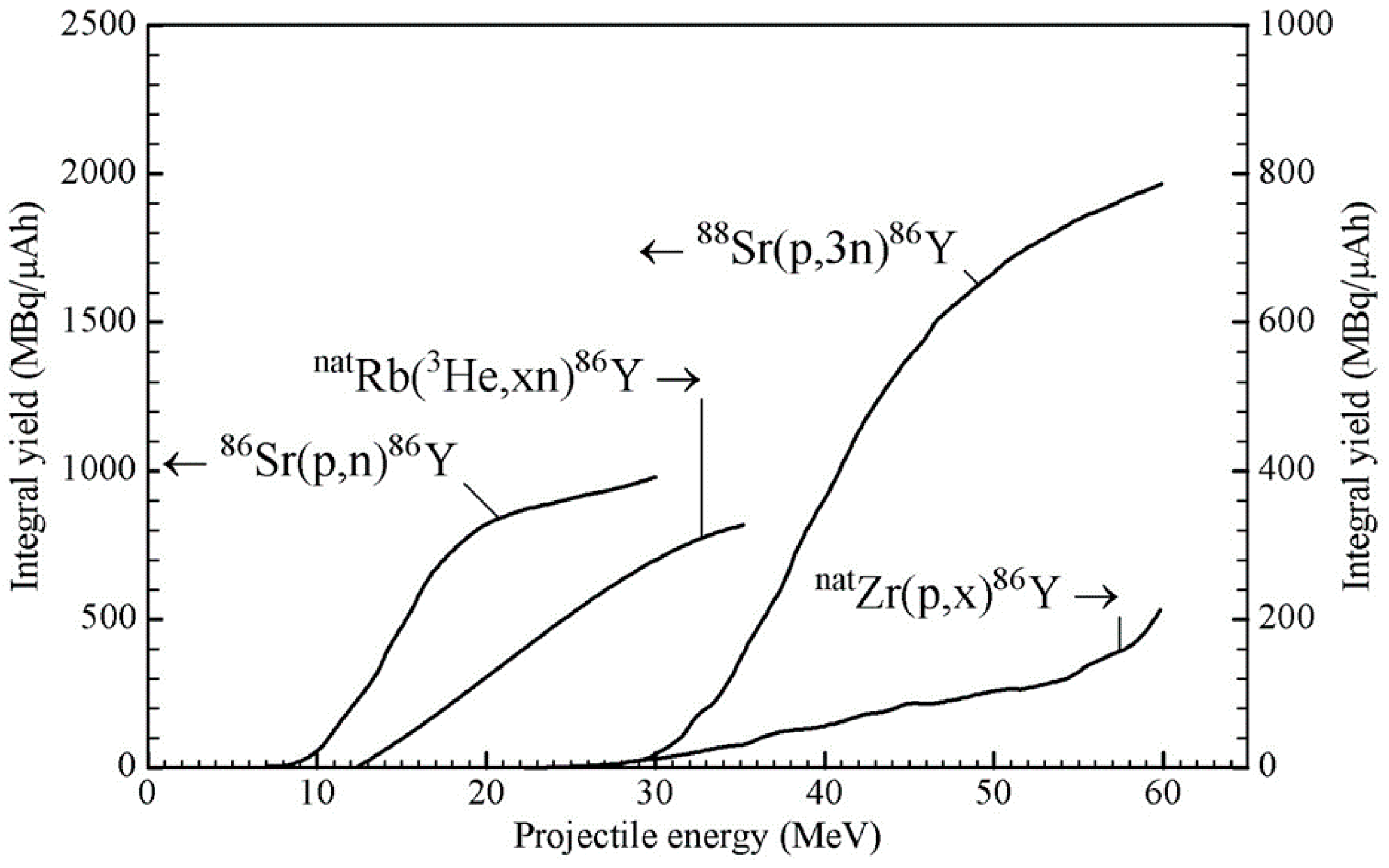
2.2. Production Data: Reaction Cross-Sections and Integral Yields
3. Production Methodologies
3.1. Targetry
3.2. Chemical Processing
3.3. Comparison of Separation Methods
3.4. Concluding Remarks about Production Methodologies
4. 86Y for PET Imaging
4.1. Radiation Emission Properties of 86Y
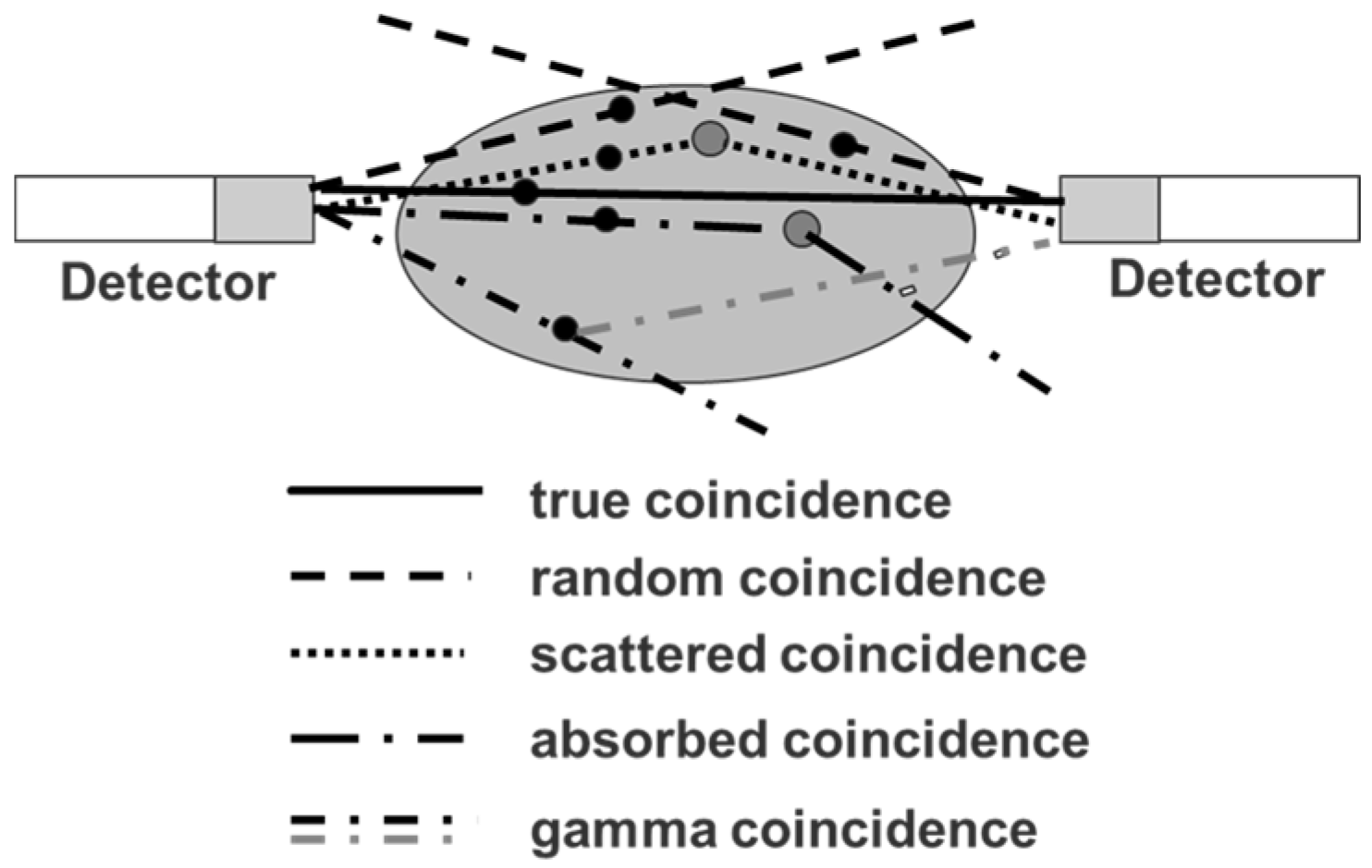
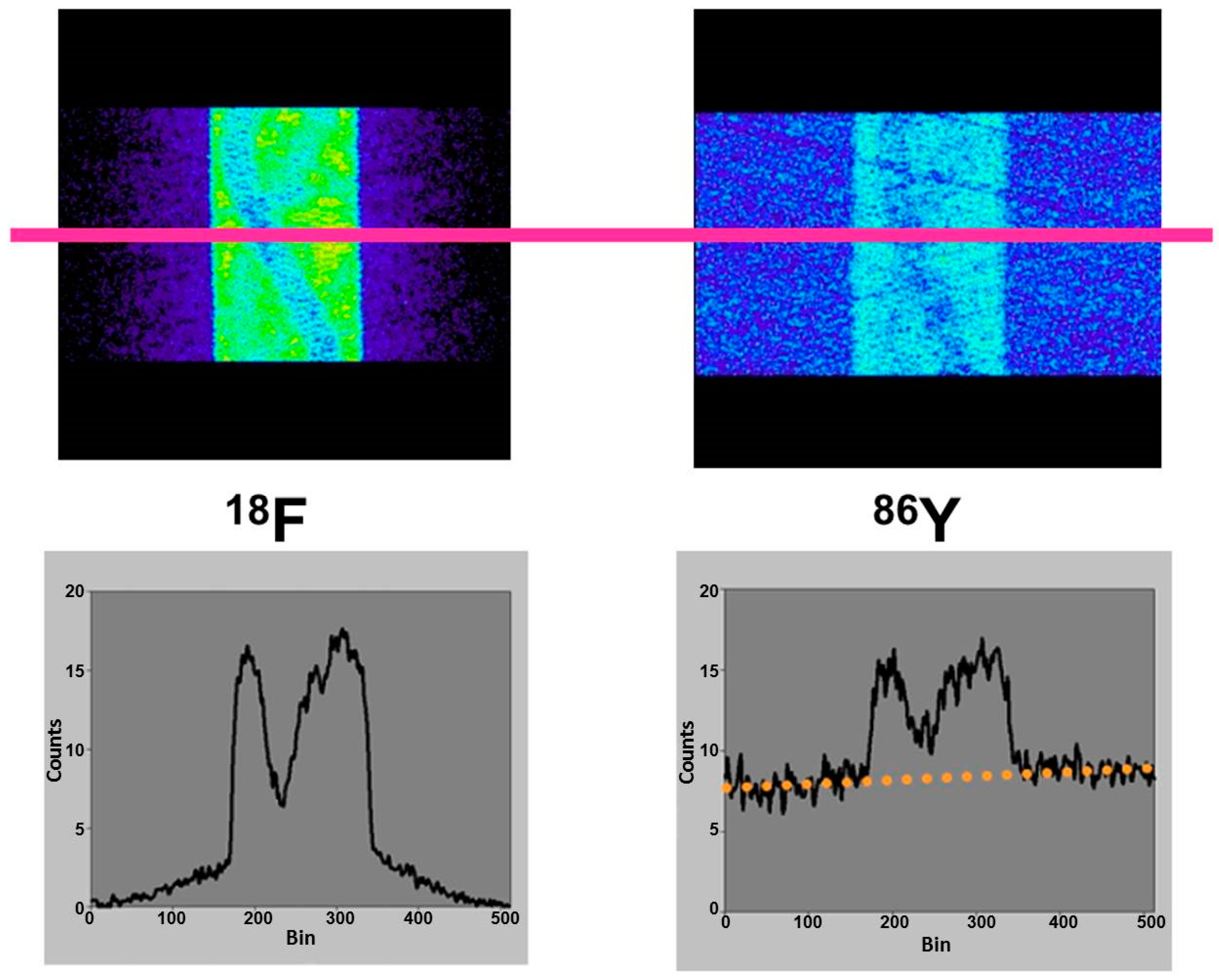
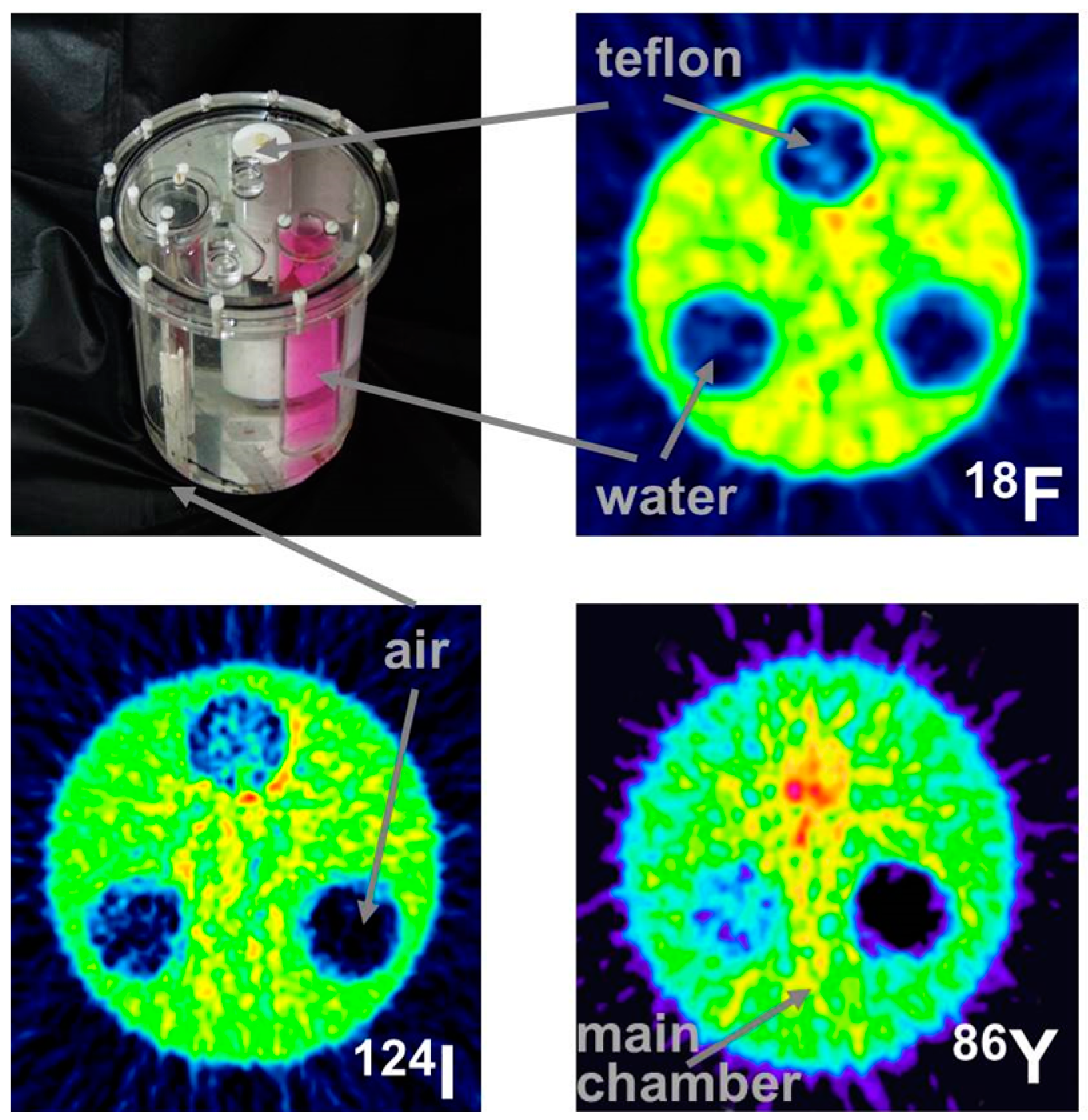
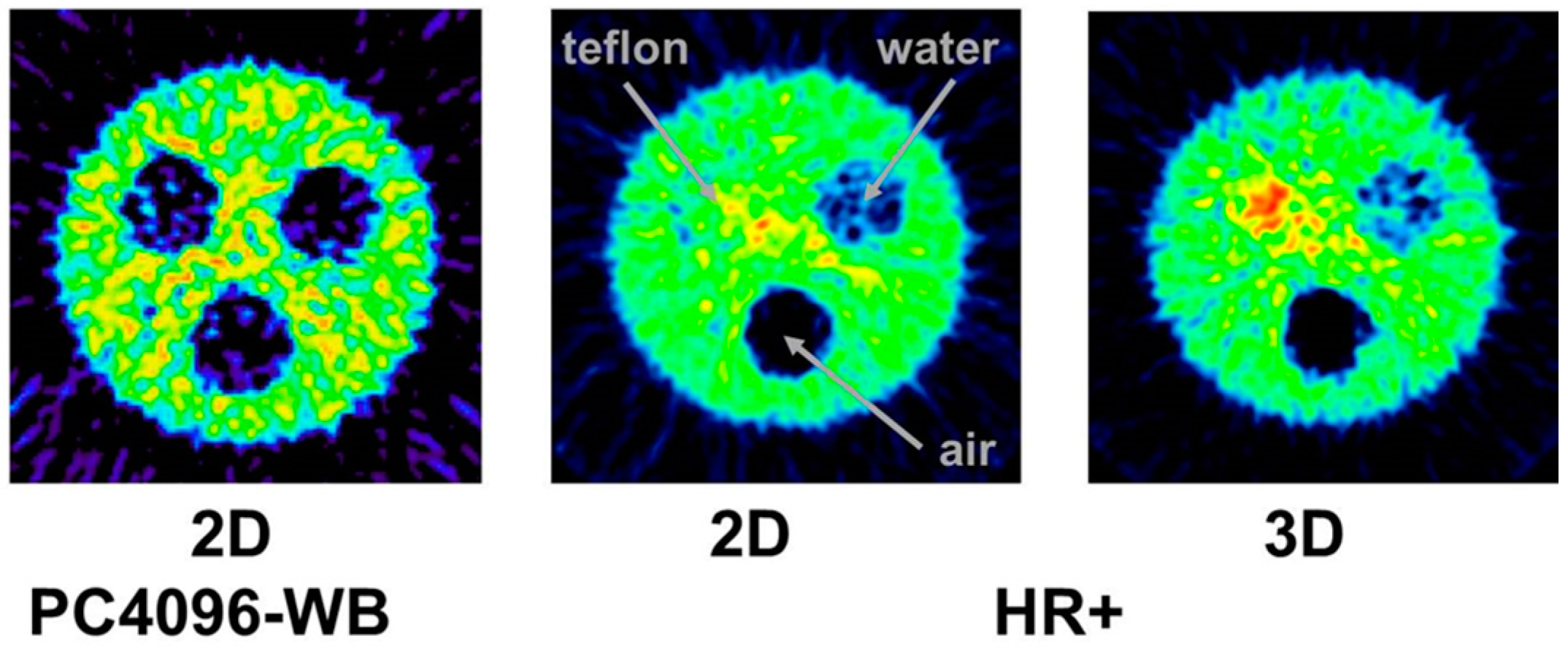
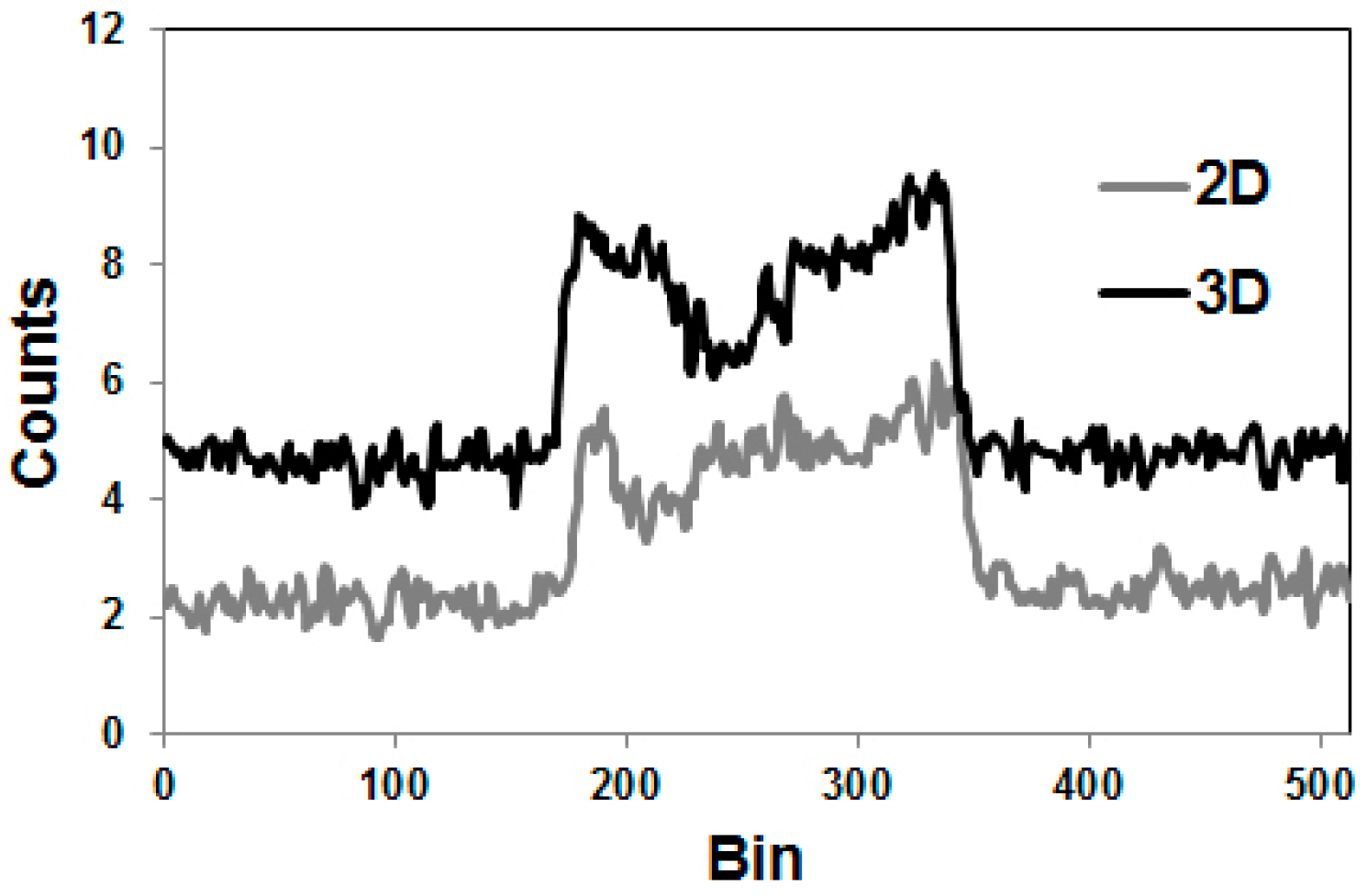
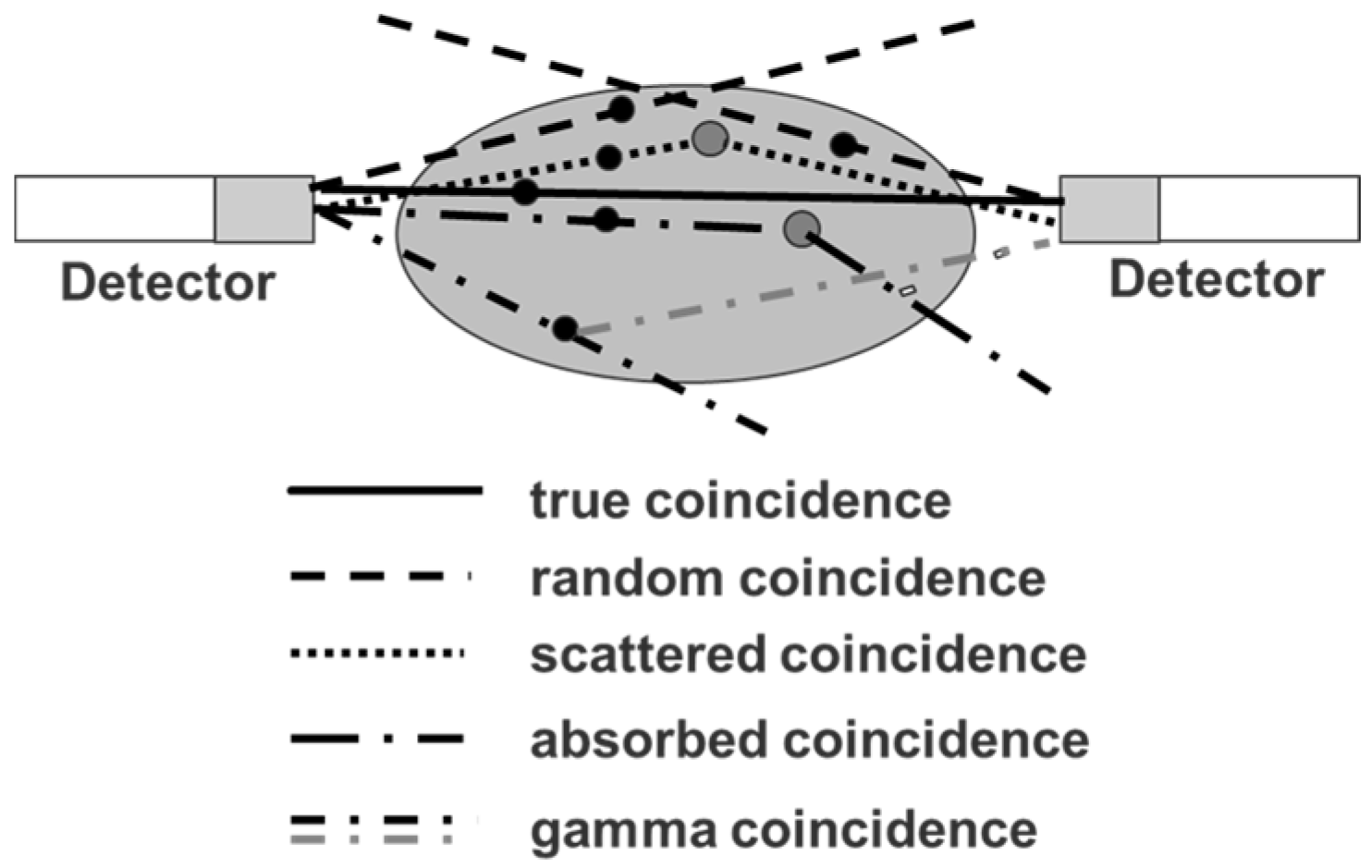
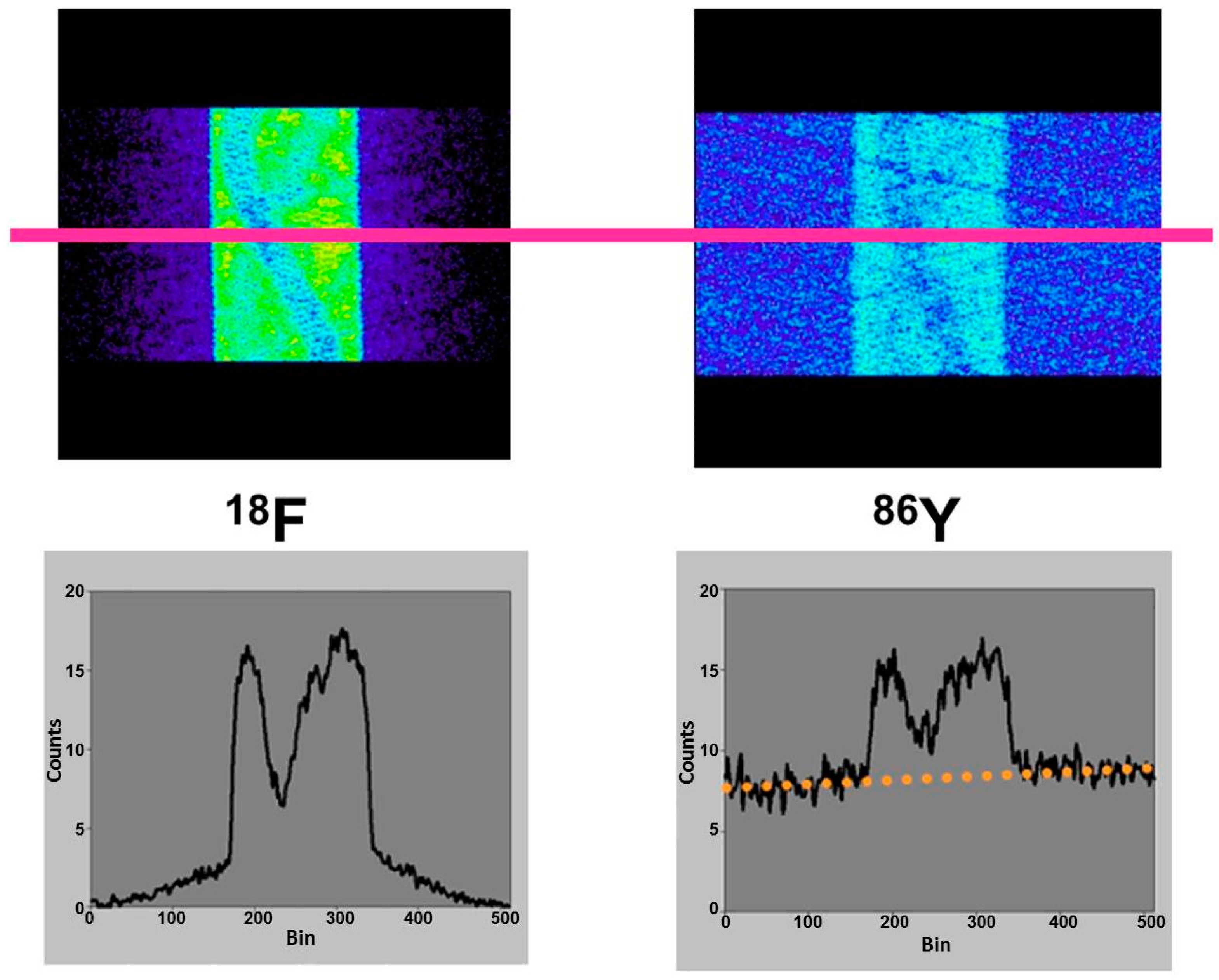
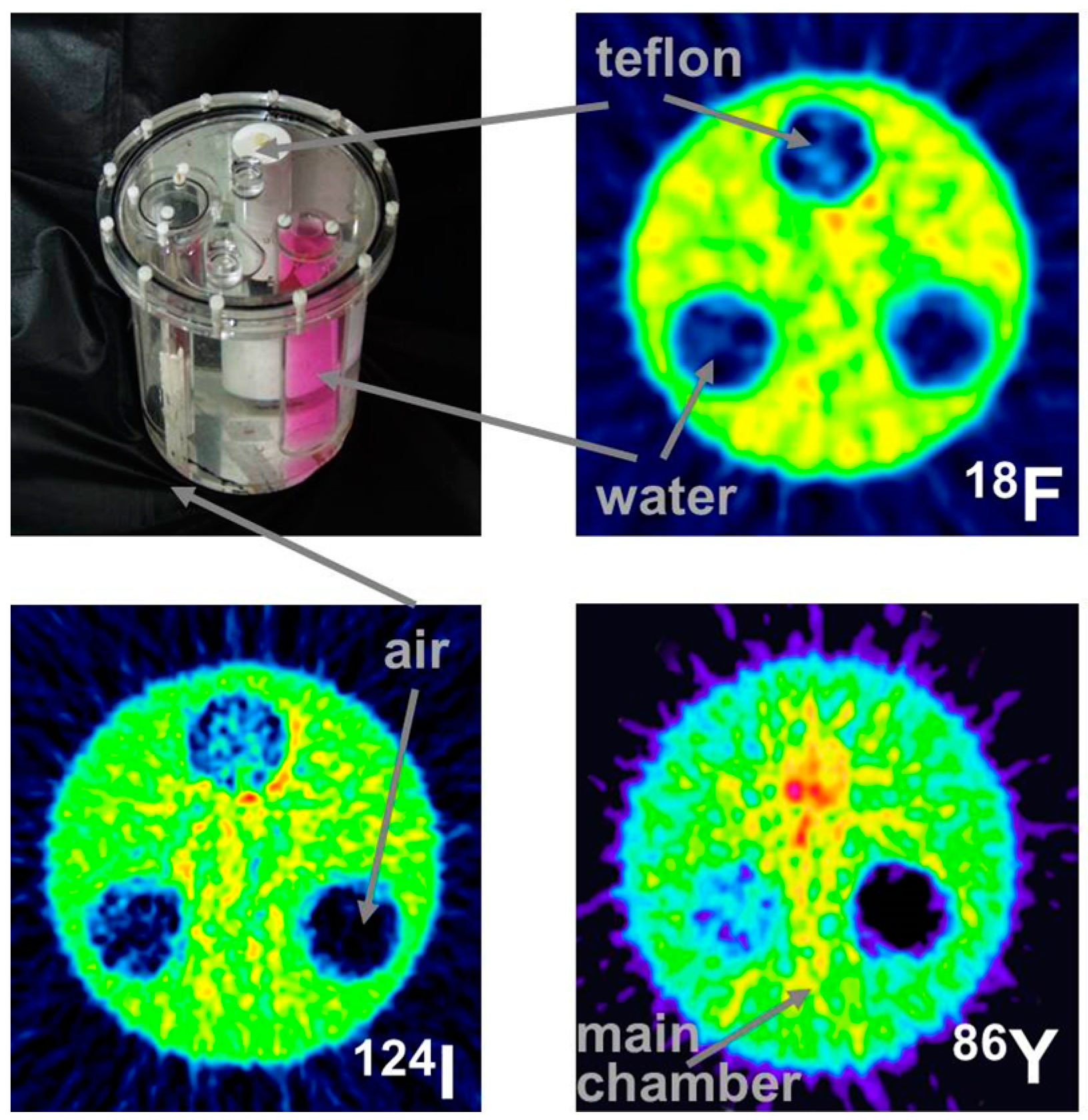
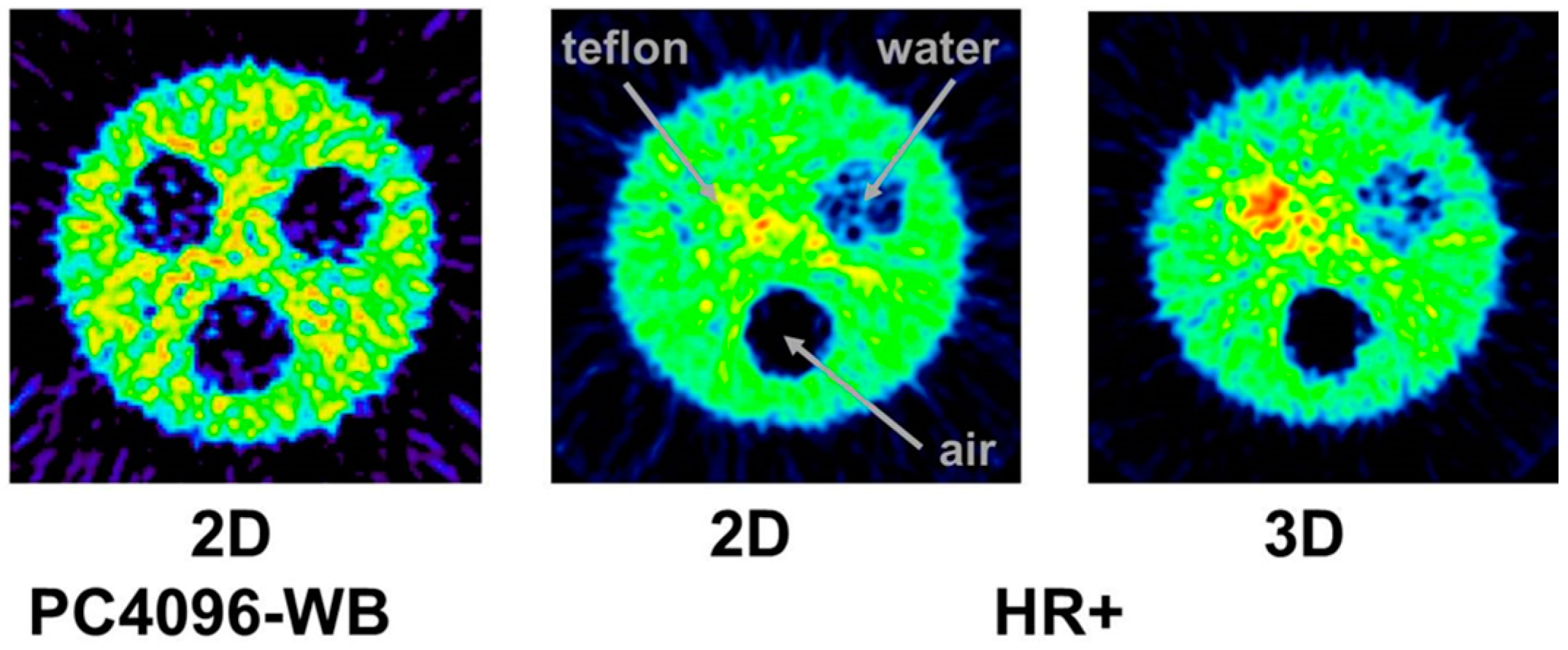
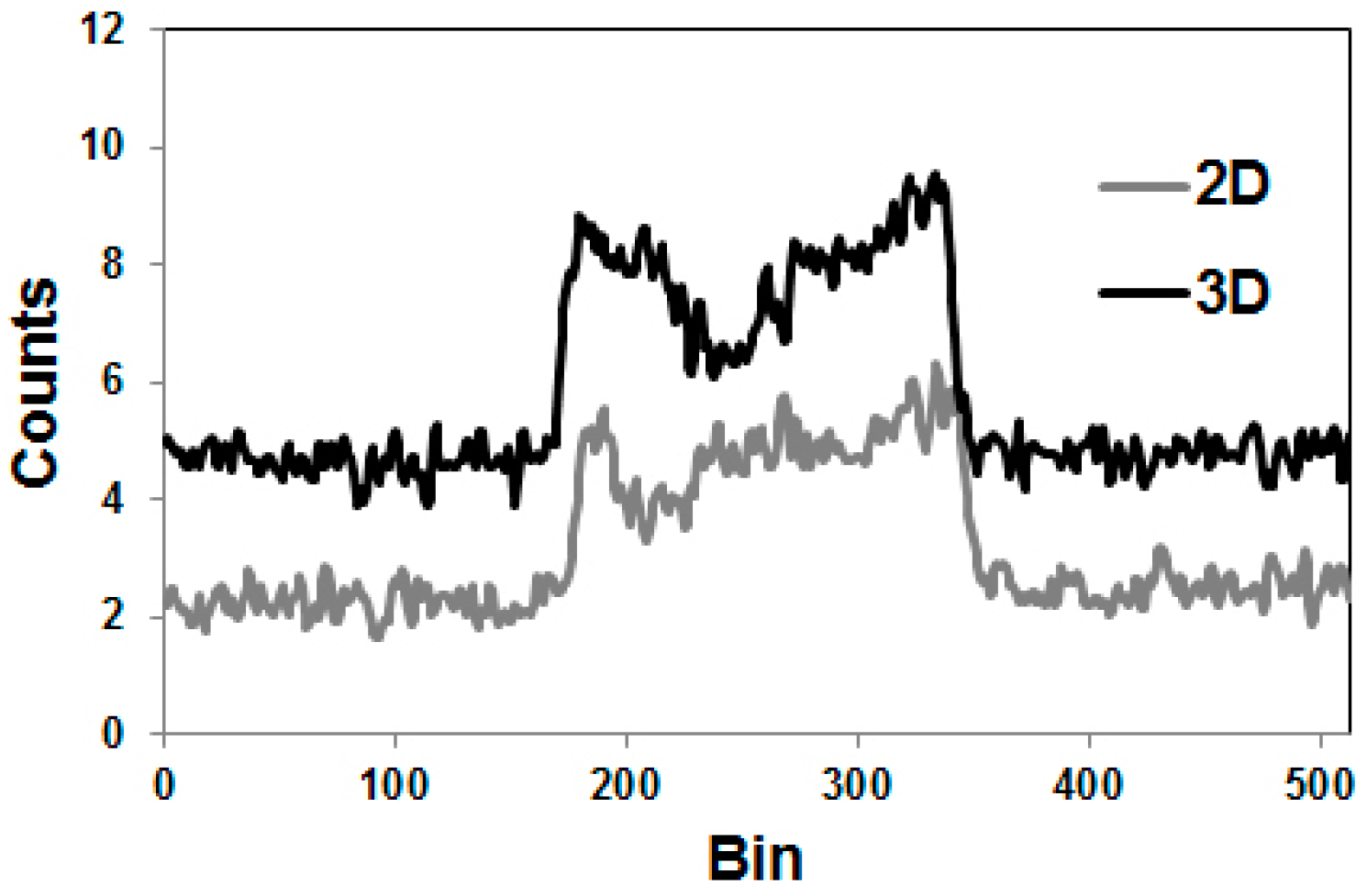
4.2. Errors Caused by Gamma Coincidences and Their Correction
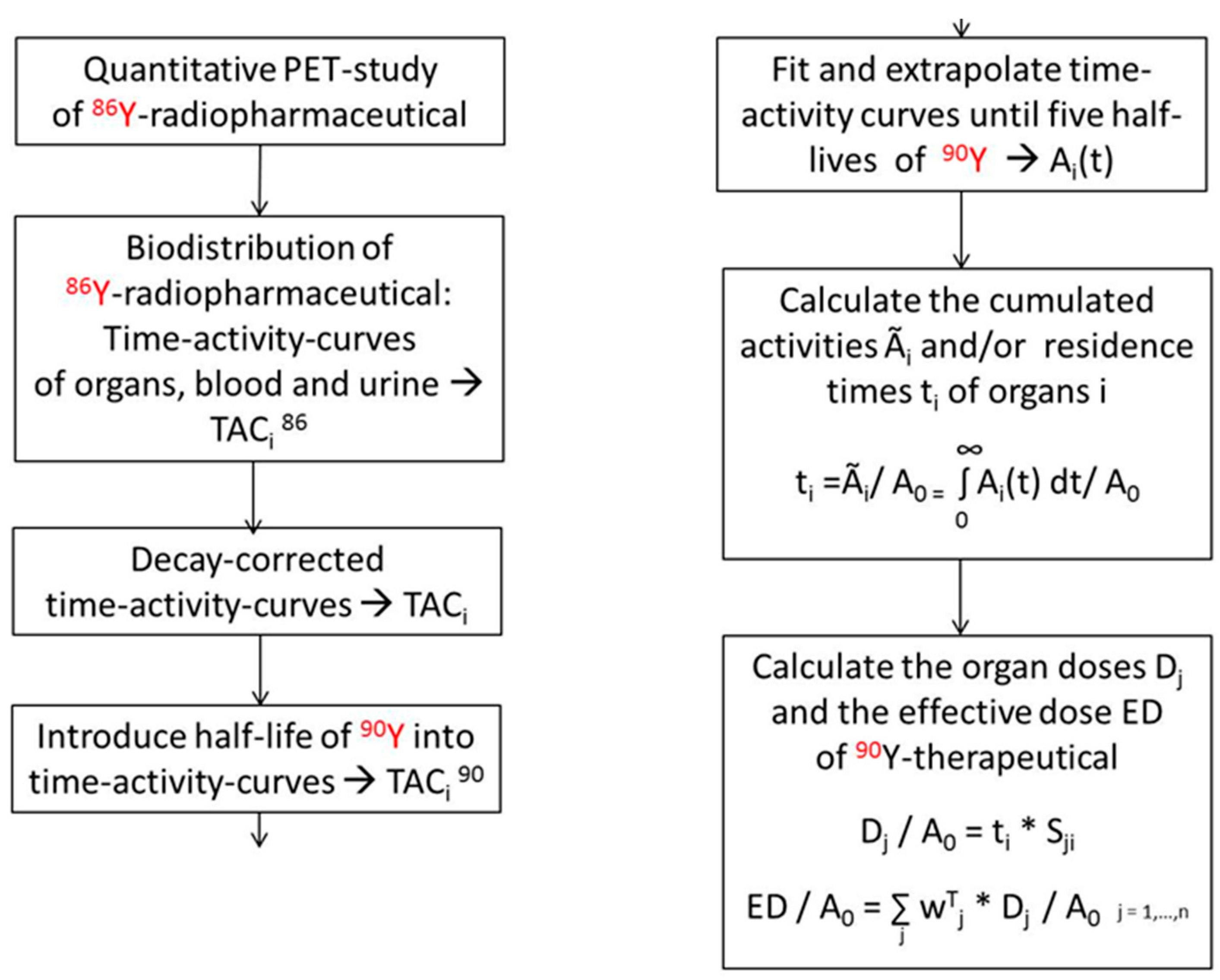
5. Radiation Doses of 90Y-Therapeuticals Extracted from 86Y PET-Imaging
5.1. PET Imaging Based on 90Y Positrons
5.2. PET Imaging with 86Y Instead of Using 90Y
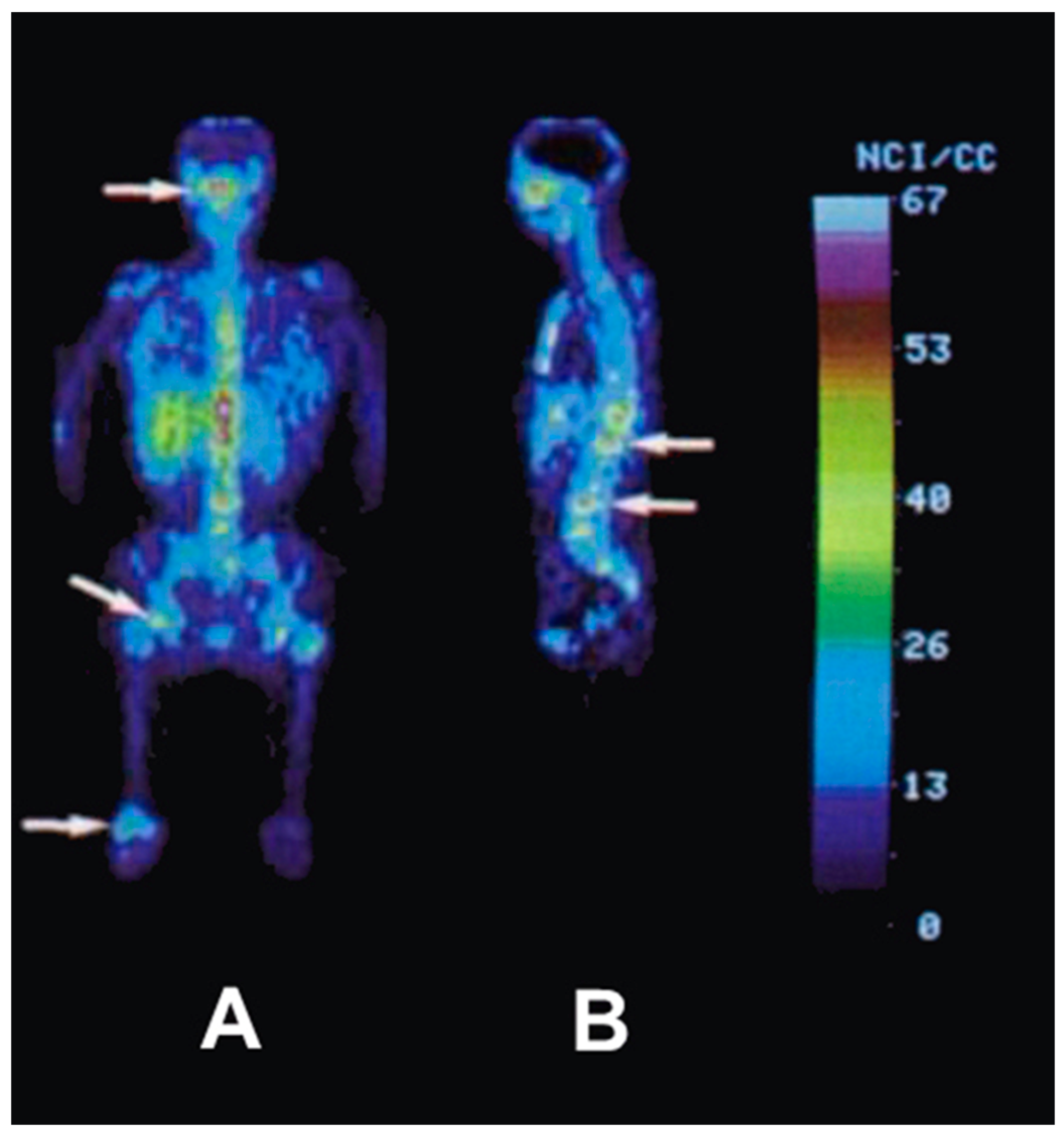
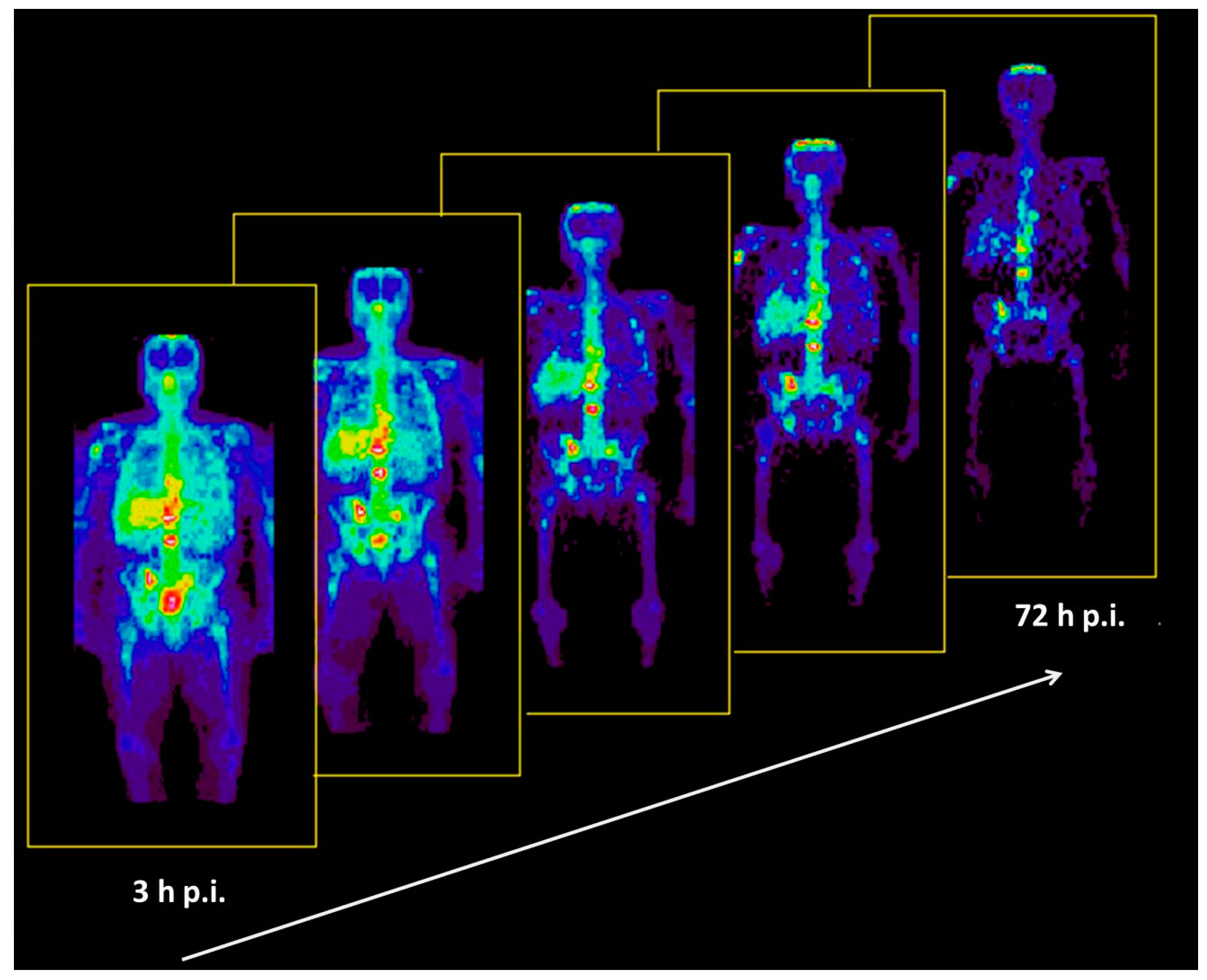
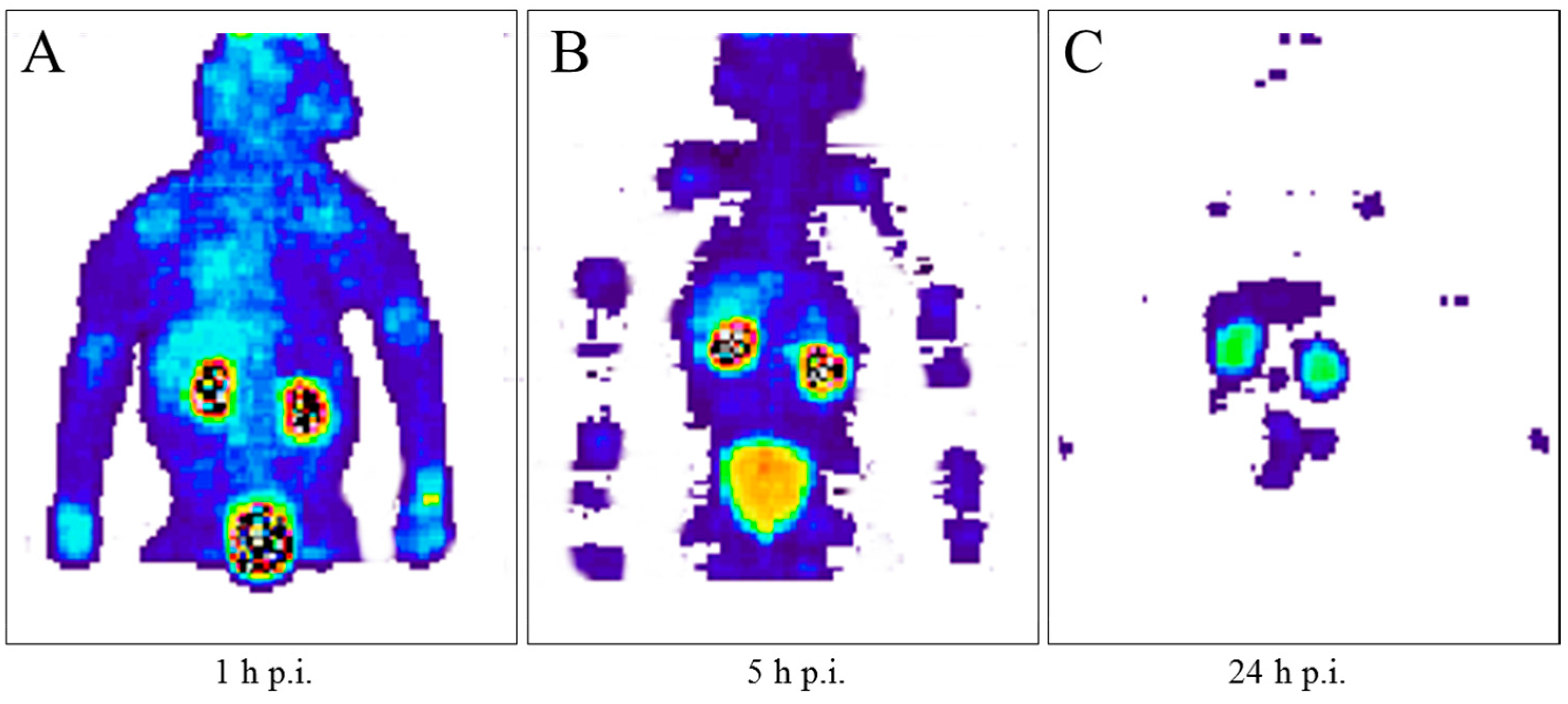
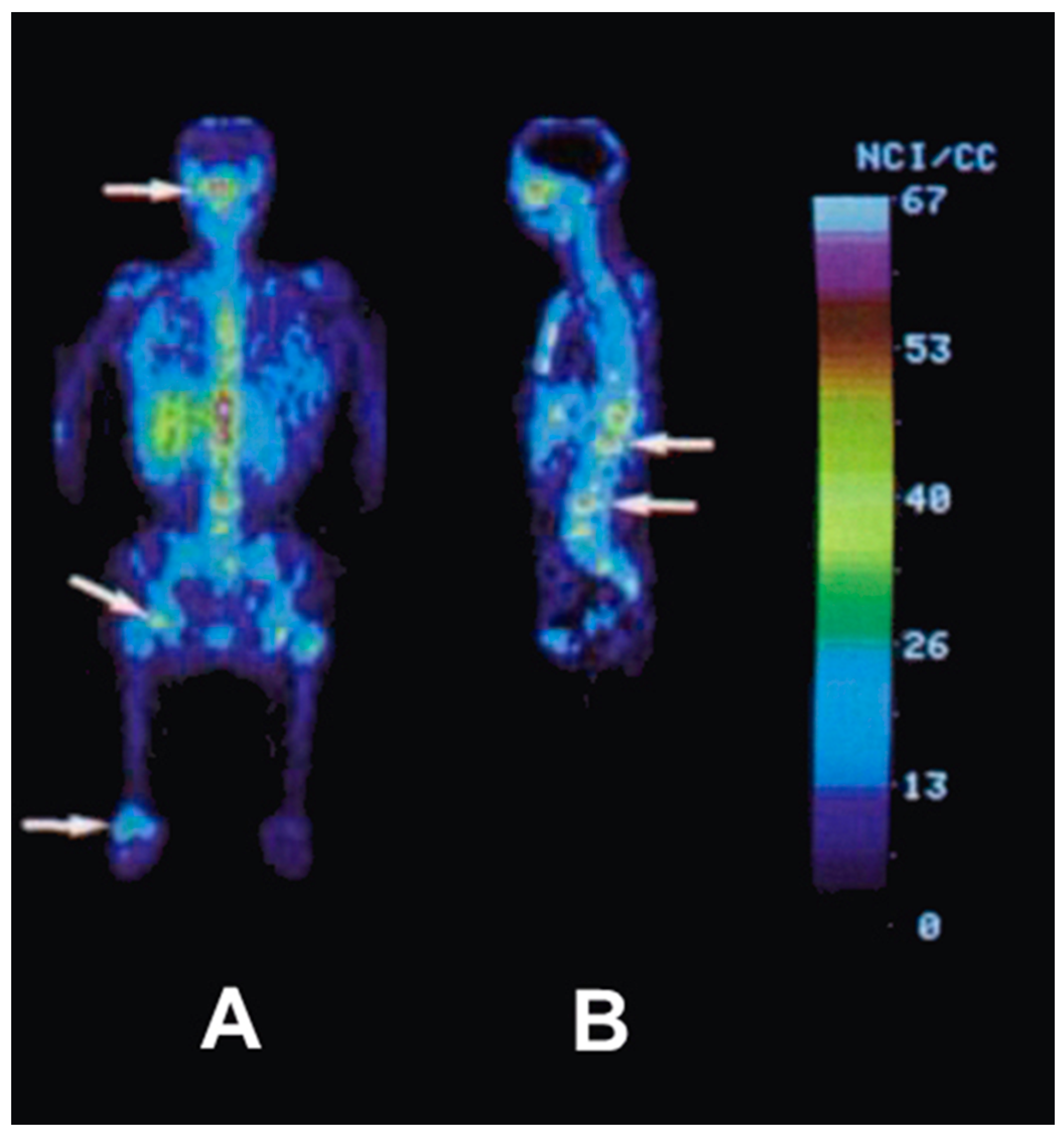
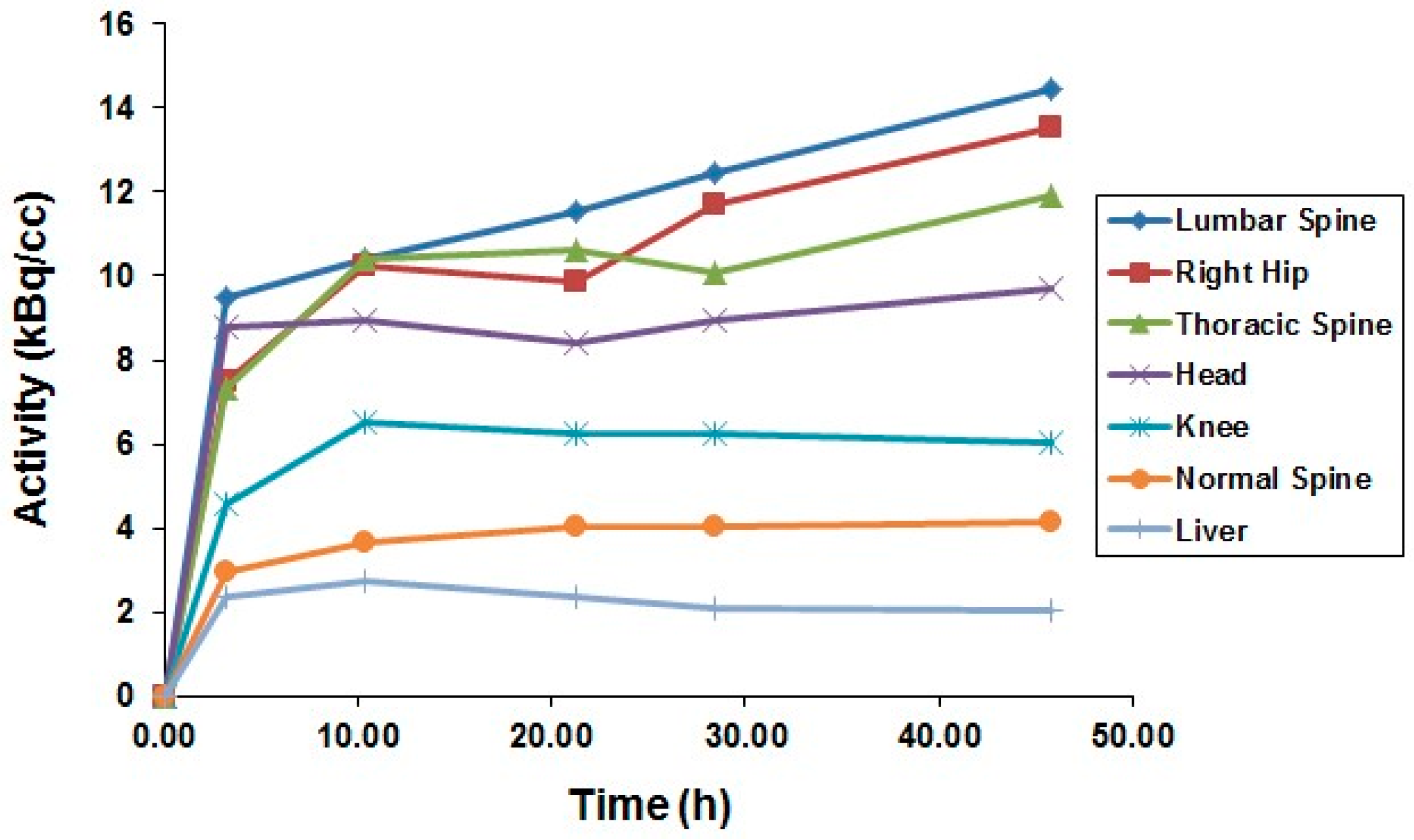
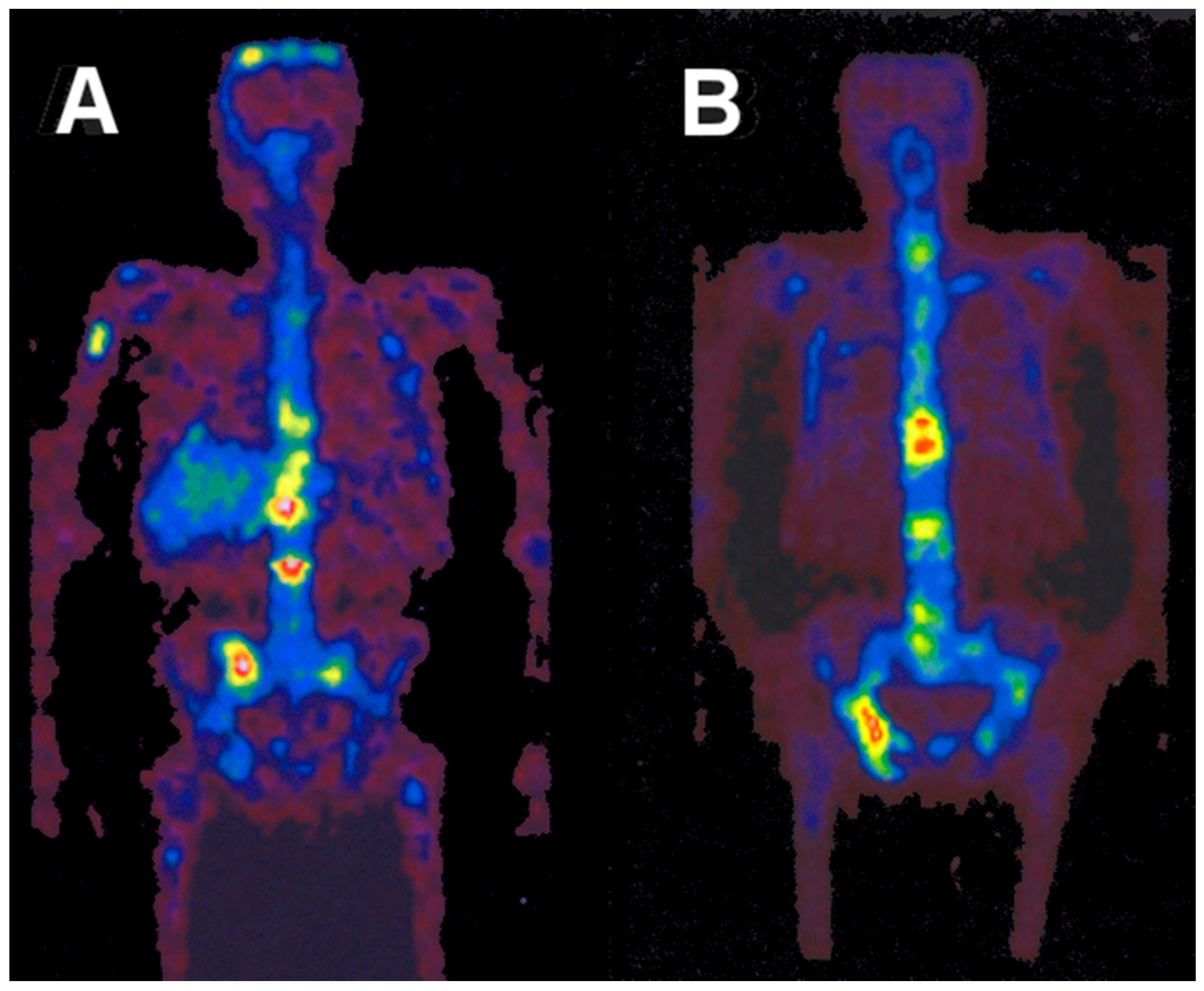
5.3. The First Human Study with 86Y
5.4. Comparative Evaluation of 86Y-Citrate and 86Y-EDTMP
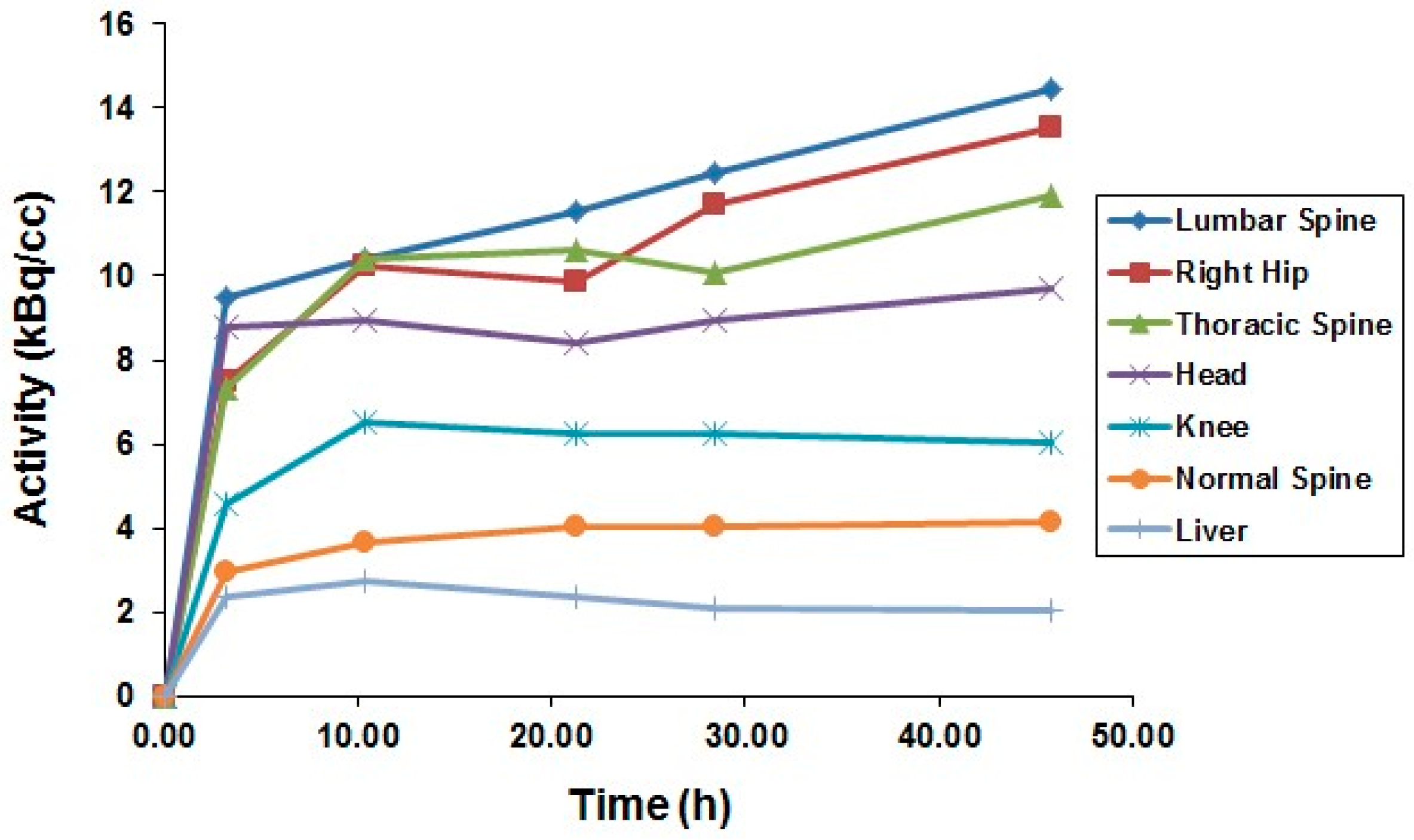
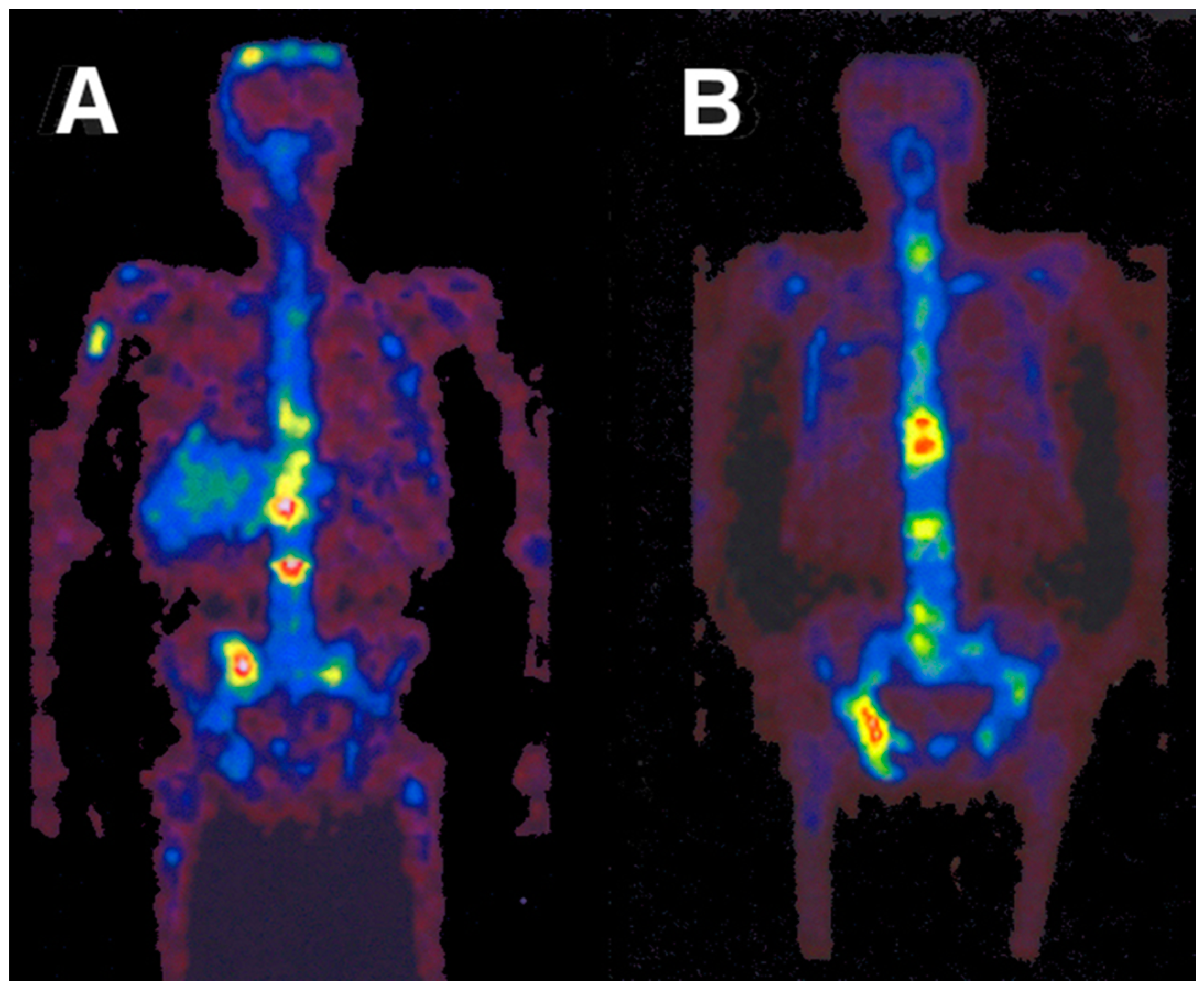
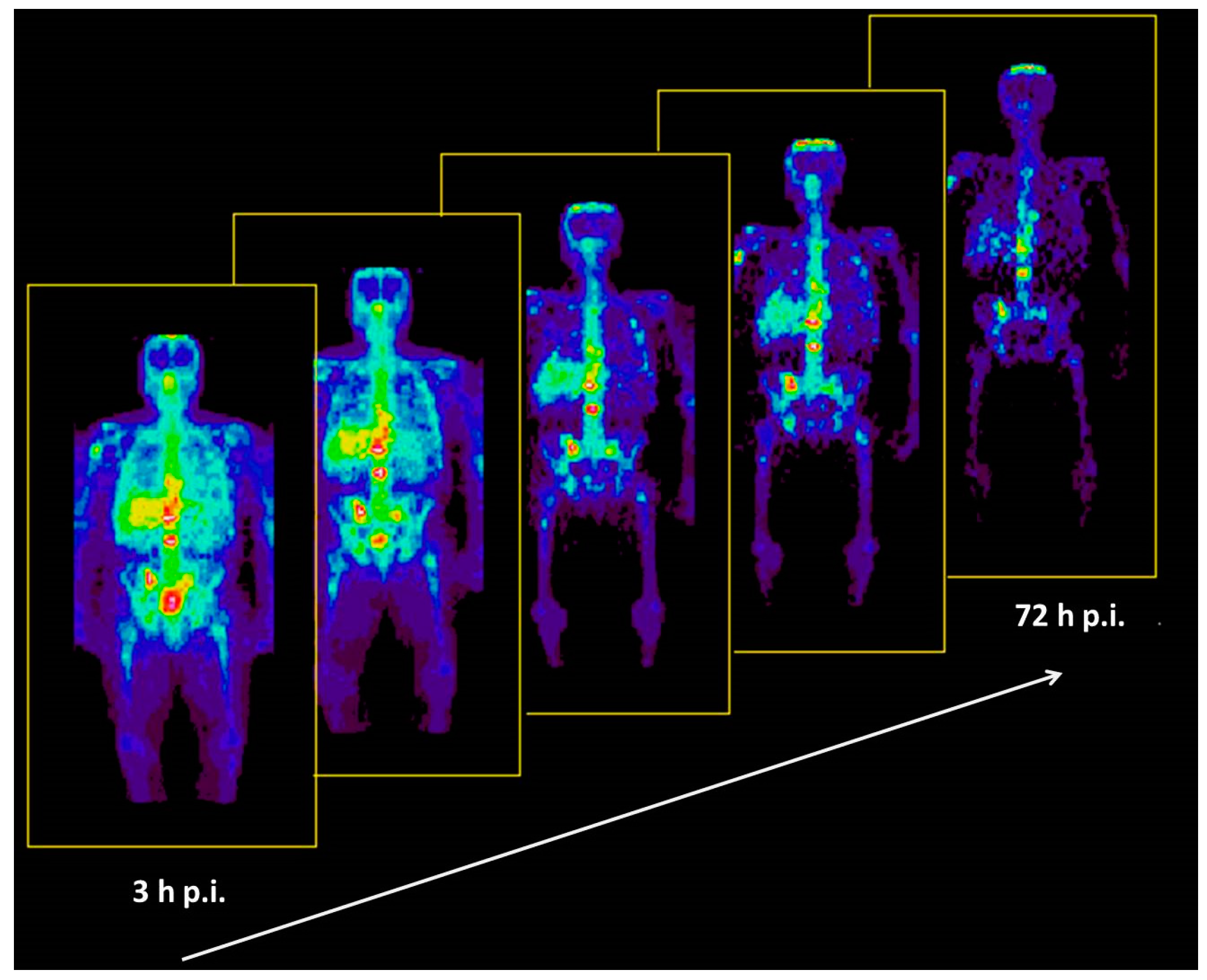
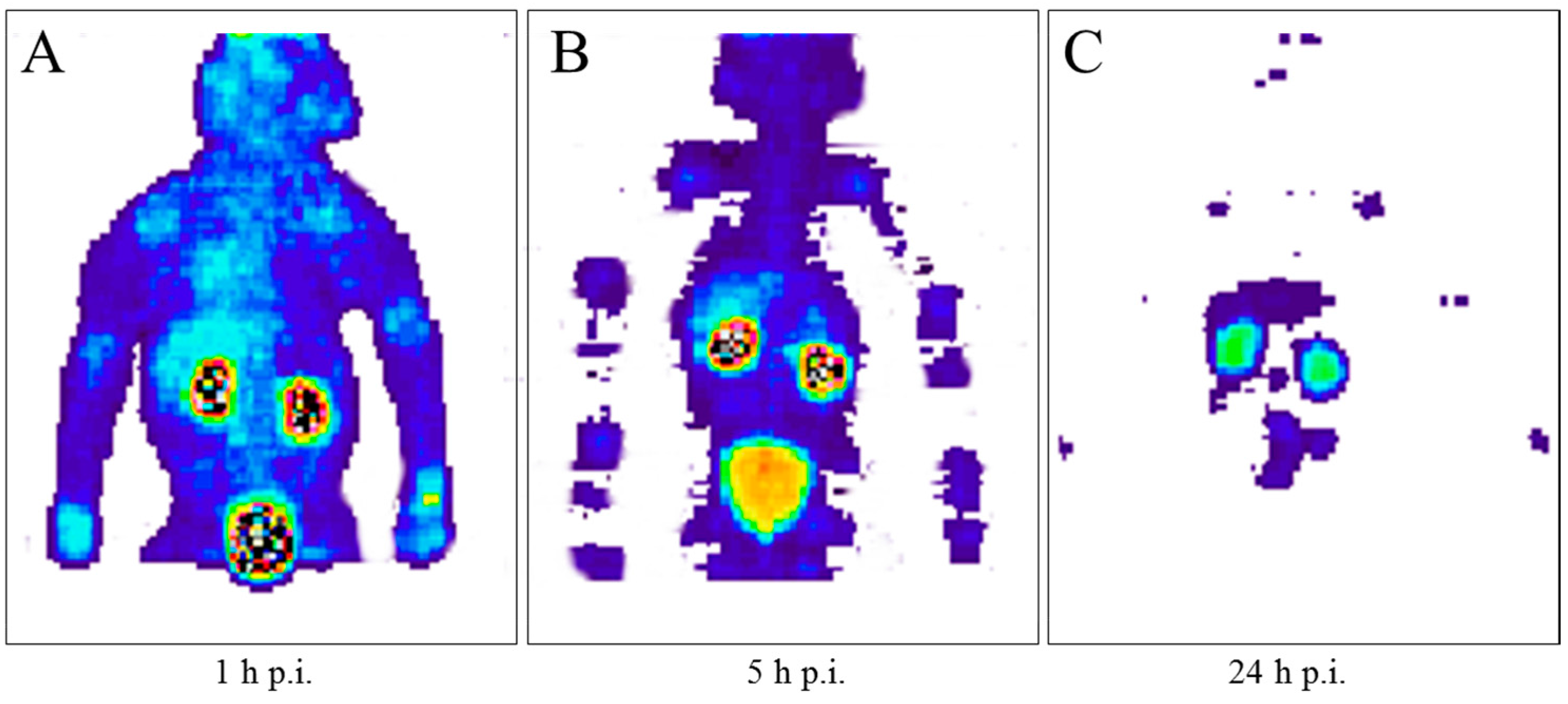
5.5. Radiation Dose of 90Y-DOTA-DPhe1-Tyr3-Octreotide Based on 86Y-DOTA-DPhe1-Tyr3-Octreotide
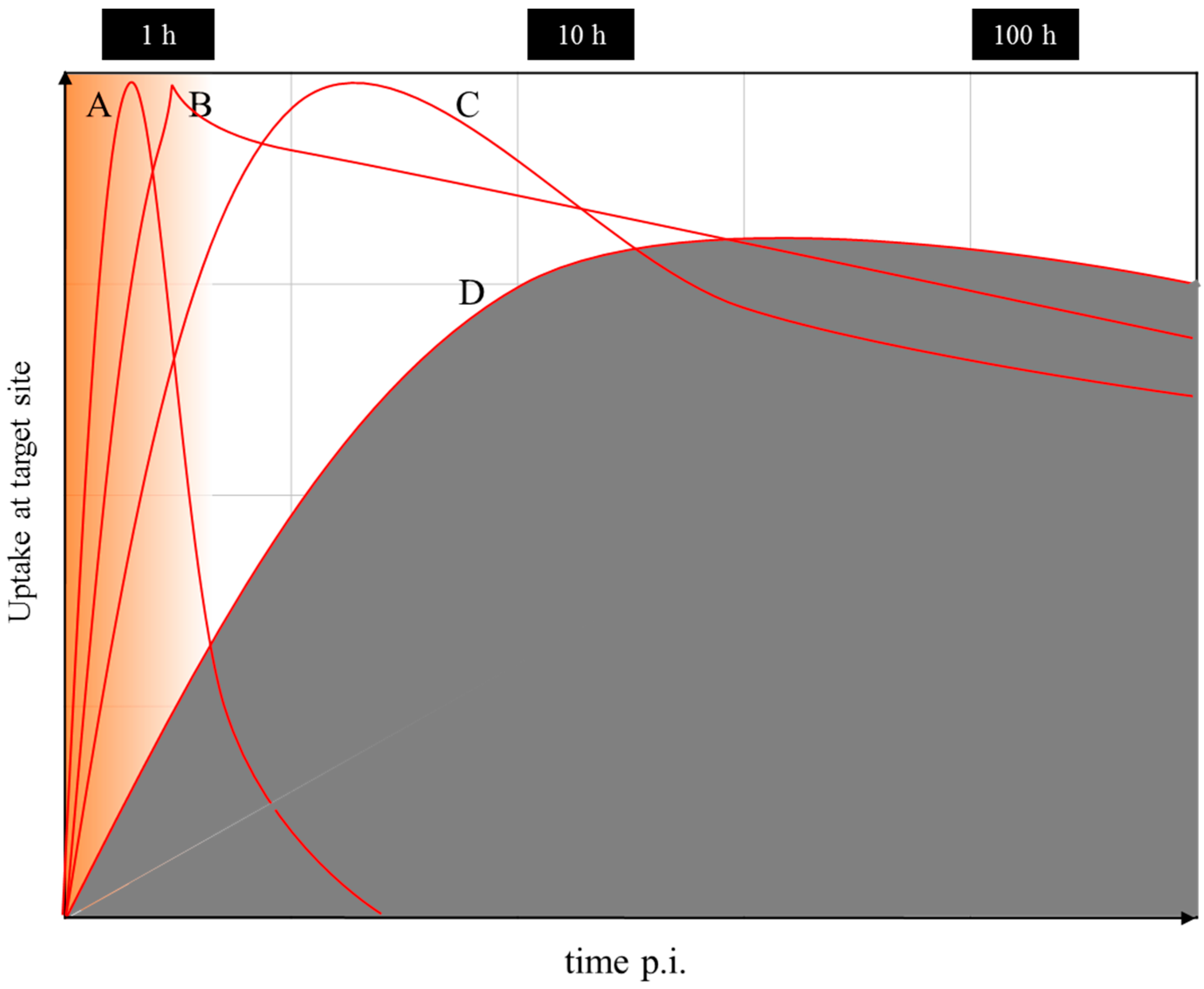
6. Discussion
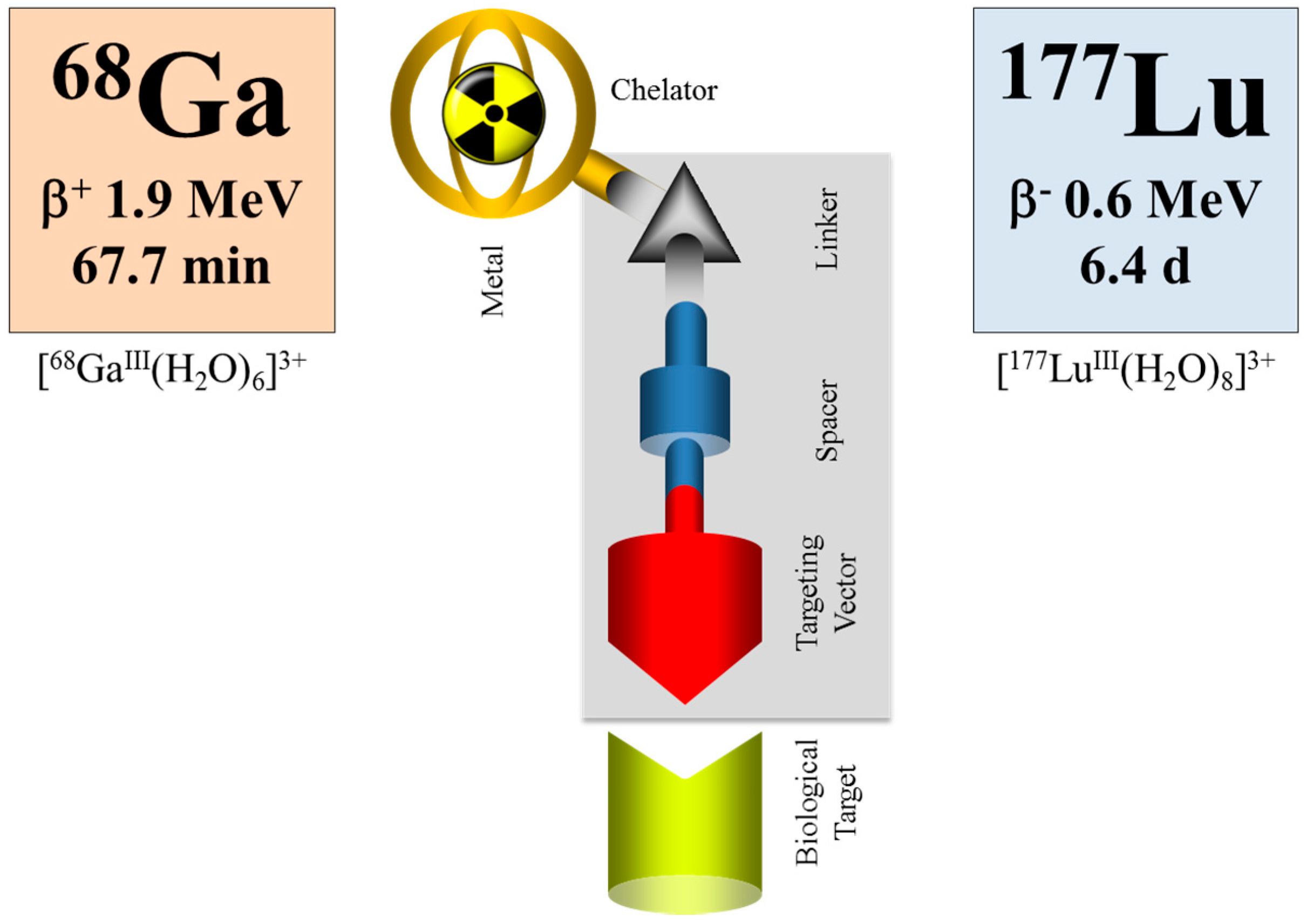
6.1. Further Use of 86Y
6.2. Alternative Approaches in Radiotheranostics
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stöcklin, G.; Qaim, S.M.; Rösch, F. The impact of radioactivity on medicine. Radiochim. Acta 1995, 70/71, 249–272. [Google Scholar]
- Zimmer, A.M.; Kuzel, T.M.; Spies, W.G.; Duda, R.B.; Webber, D.I.; Kazikiewicz, J.M.; Radosevich, J.A.; LoCicero, J.; Robinson, P.G.; Gilyon, K.A.; et al. Comparative pharmacokinetics of In-111 and Y-90 B72.3 in patients following single dose intravenous administration. Antib. Immunoconjug. Radiopharm. 1992, 5, 285–294. [Google Scholar]
- Mausner, L.F.; Srivastava, S.C. Selection of radionuclides for radioimmunotherapy. Med. Phys. 1993, 20, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.C. Paving the way to personalized medicine: Production of some theragnostic radionuclides at Brookhaven National Laboratory. Radiochim. Acta 2011, 99, 635–640. [Google Scholar] [CrossRef]
- Lederer, C.M. Table of Isotopes, 7th ed.; Shirley, V.S., Ed.; John Wiley and Sons: New York, NY, USA, 1978; Volume 99, pp. 1–1523. [Google Scholar]
- Eckerman, K.F.; Endo, A. Radionuclide Decay Data and Decay Schemes. SNM MIRD Committee: Reston, VA, USA, 2007. [Google Scholar]
- Evaluated Nuclear Structure and Decay File (ENSDF), BNL, USA. Available online: www.nndc.bnl.gov/ensdf (accessed on 6 June 2017).
- Qaim, S.M. Decay data and production yields of some non-standard positron emitters used in positron emission tomography. Q. J. Nucl. Med. Mol. Imaging 2008, 52, 111–120. [Google Scholar] [PubMed]
- Qaim, S.M. Development of novel positron emitters for medical applications: Nuclear and radiochemical aspects. Radiochim. Acta 2011, 99, 611–625. [Google Scholar] [CrossRef]
- Qaim, S.M. Nuclear data for production and medical application of radionuclides: Present status and future needs. Nucl. Med. Biol. 2017, 44, 31–49. [Google Scholar] [CrossRef] [PubMed]
- Qaim, S.M.; Tárkányi, F. Nuclear Data for the Production of Therapeutic Radionuclides; Capote, R., Ed.; IAEA Technical Reports Series No. 473; IAEA: Vienna, Austria, 2011; pp. 1–358. [Google Scholar]
