
The Good the Bad and the Ugly of Glycosaminoglycans in Tissue Engineering Applications


Abstract
:1. Introduction
2. Articular Cartilage
2.1. Formation
2.2. Disease and Trauma
2.3. Current Therapies
3. Mesenchymal Stem Cells
3.1. Isolation and Characterisation of MSCs
3.2. MSC Heterogeneity
3.3. In Vitro Chondrogenic Differentiation of MSCs
4. Growth Factors Involved in the Chondrogenic Differentiation of MSCs
4.1. Transforming Growth Factor Beta (TGFβ) Superfamily
4.1.1. TGFβ Subfamily
4.1.2. BMP Subfamily
4.1.2.1. GDF5
5. Glycosaminoglycans
Role of GAGs in Stem Cell Differentiation and Development
6. Biomaterials
6.1. Electrospun Scaffolds
6.2. Hydrogels
6.3. GAG Incorporation and Application
7. The Problems Associated with Heparin
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bhosale, M.A.; Richardson, J.B. Articular cartilage: Structure, injuries and review of management. Br. Med. Bull. 2008, 87, 77–95. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, T.E. Fell-Muir lecture: Cartilage 2010—The known unknowns. Int. J. Exp. Pathol. 2010, 91, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Correa, D.; Lietman, S.A. Articular cartilage repair: Current needs, methods and research directions. Semin. Cell Dev. Bio. 2016, 62, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Goldring, M.B. Chondrogenesis, chondrocyte differentiation, and articular cartilage metabolism in health and osteoarthritis. Ther. Adv. Musculoskelet. Dis. 2012, 4, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Schinagl, R.M.; Gurskis, D.; Chen, A.C.; Sah, R.L. Depth-dependent confined compression modulus of full-thickness bovine articular cartilage. J. Orthop. Res. 1997, 15, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Seror, J.; Zhu, L.; Goldberg, R.; Day, A.J.; Klein, J. Supramolecular synergy in the boundary lubrication of synovial joints. Nat. Commun. 2015, 6, 6497. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.H.; Kudva, A.K.; Guckert, N.L.; Linse, K.D.; Roy, K. Unique biomaterial compositions direct bone marrow stem cells into specific chondrocytic phenotypes corresponding to the various zones of articular cartilage. Biomaterials 2011, 32, 1327–1338. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.H.; Kudva, A.K.; Saxena, N.S.; Roy, K. Engineering articular cartilage with spatially-varying matrix composition and mechanical properties from a single stem cell population using a multi-layered hydrogel. Biomaterials 2011, 32, 6946–6952. [Google Scholar] [CrossRef] [PubMed]
- Boschetti, F.; Peretti, G.M. Tensile and compressive properties of healthy and osteoarthritic human articular cartilage. Biorheology 2008, 45, 337–344. [Google Scholar] [PubMed]
- Yang, N.; Meng, Q.J. Circadian Clocks in Articular Cartilage and Bone: A Compass in the Sea of Matrices. J. Biol. Rhythm. 2016, 31, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Shen, G. The role of type X collagen in facilitating and regulating endochondral ossification of articular cartilage. Orthod. Craniofac. Res. 2005, 8, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Decker, R.S.; Koyama, E.; Pacifici, M. Genesis and morphogenesis of limb synovial joints and articular cartilage. Matrix Biol. 2014, 39, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Mitrovic, D. Development of the diarthrodial joints in the rat embryo. Am. J. Anat. 1978, 151, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Hunziker, E.B. Growth plate structure and function. Pathol. Immunopathol. Res. 1988, 7, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.K.; Miyake, T. The membranous skeleton: The role of cell condensations in vertebrate skeletogenesis. Anat. Embryol. (Berl) 1992, 186, 107–124. [Google Scholar] [CrossRef] [PubMed]
- Kronenberg, H.M. Developmental regulation of the growth plate. Nature 2003, 423, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Long, F.; Ornitz, D.M. Development of the endochondral skeleton. Cold Spring Harb. Perspect. Biol. 2013, 5, a008334. [Google Scholar] [CrossRef] [PubMed]
- Kozhemyakina, E.; Lassar, A.B.; Zelzer, E. A pathway to bone: Signaling molecules and transcription factors involved in chondrocyte development and maturation. Development 2015, 142, 817–831. [Google Scholar] [CrossRef] [PubMed]
- Camarero-Espinosa, S.; Rothen-Rutishauser, B.; Foster, E.J.; Weder, C. Articular cartilage: From formation to tissue engineering. Biomater. Sci. 2016, 4, 734–767. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Huang, X.; Karperien, M.; Post, J.N. The Regulatory Role of Signaling Crosstalk in Hypertrophy of MSCs and Human Articular Chondrocytes. Int. J. Mol. Sci. 2015, 16, 19225–19247. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, M.; Ohta, Y.; Larmour, C.; Enomoto-Iwamoto, M. Toward regeneration of articular cartilage. Birth Defects Res. C Embryo Today 2013, 99, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Pacifici, M.; Koyama, E.; Shibukawa, Y.; Wu, C.; Tamamura, Y.; Enomoto-Iwamoto, M.; Iwamoto, M. Cellular and molecular mechanisms of synovial joint and articular cartilage formation. Ann. N. Y. Acad. Sci. 2006, 1068, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, M.; Tamamura, Y.; Koyama, E.; Komori, T.; Takeshita, N.; Williams, J.A.; Nakamura, T.; Enomoto-Iwamoto, M.; Pacifici, M. Transcription factor ERG and joint and articular cartilage formation during mouse limb and spine skeletogenesis. Dev. Biol. 2007, 305, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Koyama, E.; Shibukawa, Y.; Nagayama, M.; Sugito, H.; Young, B.; Yuasa, T.; Okabe, T.; Ochiai, T.; Kamiya, N.; Rountree, R.B.; et al. A distinct cohort of progenitor cells participates in synovial joint and articular cartilage formation during mouse limb skeletogenesis. Dev. Biol. 2008, 316, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Rountree, R.B.; Schoor, M.; Chen, H.; Marks, M.E.; Harley, V.; Mishina, Y.; Kingsley, D.M. BMP receptor signaling is required for postnatal maintenance of articular cartilage. PLoS Biol. 2004, 2, e355. [Google Scholar] [CrossRef] [PubMed]
- Hyde, G.; Dover, S.; Aszodi, A.; Wallis, G.A.; Boot-Handford, R.P. Lineage tracing using matrilin-1 gene expression reveals that articular chondrocytes exist as the joint interzone forms. Dev. Biol. 2007, 304, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Von der Mark, K.; Kirsch, T.; Nerlich, A.; Kuss, A.; Weseloh, G.; Glückert, K.; Stöss, H. Type X collagen synthesis in human osteoarthritic cartilage. Indication of chondrocyte hypertrophy. Arthritis Rheum. 1992, 35, 806–811. [Google Scholar] [CrossRef] [PubMed]
- St-Jacques, B.; Hammerschmidt, M.; McMahon, A.P. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999, 13, 2072–2086. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, C.A.; Yamamoto, H.; Fujita, T.; Furuichi, T.; Ito, K.; Inoue, K.; Yamana, K.; Zanma, A.; Takada, K.; Ito, Y.; Komori, T. Runx2 and Runx3 are essential for chondrocyte maturation, and Runx2 regulates limb growth through induction of Indian hedgehog. Genes Dev. 2004, 18, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.B.; Tuan, R.S. Functional characterization of hypertrophy in chondrogenesis of human mesenchymal stem cells. Arthritis Rheum. 2008, 58, 1377–1388. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, E.; Selvamurugan, N.; Westendorf, J.J.; Olson, E.N.; Partridge, N.C. HDAC4 represses matrix metalloproteinase-13 transcription in osteoblastic cells, and parathyroid hormone controls this repression. J. Biol. Chem. 2010, 285, 9616–9626. [Google Scholar] [CrossRef] [PubMed]
- Davoli, M.A.; Lamplugh, L.; Beauchemin, A.; Chan, K.; Mordier, S.; Mort, J.S.; Murphy, G.; Docherty, A.J.; Leblond, C.P.; Lee, E.R. Enzymes active in the areas undergoing cartilage resorption during the development of the secondary ossification center in the tibiae of rats aged 0–21 days: II. Two proteinases, gelatinase B and collagenase-3, are implicated in the lysis of collagen fibrils. Dev. Dyn. 2001, 222, 71–88. [Google Scholar] [PubMed]