- Sachdev, D.R.; Porile, N.T.; Yaffe, L. Reactions of 88Sr with protons of energies 7–85 MeV. Can. J. Chem. 1967, 45, 1149–1160. [Google Scholar] [CrossRef]
- Levkovskii, V.N. Activation Cross Sections for Nuclides of Average Masses (A = 40–100) by Protons and Alpha-Particles with Average Energies (E = 10–50 MeV); Experiments and systematics; Inter-Vesy: Moscow, Russia, 1991; ISBN 5-265-02732-7. [Google Scholar]
- Kantelo, M.V.; Hogan, J.J. Charged-particle emission in reaction of 90Zr with 10–86 MeV protons. Phys. Rev. 1976, C14, 64–74. [Google Scholar]
- Rösch, F.; Qaim, S.M.; Stöcklin, G. Nuclear data relevant to the production of the positron emitting radioisotope 86Y via the 86Sr(p,n)- and natRb(3He,xn)-processes. Radiochim. Acta 1993, 61, 1–8. [Google Scholar] [CrossRef]
- Rösch, F.; Qaim, S.M.; Stöcklin, G. Production of the positron emitting radioisotope 86Y for nuclear medical application. Appl. Radiat. Isot. 1993, 44, 677–681. [Google Scholar] [CrossRef]
- Uddin, M.S.; Khandaker, M.U.; Kim, K.S.; Lee, Y.S.; Kim, G.N. Excitation functions of the proton-induced nuclear reactions on natural zirconium. Nucl. Instrum. Meth. Phys. Res. 2008, B266, 13–20. [Google Scholar] [CrossRef]
- Khandaker, M.U.; Kim, K.; Lee, M.W.; Kim, K.S.; Kim, G.N.; Otuka, N. Experimental determination of proton-induced cross sections on natural zirconium. Appl. Radiat. Isot. 2009, 67, 1341–1347. [Google Scholar] [CrossRef] [PubMed]
- Szelecsényi, F.; Steyn, G.F.; Kovács, Z.; Vermeulen, C.; Nagatsu, K.; Zhang, M.R.; Suzuki, K. Exciation functions of natZr+p nuclear processes up to 70 MeV: New measurements and compilation. Nucl. Inst. Meth. Phys. Res. 2015, 343, 173–191. [Google Scholar] [CrossRef]
- Tárkányi, F.; Ditrói, F.; Takács, S.; Hermanne, A.; Al-Abyad, M.; Yamazaki, H.; Baba, M.; Mohammadi, M.A. New activation cross section data on longer lived radionuclei produced in proton induced nuclear reactions on zirconium. Appl. Radiat. Isot. 2015, 97, 149–169. [Google Scholar] [CrossRef] [PubMed]
- Zaneb, H.; Hussain, M.; Amjed, N.; Qaim, S.M. Nuclear model analysis of excitation functions of proton induced reactions on 86Sr and natZr: Evaluation of production routes of 86Y. Appl. Radiat. Isot. 2015, 104, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, D.M.; Porile, N.T. Reactions of 116Cd with intermediate energy 3He and 4He ions. Nucl. Phys. 1969, A130, 65–76. [Google Scholar] [CrossRef]
- Alfassi, Z.B.; Weinreich, R. The production of positron emitters 75Br and 76Br: Excitation functions and yields for 3He and α-particle induced reactions on arsenic. Radiochim. Acta 1980, 30, 67–71. [Google Scholar] [CrossRef]
- Scholten, B.; Lambrecht, R.M.; Cogneau, M.; Vera Ruiz, H.; Qaim, S.M. Excitation functions for the cyclotron production of 99mTc and 99Mo. Appl. Radiat. Isot. 1999, 51, 69–80. [Google Scholar] [CrossRef]
- Uddin, M.S.; Sudár, S.; Spahn, I.; Shariff, M.A.; Qaim, S.M. Excitation function of the 60Ni(p,γ)61Cu reaction from threshold to 16 MeV. Phys. Rev. 2016, C93, 044606. [Google Scholar]
- Finn, R.D.; McDevitt, M.; Ma, D.; Jurcic, J.; Scheinberg, D.; Larson, S.; Shoner, S.; Link, J.; Krohn, K.; Schlyer, D. Low energy cyclotron production and separation of yttrium-86 for evaluation of monoclonal antibody pharmacokinetics and dosimetry. In Applications of Accelerators in Research and Industry, Proceedings of the 15th International Conference, Woodbury, NY; Duggan, J.L., Morgan, I.L., Eds.; American Institute of Physics Press: College Park, MD, USA, 1999; pp. 991–993. [Google Scholar]
- Reischl, G.; Rösch, F.; Machulla, H.-J. Electrochemical separation and purification of yttrium-86. Radiochim. Acta 2002, 90, 225–228. [Google Scholar] [CrossRef]
- Kettern, K.; Linse, K.-H.; Spellerberg, S.; Coenen, H.H.; Qaim, S.M. Radiochemical studies relevant to the production of 86Y and 88Y at a small-sized cyclotron. Radiochim. Acta 2002, 90, 845–849. [Google Scholar] [CrossRef]