- Yang, L.; Tsang, K.Y.; Tang, H.C.; Chan, D.; Cheah, K.S. Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc. Natl. Acad. Sci. USA 2014, 111, 12097–12102. [Google Scholar] [CrossRef] [PubMed]
- Drissi, H.; Zuscik, M.; Rosier, R.; O'Keefe, R. Transcriptional regulation of chondrocyte maturation: Potential involvement of transcription factors in OA pathogenesis. Mol. Aspects Med. 2005, 26, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chen, J.; Zhang, S.; Ouyang, H.W. Inhibitory function of parathyroid hormone-related protein on chondrocyte hypertrophy: The implication for articular cartilage repair. Arthritis Res. Ther. 2012, 14, 221. [Google Scholar] [PubMed]
- Johnstone, B.; Hering, T.M.; Caplan, A.I.; Goldberg, V.M.; Yoo, J.U. In vitro chondrogenesis of bone marrow-derived mesenchymal progenitor cells. Exp. Cell Res. 1998, 238, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.U.; Barthel, T.S.; Nishimura, K.; Solchaga, L.; Caplan, A.I.; Goldberg, V.M.; Johnstone, B. The chondrogenic potential of human bone-marrow-derived mesenchymal progenitor cells. J. Bone Joint Surg. Am. 1998, 80, 1745–1757. [Google Scholar] [CrossRef] [PubMed]
- Pelttari, K.; Winter, A.; Steck, E.; Goetzke, K.; Hennig, T.; Ochs, B.G.; Aigner, T.; Richter, W. Premature induction of hypertrophy during in vitro chondrogenesis of human mesenchymal stem cells correlates with calcification and vascular invasion after ectopic transplantation in SCID mice. Arthritis Rheum. 2006, 54, 3254–3266. [Google Scholar] [CrossRef] [PubMed]
- Kafienah, W.; Mistry, S.; Dickinson, S.C.; Sims, T.J.; Learmonth, I.; Hollander, A.P. Three-dimensional cartilage tissue engineering using adult stem cells from osteoarthritis patients. Arthritis Rheum. 2007, 56, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Kreuz, P.C.; Steinwachs, M.; Erggelet, C.; Krause, S.J.; Ossendorf, C.; Maier, D.; Ghanem, N.; Uhl, M.; Haag, M. Classification of graft hypertrophy after autologous chondrocyte implantation of full-thickness chondral defects in the knee. Osteoarthr. Cartil. 2007, 15, 1339–1347. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, A.D.; Grady, L.M.; Ablett, M.P.; Katopodi, T.; Meadows, R.S.; Hardingham, T.E. Chondrogenic differentiation of human bone marrow stem cells in transwell cultures: Generation of scaffold-free cartilage. Stem. Cells 2007, 25, 2786–2796. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Fu, P.; Cong, R.; Wu, H.; Pei, M. Strategies to minimize hypertrophy in cartilage engineering and regeneration. Genes Dis. 2015, 2, 76–95. [Google Scholar] [CrossRef] [PubMed]
- Hunziker, E.B.; Lippuner, K.; Keel, M.J.; Shintani, N. An educational review of cartilage repair: Precepts & practice-myths & misconceptions-progress & prospects. Osteoarthr. Cartil. 2015, 23, 334–350. [Google Scholar] [PubMed]
- Guilak, F. Biomechanical factors in osteoarthritis. Best Pract. Res. Clin. Rheumatol. 2011, 25, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Antony, B.; Jones, G.; Jin, X.; Ding, C. Do early life factors affect the development of knee osteoarthritis in later life: A narrative review. Arthritis Res. Ther. 2016, 18, 202. [Google Scholar] [CrossRef] [PubMed]
- Fosang, A.J.; Beier, F. Emerging Frontiers in cartilage and chondrocyte biology. Best Pract. Res. Clin. Rheumatol. 2011, 25, 751–766. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Kovic, B.; Jin, X.; He, X.; Wang, M.; Silvestre, C. Economic and Humanistic Burden of Osteoarthritis: A Systematic Review of Large Sample Studies. Pharmacoeconomics 2016, 34, 1087–1100. [Google Scholar] [CrossRef] [PubMed]
- Osteoarthritis in General Practice; Arthritis Research UK: Scotland, UK, 2013.
- Cross, M.; Smith, E.; Hoy, D.; Nolte, S.; Ackerman, I.; Fransen, M.; Bridgett, L.; Williams, S.; Guillemin, F.; Hill, C.L.; et al. The global burden of hip and knee osteoarthritis: Estimates from the global burden of disease 2010 study. Ann. Rheum. Dis. 2014, 73, 1323–1330. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, K.; Karssiens, T.; Kumar, V.; Pandit, H. Obesity and osteoarthritis. Maturitas 2016, 89, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.D.; Siston, R.A.; Pan, X.; Flanigan, D.C. Autologous chondrocyte implantation: A systematic review. J. Bone Joint Surg. Am. 2010, 92, 2220–2233. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Merchan, E.C. Regeneration of articular cartilage of the knee. Rheumatol. Int. 2013, 33, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Tibesku, C.O.; Szuwart, T.; Kleffner, T.O.; Schlegel, P.M.; Jahn, U.R.; Van Aken, H.; Fuchs, S. Hyaline cartilage degenerates after autologous osteochondral transplantation. J. Orthop. Res. 2004, 22, 1210–1214. [Google Scholar] [CrossRef] [PubMed]
- Brittberg, M.; Lindahl, A.; Nilsson, A.; Ohlsson, C.; Isaksson, O.; Peterson, L. Treatment of deep cartilage defects in the knee with autologous chondrocyte transplantation. N. Engl. J. Med. 1994, 331, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Marlovits, S.; Striessnig, G.; Kutscha-Lissberg, F.; Resinger, C.; Aldrian, S.M.; Vecsei, V.; Trattnig, S. Early postoperative adherence of matrix-induced autologous chondrocyte implantation for the treatment of full-thickness cartilage defects of the femoral condyle. Knee Surg. Sports Traumatol. Arthrosc. 2005, 13, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Gobbi, A.; Kon, E.; Berruto, M.; Francisco, R.; Filardo, G.; Marcacci, M. Patellofemoral full-thickness chondral defects treated with Hyalograft-C: A clinical, arthroscopic, and histologic review. Am. J. Sports Med. 2006, 34, 1763–1773. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.J.; Malek, M.A.; Frassica, F.J.; Polder, J.A.; Mohan, A.K.; Bloom, E.T.; Braun, M.M.; Cote, T.R. Autologous cultured chondrocytes: Adverse events reported to the United States Food and Drug Administration. J. Bone Joint Surg. Am. 2006, 88, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Benya, P.D.; Padilla, S.R.; Nimni, M.E. Independent regulation of collagen types by chondrocytes during the loss of differentiated function in culture. Cell 1978, 15, 1313–1321. [Google Scholar] [CrossRef]
- Tew, S.R.; Murdoch, A.D.; Rauchenberg, R.P.; Hardingham, T.E. Cellular methods in cartilage research: Primary human chondrocytes in culture and chondrogenesis in human bone marrow stem cells. Methods 2008, 45, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Ebert, J.R.; Smith, A.; Fallon, M.; Butler, R.; Nairn, R.; Breidahl, W.; Wood, D.J. Incidence, degree, and development of graft hypertrophy 24 months after matrix-induced autologous chondrocyte implantation: Association with clinical outcomes. Am. J. Sports Med. 2015, 43, 2208–2215. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.A.; Buckwalter, J.A. Telomere erosion and senescence in human articular cartilage chondrocytes. J. Gerontol. A Biol. Sci. Med. Sci. 2001, 56, B172–B179. [Google Scholar] [CrossRef] [PubMed]
- Kuszel, L.; Trzeciak, T.; Richter, M.; Czarny-Ratajczak, M. Osteoarthritis and telomere shortening. J. Appl. Genet. 2015, 56, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.R. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. USA 1981, 78, 7634–7638. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.L.; Hilton, D.J.; Pease, S.; Willson, T.A.; Stewart, C.L.; Gearing, D.P.; Wagner, E.F.; Metcalf, D.; Nicola, N.A.; Gough, N.M. Myeloid leukaemia inhibitory factor maintains the developmental potential of embryonic stem cells. Nature 1988, 336, 684–687. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.T.; Southerland, S.S.; Souza, J.; Calcutt, A.F.; Cartledge, R.G. Cells isolated from adult human skeletal muscle capable of differentiating into multiple mesodermal phenotypes. Am. Surg. 1999, 65, 22–26. [Google Scholar] [PubMed]