- Garmestani, K.; Milenic, D.E.; Plascjak, P.S.; Brechbiel, W.W. A new and convenient method for purification of 86Y using a Sr(II) selective resin and comparison of biodistribution of 86Y and 111In labelled HerceptinTM. Nucl. Med. Biol. 2002, 29, 599–606. [Google Scholar] [CrossRef]
- Park, L.S.; Szajek, L.P.; Wong, K.J.; Plascjak, P.S.; Garmestani, K.; Googins, S.; Eckelman, W.C.; Carrasquillo, J.A.; Paik, C.H. Semi-automated 86Y purification using a three column system. Nucl. Med. Biol. 2004, 31, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Tang, L.; Perkins, T.A.; Rowland, D.J.; Laforest, R.; Lewis, J.S.; Welch, M.J. Preparation of high specific activity 86Y using a small biomedical cyclotron. Nucl. Med. Biol. 2005, 32, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Avila-Rodriguez, M.A.; Nye, J.A.; Nickles, R.J. Production and separation of non-carrier-added 86Y from enriched 86Sr targets. Appl. Radiat. Isot. 2008, 66, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Lukic, D.; Tamburella, C.; Buchegger, F.; Beyer, G.-J.; Comor, J.J.; Seimbille, Y. High efficient production and purification of 86Y based on electrochemical separation. Appl. Radiat. Isot. 2009, 67, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Kandil, S.A.; Scholten, B.; Hassan, K.F.; Hanafi, H.A.; Qaim, S.M. A comparative study on the separation of radioyttrium from Sr- and Rb-targets via ion-exchange and solvent extraction techniques, with special reference to the production of no-carrier-added 86Y, 87Y and 88Y using a cyclotron. J. Radioanal. Nucl. Chem. 2009, 279, 823–832. [Google Scholar] [CrossRef]
- Sadeghi, M.; Aboudzadeh, M.; Zali, A.; Boulourinovin, F. Radiochemical studies relevant to 86Y production via 86Sr(p,n)86Y for PET imaging. Appl. Radiat. Isot. 2009, 67, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, M.; Aboudzadeh, M.; Zali, A.; Zeinali, B. 86Y-production via 86Sr(p,n) for PET imaging at a cyclotron. Appl. Radiat. Isot. 2009, 67, 1392–1396. [Google Scholar] [CrossRef] [PubMed]
- Michael, H.; Rosezin, H.; Apelt, H.; Blessing, G.; Knieper, J.; Qaim, S.M. Some technical improvements in the production of 123I via the 124Te(p,2n)123I reaction at a compact cyclotron. Int. J. Appl. Radiat. Isot. 1981, 32, 581–587. [Google Scholar] [CrossRef]
- Spellerberg, S.; Scholten, B.; Spahn, I.; Bolten, W.; Holzgreve, M.; Coenen, H.H.; Qaim, S.M. Target development for diversified irradiations at a medical cyclotron. Appl. Radiat. Isot. 2015, 104, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Vogg, A.T.J.; Lang, R.; Meier-Boeke, P.; Scheel, W.; Reske, S.N.; Neumaier, B. Cyclotron production of radionuclides in aqueous targets matrices as alternative to solid targets. Production of 86Y as example. In Advances in Nuclear and Radiochemistry Based on NRC-6; Qaim, S.M., Coenen, H.H., Eds.; Forschungszentrum Jülich, 2004; pp. 318–320. ISBN 3-89336-362-9. [Google Scholar]
- Oehlke, E.; Hoehr, C.; Hou, X.; Hanemaayer, V.; Zeisler, S.; Adam, M.J.; Ruth, T.J.; Celler, A.; Buckley, K.; Benard, F.; et al. Production of 86Y and other radiometals for research purposes using a solution target system. Nucl. Med. Biol. 2015, 42, 842–849. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, D.G.; Mausner, L.F.; Srivastava, S.C. Irradiation of strontium chloride targets at proton energies above 35 MeV to produce PET radioisotope Y-86. Radiochim. Acta 2011, 99, 755–761. [Google Scholar] [CrossRef]
- Sadeghi, M.; Zali, A.; Avila, M. A novel method for radiochemical separation of radioyttrium from Sr targets using precipitation technique. Radiochim. Acta 2010, 98, 437–439. [Google Scholar] [CrossRef]
- Herzog, H.; Tellmann, L.; Scholten, B.; Coenen, H.H.; Qaim, S.M. PET imaging problems with the non-standard positron emitters yttrium-86 and iodine-124. Q. J. Nucl. Med. Mol. Imaging 2008, 52, 159–165. [Google Scholar] [PubMed]
- Lubberink, M.; Herzog, H. Quantitative imaging of 124I and 86Y with PET. Eur. J. Nucl. Med. Mol. Imaging 2011, 38 (Suppl. 1), 10. [Google Scholar] [CrossRef] [PubMed]
- Herzog, H.; Rösch, F.; Stöcklin, G.; Lueders, C.; Qaim, S.M.; Feinendegen, L.E. Measurement of pharmacokinetics of yttrium-86 radiopharmaceuticals with PET and radiation dose calculation of analogous yttrium-90 radiotherapeutics. J. Nucl. Med. 1993, 34, 2222–2226. [Google Scholar] [PubMed]
- Rösch, F.; Herzog, H.; Plag, C.; Neumaier, B.; Braun, U.; Müller-Gärtner, H.W.; Stöcklin, G. Radiation doses of yttrium-90 citrate and yttrium-90 EDTMP as determined via analogous yttrium-86 complexes and positron emission tomography. Eur. J. Nucl. Med. 1996, 23, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Rösch, F.; Herzog, H.; Stolz, B.; Brockmann, J.; Köhle, M.; Mühlensiepen, H.; Marbach, P.; Müller-Gärtner, H.W. Uptake kinetics of the somatostatin receptor ligand [86Y]DOTA-DPhe1-Tyr3-octreotide ([86Y]SMT487) using positron emission tomography in non-human primates and calculation of radiation doses of the 90Y-labelled analogue. Eur. J. Nucl. Med. 1999, 26, 358–366. [Google Scholar] [PubMed]
- Pentlow, K.S.; Finn, R.D.; Larson, S.M.; Erdi, Y.E.; Beattie, B.J.; Humm, J.L. Quantitative imaging of yttrium-86 with PET. The occurrence and correction of anomalous apparent activity in high density regions. Clin. Positron Imaging 2000, 3, 85–90. [Google Scholar] [CrossRef]
- Walrand, S.; Jamar, F.; Mathieu, I.; De Camps, J.; Lonneux, M.; Sibomana, M.; Labar, D.; Michel, C.; Pauwels, S. Quantitation in PET using isotopes emitting prompt single gammas: Application to yttrium-86. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Kull, T.; Ruckgaber, J.; Weller, R.; Reske, S.; Glatting, G. Quantitative imaging of yttrium-86 PET with the ECAT EXACT HR+ in 2D mode. Cancer Biother. Radiopharm. 2004, 19, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, H.; Herzog, H.; Förster, G.; Reber, H.; Nickel, O.; Rösch, F.; Bartenstein, P. PET imaging with yttrium-86: Comparison of phantom measurements acquired with different PET scanners before and after applying background. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 716–720. [Google Scholar] [CrossRef] [PubMed]
- IEC IS. Radionuclide imaging devices characteristics and test conditions Part 1: Positron emission tomographs. Int. Electrotech. Comm. 1998. Available online: https://webstore.iec.ch/p-preview/info_iec61675-1%7Bed1.1%7Den.pdf (accessed on 20 June 2017).
- Walrand, S.; Flux, G.D.; Konijnenberg, M.W.; Valkema, R.; Krenning, E.P.; Lhommel, R.; Pauwels, S.; Jamar, F. Dosimetry of yttrium-labelled radiopharmaceuticals for internal therapy: 86Y or 90Y imaging? Eur. J. Nucl. Med. Mol. Imaging 2011, 38 (Suppl. 1), 57–68. [Google Scholar] [CrossRef] [PubMed]
- Beattie, B.J.; Finn, R.D.; Rowland, D.J.; Pentlow, K.S. Quantitative imaging of bromine-76 and yttrium-86 with PET: A method for the removal of spurious activity introduced by cascade gamma rays. Med. Phys. 2003, 30, 2410–2423. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, N.W. Retention of 90Y in patients with rheumatoid arthritis. Ann. Rheum. Dis. 1973, 32, 38–40. [Google Scholar] [CrossRef]
- Donaldson, S.S.; Chassagne, D.; Sancho-Garnier, H.; Beyer, H.P. Hemangiomas of infancy: Results of 90Y interstitial therapy: A retrospective study. Int. J. Radiat. Oncol. Biol. Phys. 1979, 5, 1–11. [Google Scholar] [CrossRef]
- Washburn, L.C.; Hwa Sun, T.T.; Crook, J.E.; Byrd, B.L.; Carlton, J.E.; Hung, Y.W.; Steplewski, Z.S. 90Y-labeled monoclonal antibodies for cancer therapy. Int. J. Radiat. Appl. Instrum. B 1986, 13, 453–456. [Google Scholar] [CrossRef]