- Beane, O.S.; Darling, E.M. Isolation, characterization, and differentiation of stem cells for cartilage regeneration. Ann. Biomed. Eng. 2012, 40, 2079–2097. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Ichisaka, T.; Yamanaka, S. Generation of germline-competent induced pluripotent stem cells. Nature 2007, 448, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Shih, C.C.; Forman, S.J.; Chu, P.; Slovak, M. Human embryonic stem cells are prone to generate primitive, undifferentiated tumors in engrafted human fetal tissues in severe combined immunodeficient mice. Stem. Cells Dev. 2007, 16, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Swijnenburg, R.J.; Schrepfer, S.; Govaert, J.A.; Cao, F.; Ransohoff, K.; Sheikh, A.Y.; Haddad, M.; Connolly, A.J.; Davis, M.M.; Robbins, R.C.; et al. Immunosuppressive therapy mitigates immunological rejection of human embryonic stem cell xenografts. Proc. Natl. Acad. Sci. USA 2008, 105, 12991–12996. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Aranda, I.; Ramos-Mejia, V.; Bueno, C.; Munoz-Lopez, M.; Real, P.J.; Macia, A.; Sanchez, L.; Ligero, G.; Garcia-Parez, J.L.; Menendez, P. Human induced pluripotent stem cells develop teratoma more efficiently and faster than human embryonic stem cells regardless the site of injection. Stem. Cells 2010, 28, 1568–1570. [Google Scholar] [CrossRef] [PubMed]
- Polo, J.M.; Liu, S.; Figueroa, M.E.; Kulalert, W.; Eminli, S.; Tan, K.Y.; Apostolou, E.; Stadtfeld, M.; Li, Y.; Shioda, T.; et al. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat. Biotechnol. 2010, 28, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, B. Reprogramming barriers and enhancers: Strategies to enhance the efficiency and kinetics of induced pluripotency. Cell Regen. (Lond.) 2015, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Vonk, L.A.; de Windt, T.S.; Slaper-Cortenbach, I.C.; Saris, D.B. Autologous, allogeneic, induced pluripotent stem cell or a combination stem cell therapy? Where are we headed in cartilage repair and why: A concise review. Stem. Cell Res. Ther. 2015, 6, 94. [Google Scholar] [CrossRef] [PubMed]
- Martin, I.; Ireland, H.; Baldomero, H.; Dominici, M.; Saris, D.B.; Passweg, J. The Survey on Cellular and Engineered Tissue Therapies in Europe in 2013. Tissue Eng. Part A 2016, 22, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Jahagirdar, B.N.; Reinhardt, R.L.; Schwartz, R.E.; Keene, C.D.; Ortiz-Gonzalez, X.R.; Reyes, M.; Lenvik, T.; Lund, T.; Blackstad, M.; et al. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature 2002, 418, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Ham, O.; Song, B.W.; Lee, S.Y.; Choi, E.; Cha, M.J.; Lee, C.Y.; Park, J.H.; Kim, I.K.; Chang, W.; Lim, S.; et al. The role of microRNA-23b in the differentiation of MSC into chondrocyte by targeting protein kinase A signaling. Biomaterials 2012, 33, 4500–4507. [Google Scholar] [CrossRef] [PubMed]
- Monsel, A.; Zhu, Y.G.; Gennai, S.; Hao, Q.; Liu, J.; Lee, J.W. Cell-based therapy for acute organ injury: Preclinical evidence and ongoing clinical trials using mesenchymal stem cells. Anesthesiology 2014, 121, 1099–1121. [Google Scholar] [CrossRef] [PubMed]
- Le Blanc, K.; Tammik, C.; Rosendahl, K.; Zetterberg, E.; Ringden, O. HLA expression and immunologic properties of differentiated and undifferentiated mesenchymal stem cells. Exp. Hematol. 2003, 31, 890–896. [Google Scholar] [CrossRef]
- Chamberlain, G.; Fox, J.; Ashton, B.; Middleton, J. Concise review: Mesenchymal stem cells: Their phenotype, differentiation capacity, immunological features, and potential for homing. Stem. Cells. 2007, 25, 2739–2749. [Google Scholar] [CrossRef] [PubMed]