- ICRP Publication. Radionuclide Transformations—Energy and Intensity of Emissions; Pergamon Press: Oxford, UK, 1983; Volume 38. [Google Scholar]
- Ford, K. Predicted 0+ level in 90Zr. Phys. Rev. 1955, 98, 1516–1517. [Google Scholar] [CrossRef]
- Johnson, O.E.; Johnson, R.G.; Langer, L.M. Evidence for a 0+ first excited state in 90Zr. Phys. Rev. 1955, 98, 1517. [Google Scholar] [CrossRef]
- Greenberg, J.; Deutsch, M. Positrons from the decay of 32P and 90Y. Phys. Rev. 1956, 102, 415–421. [Google Scholar] [CrossRef]
- Selwyn, R.G.; Nickles, R.J.; Thomadsen, B.R.; DeWerd, L.A.; Micka, J.A. A new internal pair production branching ratio of 90Y: The development of a non-destructive assay of 90Y and 90Sr. Appl. Radiat. Isot. 2007, 63, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Nickles, R.J.; Roberts, A.D.; Nye, J.A.; Converse, A.K.; Barnhart, T.E.; Avila-Rodriguez, M.A.; Sudaresan, R.; Dick, D.W.; Hammas, R.J.; Thomadsen, B.R. Assaying and PET imaging of yttrium-90: 1>>34 ppm>0. IEEE Nucl. Sci. Symp. Conf. Rec. 2004, 6, 3412–3414. [Google Scholar]
- Lhommel, R.; Goffette, P.; Van den Eynde, M.; Jamar, F.; Pauwels, S.; Bilbao, J.I.; Walrand, S. Yttrium-90 TOF PET scan demonstrates high resolution biodistribution after liver SIRT. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 1696. [Google Scholar] [CrossRef] [PubMed]
- Werner, M.K.; Brechtel, K.; Beyer, T.; Dittmann, K.; Pfannenberg, C.; Kupferschläger, J. PET/CT for the assessment and quantification of 90Y biodistribution after selective internal radiotherapy (SIRT) of liver metastases. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 407–408. [Google Scholar] [CrossRef] [PubMed]
- Lhommel, R.; van Elmbt, L.; Goffette, P.; Van den Eynde, M.; Jamar, F.; Pauwels, S.; Walrand, S. Feasibility of 90Y TOF PET-based dosimetry in liver metastasis therapy using SIR-Spheres. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 1654–1662. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, C.; Bartolomei, M.; Mattone, V.; Casi, M.; De Lauro, F.; Bartolini, N.; Gentili, G.; Amadori, S.; Agostini, M.; Sarti, G. 90Y-PET/CT imaging quantification for dosimetry in peptide receptor radionuclide therapy: Analysis and corrections of the impairing factors. Cancer Biother. Radiopharm. 2015, 30, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Bolch, W.E.; Eckerman, K.F.; Sgouros, G.; Thomas, S.R. MIRD Pamphlet No. 21: A generalized schema for radiopharmaceutical dosimetry—Standardization of nomenclature. J. Nucl. Med. 2009, 50, 477–484. [Google Scholar] [CrossRef] [PubMed]
- ICRP. The 2007 Recommendations of the International Commission on Radiological Protection. Ann. ICRP 2007, 37, 2–4. [Google Scholar]
- Snyder, W.S.; Ford, M.R.; Warner, O.G.; Watson, S.B. “S,” Absorbed Dose Per Unit Cumulated Activity for Selected Radionuclides and Organs; MIRD pamphlet no. 11; Society of Nuclear Medicine: New York, NY, USA, 1975. [Google Scholar]
- Otte, A.; Mueller-Brand, J.; Dellas, S.; Nitzsche, E.U.; Herrmann, R.; Maecke, H.R. Yttrium-90-labelled somatostatin analogue for cancer treatment. Lancet 1998, 7, 417–418. [Google Scholar] [CrossRef]
- Förster, G.J.; Engelbach, M.J.; Brockmann, J.J.; Reber, H.J.; Buchholz, H.G.; Mäcke, H.R.; Rösch, F.; Herzog, H.R.; Bartenstein, P.R. Preliminary data on biodistribution and dosimetry for therapy planning of somatostatin receptor positive tumours: Comparison of 86Y-DOTATOC and 111In-DTPA-octreotide. Eur. J. Nucl. Med. 2001, 28, 1743–1750. [Google Scholar] [CrossRef] [PubMed]
- Helisch, A.; Förster, G.J.; Reber, H.; Buchholz, H.G.; Arnold, R.; Göke, B.; Weber, M.M.; Wiedenmann, B.; Pauwels, S.; Haus, U.; et al. Pre-therapeutic dosimetry and biodistribution of 86Y-DOTA-Phe1-Tyr3-octreotide versus 111In-pentetreotide in patients with advanced neuroendocrine tumours. Eur. J. Nucl. Med. Mol. Imaging 2004, 31, 1386–1392. [Google Scholar] [CrossRef] [PubMed]