- Uccelli, A.; Moretta, L.; Pistoia, V. Mesenchymal stem cells in health and disease. Nat. Rev. Immunol. 2008, 8, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Atoui, R.; Chiu, R.C. Mesenchymal stromal cells as universal donor cells. Exp. Opin. Biol. Ther. 2012, 12, 1293–1297. [Google Scholar] [CrossRef] [PubMed]
- Le Blanc, K.; Rasmusson, I.; Sundberg, B.; Gotherstrom, C.; Hassan, M.; Uzunel, M.; Ringden, O. Treatment of severe acute graft-versus-host disease with third party haploidentical mesenchymal stem cells. Lancet 2004, 363, 1439–1441. [Google Scholar] [CrossRef]
- Ra, J.C.; Kang, S.K.; Shin, I.S.; Park, H.G.; Joo, S.A.; Kim, J.G.; Kang, B.C.; Lee, Y.S.; Nakama, K.; Piao, M.; et al. Stem cell treatment for patients with autoimmune disease by systemic infusion of culture-expanded autologous adipose tissue derived mesenchymal stem cells. J. Transl. Med. 2011, 9, 181. [Google Scholar] [PubMed]
- Wang, L.; Wang, L.; Cong, X.; Liu, G.; Zhou, J.; Bai, B.; Li, Y.; Bai, W.; Li, M.; Ji, H.; et al. Human umbilical cord mesenchymal stem cell therapy for patients with active rheumatoid arthritis: Safety and efficacy. Stem. Cells Dev. 2013, 22, 3192–3202. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, H.; Guan, L.; Zhao, S.; Gu, Z.; Wei, H.; Gao, Z.; Wang, F.; Yang, N.; Luo, L.; et al. Mesenchymal stem cells provide prophylaxis against acute graft-versus-host disease following allogeneic hematopoietic stem cell transplantation: A meta-analysis of animal models. Oncotarget 2016, 7, 61764–61774. [Google Scholar] [PubMed]
- Dennis, J.E.; Charbord, P. Origin and differentiation of human and murine stroma. Stem. Cells 2002, 20, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Wakitani, S.; Mitsuoka, T.; Nakamura, N.; Toritsuka, Y.; Nakamura, Y.; Horibe, S. Autologous bone marrow stromal cell transplantation for repair of full-thickness articular cartilage defects in human patellae: Two case reports. Cell Trans. 2004, 13, 595–600. [Google Scholar] [CrossRef]
- Giannini, S.; Buda, R.; Battaglia, M.; Cavallo, M.; Ruffilli, A.; Ramponi, L.; Pagliazzi, G.; Vannini, F. One-step repair in talar osteochondral lesions: 4-year clinical results and t2-mapping capability in outcome prediction. Am. J. Sports Med. 2013, 41, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Skowronski, J.; Rutka, M. Osteochondral lesions of the knee reconstructed with mesenchymal stem cells—Results. Ortop. Traumatol. Rehabil. 2013, 15, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Vega, A.; Martin-Ferrero, M.A.; Del Canto, F.; Alberca, M.; Garcia, V.; Munar, A.; Orozco, L.; Soler, R.; Fuertes, J.J.; Huguet, M.; et al. Treatment of Knee Osteoarthritis With Allogeneic Bone Marrow Mesenchymal Stem Cells: A Randomized Controlled Trial. Transplantation 2015, 99, 1681–1690. [Google Scholar] [CrossRef] [PubMed]
- Soler, R.; Orozco, L.; Munar, A.; Huguet, M.; Lopez, R.; Vives, J.; Coll, R.; Codinach, M.; Garcia-Lopez, J. Final results of a phase I-II trial using ex vivo expanded autologous Mesenchymal Stromal Cells for the treatment of osteoarthritis of the knee confirming safety and suggesting cartilage regeneration. Knee 2016, 23, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Vodyanik, M.A.; Yu, J.; Zhang, X.; Tian, S.; Stewart, R.; Thomson, J.A.; Slukvin, I.I. A mesoderm-derived precursor for mesenchymal stem and endothelial cells. Cell Stem. Cell 2010, 7, 718–729. [Google Scholar] [CrossRef] [PubMed]
- Alrefaei, G.I.; Ayuob, N.N.; Ali, S.S.; Al-Karim, S. Effects of maternal age on the expression of mesenchymal stem cell markers in the components of human umbilical cord. Folia Histochem. Cytobiol. 2015, 53, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Friedenstein, A.J.; Gorskaja, J.F.; Kulagina, N.N. Fibroblast precursors in normal and irradiated mouse hematopoietic organs. Exp. Hematol. 1976, 4, 267–274. [Google Scholar] [PubMed]