- Jamar, F.; Barone, R.; Mathieu, I.; Walrand, S.; Labar, D.; Carlier, P.; de Camps, J.; Schran, H.; Chen, T.; Smith, M.C.; et al. 86Y-DOTA-D-Phe1-Tyr3-octreotide (SMT487)—A phase 1 clinical study: Pharmacokinetics, biodistribution and renal protective effect of different regimens of amino acid co-infusion. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Barone, R.; Walrand, S.; Konijnenberg, M.; Valkema, R.; Kvols, L.K.; Krenning, E.P.; Pauwels, S.; Jamar, F. Therapy using labelled somatostatin analogues: Comparison of the absorbed doses with 111In-DTPA-D-Phe1-octreotide and yttrium-labelled DOTA-D-Phe1-Tyr3-octreotide. Nucl. Med. Commun. 2008, 29, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Stolz, B.; Bruns, C.; Albert, R.; Rösch, F.; Smith-Jones, P.; Raulf, F.; Hoyer, D.; Weckbecker, G. Somatostatin receptor-targeted radiotherapy—Preclinical proof of concept. In Octreotide: The Next Decade; BioScientifica Ltd.: Lamberts, UK, 1999; p. 1. [Google Scholar]
- Forrer, F.; Waldherr, C.; Maecke, H.R.; Mueller-Brand, J. Targeted radionuclide therapy with 90Y-DOTATOC in patients with neuroendocrine tumors. Anticancer Res. 2006, 26, 703–707. [Google Scholar] [PubMed]
- Vinjamuri, S.; Gilbert, T.M.; Banks, M.; McKane, G.; Maltby, P.; Poston, G.; Weissman, H.; Palmer, D.H.; Vora, J.; Pritchard, D.M.; et al. Peptide receptor radionuclide therapy with 90Y-DOTATATE/90Y-DOTATOC in patients with progressive metastatic neuroendocrine tumours: Assessment of response, survival and toxicity. Br. J. Cancer 2013, 16, 1440–1448. [Google Scholar] [CrossRef] [PubMed]
- Nayak, T.K.; Brechbiel, M.W. 86Y based PET radiopharmaceuticals: Radiochemistry and biological applications. Med. Chem. 2011, 7, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Cui, L.; Wang, F.; Liu, Z. PET tracers based on 86Y. Curr. Radiopharm. 2011, 4, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Biddlecombe, G.B.; Rogers, B.E.; de Visser, M.; Parry, J.J.; de Jong, M.; Erion, J.L.; Lewis, J.S. Molecular imaging of gastrin-releasing peptide receptor-positive tumors in mice using 64Cu and 86Y-DOTA-(Pro1, Tyr4)-bombesin (1–14). Bioconj. Chem. 2007, 18, 724–730. [Google Scholar] [CrossRef] [PubMed]
- McQuade, P.; Miao, Y.; Yoo, J.; Quinn, T.P.; Welch, M.J.; Lewis, J.S. Imaging of melanoma using 64Cu- and 86Y-DOTA-ReCCMSH(Arg11), a cyclized peptide analogue of alpha-MSH. J. Med. Chem. 2005, 48, 2985–2992. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Zhang, X.; Gallazzi, F.; Miao, Y.; Jin, X.; Brechbiel, M.W.; Xu, H.; Clifford, T.; Welch, M.J.; Lewis, J.S.; et al. Melanoma imaging using 111In-, 86Y- and 68Ga-labeled CHX-A″-Re(Arg11)CCMSH. Nucl. Med. Biol. 2009, 36, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.R.; Foss, C.A.; Pullambhatla, M.; Wang, Y.; Srinivasan, S.; Hobbs, R.F.; Baidoo, K.E.; Brechbiel, M.W.; Nimmagadda, S.; Mease, R.C.; et al. Preclinical evaluation of 86Y-labeled inhibitors of prostate- specific membrane antigen for dosimetry estimates. J. Nucl. Med. 2015, 56, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Lovqvist, A.; Humm, J.L.; Sheikh, A.; Finn, R.D.; Koziorowski, J.; Ruan, S.; Pentlow, K.S.; Jungbluth, A.; Welt, S.; Lee, F.T.; et al. PET imaging of 86Y-labeled anti-lewis Y monoclonal antibodies in a nude mouse model: Comparison between 86Y and 111In radiolabels. J. Nucl. Med. 2001, 42, 1281–1287. [Google Scholar] [PubMed]
- Nayak, T.K.; Regino, C.A.; Wong, K.J.; Milenic, D.E.; Garmestani, K.; Baidoo, K.E.; Szajek, L.P.; Brechbiel, M.W. PET imaging of HER1-expressing xenografts in mice with 86Y-CHX-A″-DTPA-cetuximab. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 1368–1376. [Google Scholar] [CrossRef] [PubMed]