- Caplan, A.I. Mesenchymal stem cells. J. Orthop. Res. 1991, 9, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Zuk, P.A.; Zhu, M.; Ashjian, P.; De Ugarte, D.A.; Huang, J.I.; Mizuno, H.; Alfonso, Z.C.; Fraser, J.K.; Benhaim, P.; Hedrick, M.H. Human adipose tissue is a source of multipotent stem cells. Mol. Biol. Cell 2002, 13, 4279–4295. [Google Scholar] [CrossRef] [PubMed]
- De Bari, C.; Dell’Accio, F.; Tylzanowski, P.; Luyten, F.P. Multipotent mesenchymal stem cells from adult human synovial membrane. Arthritis Rheum 2001, 44, 1928–1942. [Google Scholar] [CrossRef]
- Erices, A.; Conget, P.; Minguell, J.J. Mesenchymal progenitor cells in human umbilical cord blood. Br. J. Haematol. 2000, 109, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Arufe, M.C.; De la Fuente, A.; Fuentes-Boquete, I.; De Toro, F.J.; Blanco, F.J. Differentiation of synovial CD-105(+) human mesenchymal stem cells into chondrocyte-like cells through spheroid formation. J. Cell Biochem. 2009, 108, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.S.; Choi, Y.; Kim, H.S.; Kim, H.O. Comparison of molecular profiles of human mesenchymal stem cells derived from bone marrow, umbilical cord blood, placenta and adipose tissue. Int. J. Mol. Med. 2016, 37, 115–125. [Google Scholar] [PubMed]
- Nazempour, A.; Van Wie, B.J. Chondrocytes, Mesenchymal Stem Cells, and Their Combination in Articular Cartilage Regenerative Medicine. Ann. Biomed. Eng. 2016, 44, 1325–1354. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, Y.; Sekiya, I.; Yagishita, K.; Muneta, T. Comparison of human stem cells derived from various mesenchymal tissues: Superiority of synovium as a cell source. Arthritis Rheum. 2005, 52, 2521–2529. [Google Scholar] [CrossRef] [PubMed]
- Koga, H.; Muneta, T.; Ju, Y.J.; Nagase, T.; Nimura, A.; Mochizuki, T.; Ichinose, S.; von der Mark, K.; Sekiya, I. Synovial stem cells are regionally specified according to local microenvironments after implantation for cartilage regeneration. Stem. Cells 2007, 25, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.A.; Pei, M. Synovium-derived stem cells: A tissue-specific stem cell for cartilage engineering and regeneration. Tissue Eng. Part B 2012, 18, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Cai, X.; Zhang, S.; Karperien, M.; Lin, Y. Regeneration of articular cartilage by adipose tissue derived mesenchymal stem cells: Perspectives from stem cell biology and molecular medicine. J. Cell Physiol. 2013, 228, 938–944. [Google Scholar] [CrossRef] [PubMed]
- Perdisa, F.; Gostynska, N.; Roffi, A.; Filardo, G.; Marcacci, M.; Kon, E. Adipose-Derived Mesenchymal Stem Cells for the Treatment of Articular Cartilage: A Systematic Review on Preclinical and Clinical Evidence. Stem. Cells Int. 2015, 2015, 597652. [Google Scholar] [CrossRef] [PubMed]
- Caterson, E.J.; Nesti, L.J.; Danielson, K.G.; Tuan, R.S. Human marrow-derived mesenchymal progenitor cells: Isolation, culture expansion, and analysis of differentiation. Mol. Biotechnol. 2002, 20, 245–256. [Google Scholar] [CrossRef]
- Hung, S.C.; Chen, N.J.; Hsieh, S.L.; Li, H.; Ma, H.L.; Lo, W.H. Isolation and characterization of size-sieved stem cells from human bone marrow. Stem. Cells 2002, 20, 249–258. [Google Scholar] [CrossRef] [PubMed]
- De Ugarte, D.A.; Alfonso, Z.; Zuk, P.A.; Elbarbary, A.; Zhu, M.; Ashjian, P.; Benhaim, P.; Hedrick, M.H.; Fraser, J.K. Differential expression of stem cell mobilization-associated molecules on multi-lineage cells from adipose tissue and bone marrow. Immunol. Lett. 2003, 89, 267–270. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.J.; Wang, J.A.; Cao, J.; Zhang, X. Differentiation of bone marrow mesenchymal stem cells induced by myocardial medium under hypoxic conditions. Acta Pharmacol. Sin. 2006, 27, 1153–1158. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Liu, G.; Bharadwaj, S.; Zhang, Y. Isolation and myogenic differentiation of mesenchymal stem cells for urologic tissue engineering. Methods Mol. Biol. 2013, 1001, 65–80. [Google Scholar] [PubMed]