- Nayak, T.K.; Garmestani, K.; Milenic, D.E.; Baidoo, K.E.; Brechbiel, M.W. HER1-targeted 86Y-panitumumab possesses superior targeting characteristics than 86Y-cetuximab for PET imaging of human malignant mesothelioma tumors xenografts. PLoS ONE 2011, 6, e18198. [Google Scholar] [CrossRef] [PubMed]
- Nayak, T.K.; Garmestani, K.; Baidoo, K.E.; Milenic, D.E.; Brechbiel, M.W. Preparation, biological evaluation, and pharmacokinetics of the human anti-HER1 monoclonal antibody panitumumab labeled with 86Y for quantitative PET of carcinoma. J. Nucl. Med. 2010, 51, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Schneider, D.W.; Heitner, T.; Alicke, B.; Light, D.R.; McLean, K.; Satozawa, N.; Perry, G.K.; Yoo, J.; Lewis, J.S.; Parry, R. In vivo biodistribution, PET imaging, and tumor accumulation of Y-86- and In-111-antimindin/RG-1, engineered antibody fragments in LNCaP tumor-bearing nude mice. Int. J. Cancer 2011, 128, 920–926. [Google Scholar]
- Wong, K.J.; Baidoo, K.E.; Nayak, T.K.; Garmestani, K.; Brechbiel, M.W.; Milenic, D.E. In vitro and in vivo pre-clinical analysis of a F(ab′)2 fragment of panitumumab for molecular imaging and therapy of HER1 positive cancers. EJNMMI Res. 2011, 1. [Google Scholar] [CrossRef] [PubMed]
- McDevitt, M.R.; Chattopadhyay, D.; Jaggi, J.S.; Finn, R.D.; Zanzonico, P.B.; Rey, D.; Mendenhall, J.; Batt, C.A.; Njardson, J.T.; Scheinberg, D.A. PET imaging of soluble yttrium-86-labeled carbon nanotubes in mice. PLoS ONE 2007, 19, e907. [Google Scholar] [CrossRef] [PubMed]
- Cheal, S.M.; Xu, H.; Guo, H.-F.; Lee, S.-G.; Punzalan, B.; Chalasani, S.; Fung, E.K.; Jungbluth, A.; Zanzonico, P.B.; Carrasquillo, J.A.; et al. Theranostic pretargeted radioimmunotherapy of colorectal cancer xenografts in mice using picomolar affinity 86Y-or 177Lu-DOTA-Bn binding scFv C825/GPA33 IgG bispecific immunoconjugates. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 925–937. [Google Scholar] [CrossRef] [PubMed]
- Palm, S.; Enmon, R.M., Jr.; Matei, C.; Kolbert, K.S.; Xu, S.; Zanzonico, P.B.; Finn, R.L.; Koutcher, J.A.; Larson, S.M.; Sgouros, G. Pharmacokinetics and biodistribution of 86Y-trastuzumab for 90Y dosimetry in an ovarian carcinoma model: Correlative MicroPET and MRI. J. Nucl. Med. 2003, 44, 1148–1155. [Google Scholar] [PubMed]
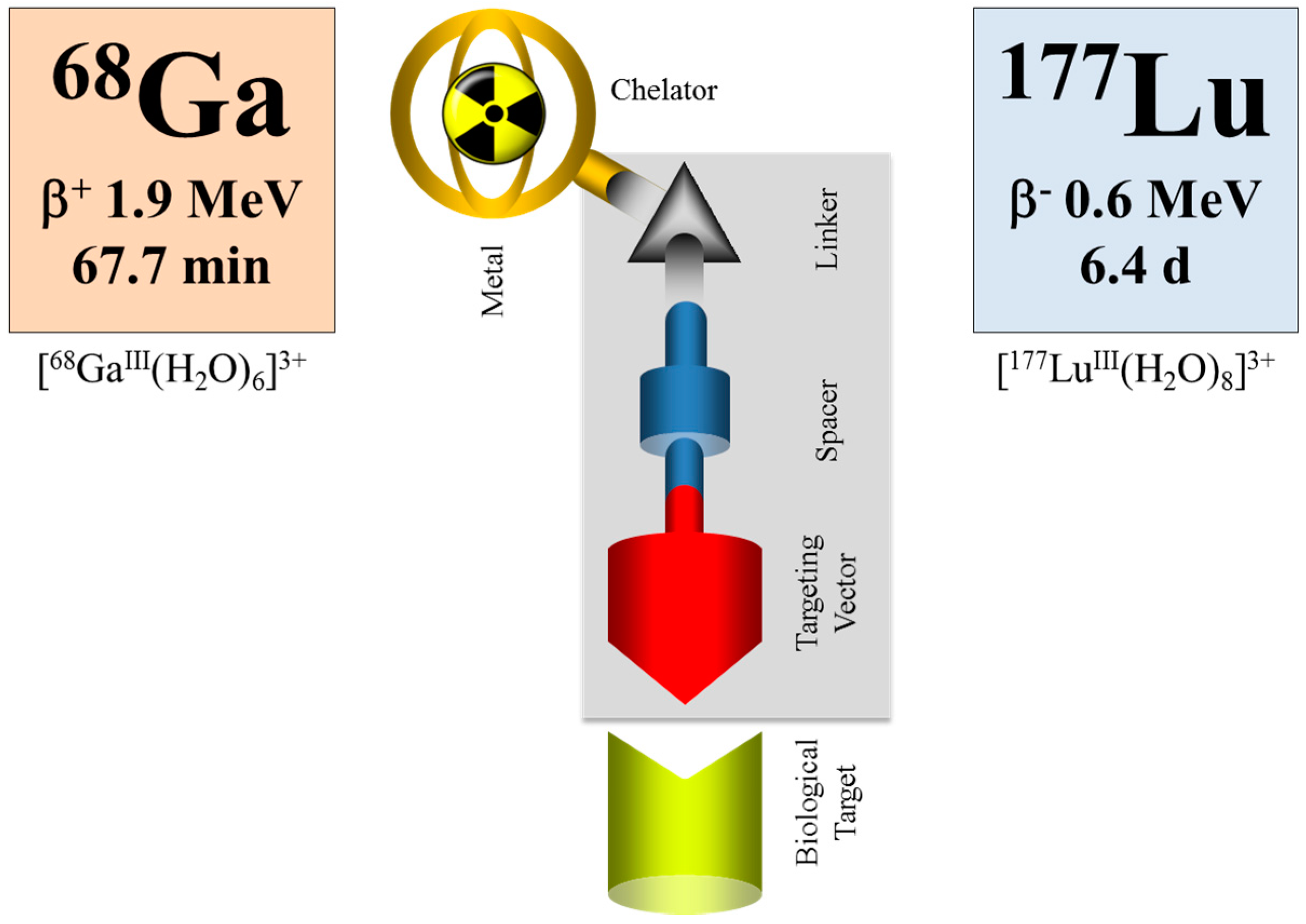
- Baum, R.B.; Rösch, F. Theranostics, Gallium-68, and Other Radionuclides: A Pathway to Personalized Diagnosis and Treatment; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Rösch, F.; Baum, R.P. Generator-based PET radiopharmaceuticals for molecular imaging of tumours: On the way to THERANOSTICS. Dalton Trans. 2011, 40, 6104–6111. [Google Scholar] [CrossRef] [PubMed]
- Velikyan, I. Molecular imaging and radiotherapy: Theranostics for personalized patient management. Theranostics 2012, 2, 424–426. [Google Scholar] [CrossRef] [PubMed]
- Fani, M.; Pozzo, L.D.; Abiraj, K.; Mansi, R.; Tamma, M.L.; Cescato, R.; Waser, B.; Weber, W.A.; Reubi, J.C.; Mäcke, H.R. PET of Somatostatin Receptor-Positive Tumors using 64Cu- and 68Ga-somatostatin antagonists: The chelate makes the difference. J. Nucl. Med. 2011, 52, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Poschenrieder, A.; Schottelius, M.; Schwaiger, M.; Kessler, H.; Wester, H.-J. The influence of diferent metal-chelate conjugates of pentixafor on the CXCR4 affinity. EJNMMI Res. 2016, 16, 36. [Google Scholar] [CrossRef] [PubMed]
- Filosofov, D.V.; Loktionova, N.S.; Rösch, F. A 44Ti/44Sc radionuclide generator for potential application of 44Sc-based PET-radiopharmaceuticals. Radiochim. Acta 2010, 98, 149–156. [Google Scholar] [CrossRef]
- Rösch, F. Scandium-44: Benefits of a long-lived PET radionuclide available from the 44Ti/44Sc generator system. Curr. Radiopharm. 2012, 5, 187–201. [Google Scholar] [CrossRef]
- Müller, C.; Bunka, M.; Haller, S.; Köster, U.; Groehn, V.; Bernhardt, P.; van der Meulen, N.; Türler, A.; Schibli, R. Promising prospects for 44Sc-/47Sc-based theragnostics: Application of 47Sc for radionuclide tumor therapy in mice. J. Nucl. Med. 2014, 55, 1658–1664. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Radionuclide | t½ | Mode of Decay (%) | Eβ(max) [keV] | Iβ [%] | Eγ [keV] | Iγ [%] |
|---|---|---|---|---|---|---|
| 90Y | 2.7 d | βˉ (100) | 2290 | 100 | ||
| 86Y | 14.7 h | EC (67) β+ (33) | 1043 1248 1603 2019 2335 3153 | 4.6 16.0 6.2 4.4 1.3 0.6  Σ ca. 33 | 443.1 515.4 580.5 627.8 645.8 703.3 777.6 1076.7 1153.2 1854.3 1920.8 | 16.9 4.9 4.8 32.6 9.2 15.4 22.4 82.5 30.5 17.2 20.8 |
| Nuclear Reaction | Energy Range | Thick Target Yield of 86Y | Radionuclidic Impurities (%) | ||||
|---|---|---|---|---|---|---|---|
| (MeV) | (MBq/μAh) | 85Y (t½ = 2.7 h) | 85mY (t½ = 4.9 h) | 87gY (t½ = 3.1 d) | 87mY (t½ = 13.4 h) | 88Y (t½ = 106.6 d) | |
| 86Sr(p,n)86Y a | 14→7 | 371 | <0.1 | <0.1 | 0.4 d | 1–3 d | 0.1 d |
| 88Sr(p,3n)86Y a | 43→33 | 1005 | 35.5 | 2.8 | 5.4 | 27.7 | 0.04 |
| natRb(3He,xn)86Y b | 24→12 | 190 | 71 | 208 | 12 | 46 | 0.2 d |
| natZr(p,x)86Y c | 40→25 | 50 | unknown | unknown | 46 | 6 | 0.3 |
| Method | Typical Irradiation Conditions | Efficiency of Chemical Separation (%) | Typical Production Batch Yield (GBq) | Chemical Impurities (ng/mL) | Ref. | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Energy Range (MeV) | Beam Current (μA) | Time of Irradiation (h) | Fe | Sr | Y | La | ||||
| Coprecipitation and ion-exchange | 12→8 16→10 15→6 | 4 6 - | 3 4 - | 90 90 - | 1.4 3.5 1.3 | 2.3 | 2.6 | <0.02 | <0.1 | [16] [28] [26] |
| Electrolysis | 15.1→0 14.5→6 14.5→6 14.2→8 | 10 2 4 5 | 2.5 4 1 0.5 | 90 90 90 91 | 1.2 1.3 a 0.34 0.36 | <100 700 | [27] [31] [31] [33] | |||
| Single column cation-exchange | 16→12 45→39 | 1 25 | 3 0.5 | 91 90 | 0.45 5.0 b | 2 × 105 | 500 2 × 106 | [34] [41] | ||
| Multiple column chromatography | 13.8→6.1 | 10 | 1 | 80 | 0.70 | [29] | ||||
| Solvent extraction | 16→10 | 1 | 3 | 89 | 0.48 c | 1 × 103 | [34] | |||
| Precipitation | 11→6.1 | 10 | 2 | 88 | 0.89 | 1.5 × 104 | [32] | |||
| Organ | Dose per Organ (mGy/MBq) | |||
|---|---|---|---|---|
| Baboon Study Rösch et al., [47] | Baboon Study Rösch et al., with Amino Acid Cocktail Co-infusion [47] | Human Study Förster et al., [72] | Human Study Helisch et al., [73] | |
| Intestine wall | - | - | 0.05 ± 0.002 | - |
| Kidneys | 2.81 | 2.11 | 2.73 ± 1.41 | 1.71 ± 0.89 |
| Liver | 0.32 | 0.34 | 0.660 ± 0.002 | 0.72 ± 0.04 |
| Other tissue | - | - | 0.05 ± 0.002 | - |
| Red marrow | 0.07 | 0.03 | 0.05 ± 0.002 | 0.60 ± 0.02 |
| Spleen | - | - | 2.32 ± 1.97 | 2.19 ± 1.11 |
| Urinary bladder wall | 0.64 | 0.65 | 1.03 ± 0.23 | - |
| Effective dose (mSv/MBq) | 0.25 | 0.23 | 0.22 ± 0.07 | 0.17 ± 0.10 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rösch, F.; Herzog, H.; Qaim, S.M. The Beginning and Development of the Theranostic Approach in Nuclear Medicine, as Exemplified by the Radionuclide Pair 86Y and 90Y. Pharmaceuticals 2017, 10, 56. https://doi.org/10.3390/ph10020056
Rösch F, Herzog H, Qaim SM. The Beginning and Development of the Theranostic Approach in Nuclear Medicine, as Exemplified by the Radionuclide Pair 86Y and 90Y. Pharmaceuticals. 2017; 10(2):56. https://doi.org/10.3390/ph10020056
Chicago/Turabian StyleRösch, Frank, Hans Herzog, and Syed M. Qaim. 2017. "The Beginning and Development of the Theranostic Approach in Nuclear Medicine, as Exemplified by the Radionuclide Pair 86Y and 90Y" Pharmaceuticals 10, no. 2: 56. https://doi.org/10.3390/ph10020056
APA StyleRösch, F., Herzog, H., & Qaim, S. M. (2017). The Beginning and Development of the Theranostic Approach in Nuclear Medicine, as Exemplified by the Radionuclide Pair 86Y and 90Y. Pharmaceuticals, 10(2), 56. https://doi.org/10.3390/ph10020056