- Dugan, J.M.; Cartmell, S.H.; Gough, J.E. Uniaxial cyclic strain of human adipose-derived mesenchymal stem cells and C2C12 myoblasts in coculture. J. Tissue Eng. 2014, 5, 2041731414530138. [Google Scholar] [CrossRef] [PubMed]
- Mu, M.W.; Zhao, Z.Y.; Li, C.G. Comparative study of neural differentiation of bone marrow mesenchymal stem cells by different induction methods. Genet Mol. Res. 2015, 14, 14169–14176. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.A.; Kinsey, S.E.; English, A.; Jones, R.A.; Straszynski, L.; Meredith, D.M.; Markham, A.F.; Jack, A.; Emery, P.; McGonagle, D. Isolation and characterization of bone marrow multipotential mesenchymal progenitor cells. Arthritis Rheum. 2002, 46, 3349–3360. [Google Scholar] [CrossRef] [PubMed]
- Kaltz, N.; Ringe, J.; Holzwarth, C.; Charbord, P.; Niemeyer, M.; Jacobs, V.R.; Peschel, C.; Haupl, T.; Oostendorp, R.A. Novel markers of mesenchymal stem cells defined by genome-wide gene expression analysis of stromal cells from different sources. Exp. Cell Res. 2010, 316, 2609–2617. [Google Scholar] [CrossRef] [PubMed]
- Mendicino, M.; Bailey, A.M.; Wonnacott, K.; Puri, R.K.; Bauer, S.R. MSC-based product characterization for clinical trials: An FDA perspective. Cell Stem. Cell 2014, 14, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, G.; Suchanek, M.; Horn, P.; Graf, F.; Volz, C.; Horn, T.; Zhang, X.; Wagner, W.; Ho, A.D.; Boutros, M. Functional fingerprinting of human mesenchymal stem cells using high-throughput RNAi screening. Genome Med. 2015, 7, 46. [Google Scholar] [CrossRef] [PubMed]
- Samsonraj, R.M.; Rai, B.; Sathiyanathan, P.; Puan, K.J.; Rotzschke, O.; Hui, J.H.; Raghunath, M.; Stanton, L.W.; Nurcombe, V.; Cool, S.M. Establishing criteria for human mesenchymal stem cell potency. Stem. Cells 2015, 33, 1878–1891. [Google Scholar] [CrossRef] [PubMed]
- Sacchetti, B.; Funari, A.; Remoli, C.; Giannicola, G.; Kogler, G.; Liedtke, S.; Cossu, G.; Serafini, M.; Sampaolesi, M.; Tagliafico, E.; et al. No Identical “Mesenchymal Stem Cells” at Different Times and Sites: Human Committed Progenitors of Distinct Origin and Differentiation Potential Are Incorporated as Adventitial Cells in Microvessels. Stem. Cell Reports 2016, 6, 897–913. [Google Scholar] [CrossRef] [PubMed]
- Shahdadfar, A.; Fronsdal, K.; Haug, T.; Reinholt, F.P.; Brinchmann, J.E. In vitro expansion of human mesenchymal stem cells: Choice of serum is a determinant of cell proliferation, differentiation, gene expression, and transcriptome stability. Stem. Cells 2005, 23, 1357–1366. [Google Scholar] [CrossRef] [PubMed]
- Avanzini, M.A.; Bernardo, M.E.; Cometa, A.M.; Perotti, C.; Zaffaroni, N.; Novara, F.; Visai, L.; Moretta, A.; Del Fante, C.; Villa, R.; et al. Generation of mesenchymal stromal cells in the presence of platelet lysate: A phenotypic and functional comparison of umbilical cord blood- and bone marrow-derived progenitors. Haematologica 2009, 94, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Torensma, R.; Prins, H.J.; Schrama, E.; Verwiel, E.T.; Martens, A.C.; Roelofs, H.; Jansen, B.J. The impact of cell source, culture methodology, culture location, and individual donors on gene expression profiles of bone marrow-derived and adipose-derived stromal cells. Stem. Cells Dev. 2013, 22, 1086–1096. [Google Scholar] [CrossRef] [PubMed]
- Steinert, A.F.; Ghivizzani, S.C.; Rethwilm, A.; Tuan, R.S.; Evans, C.H.; Noth, U. Major biological obstacles for persistent cell-based regeneration of articular cartilage. Arthritis Res. Ther. 2007, 9, 213. [Google Scholar] [CrossRef] [PubMed]
- Wakitani, S.; Nawata, M.; Tensho, K.; Okabe, T.; Machida, H.; Ohgushi, H. Repair of articular cartilage defects in the patello-femoral joint with autologous bone marrow mesenchymal cell transplantation: Three case reports involving nine defects in five knees. J. Tissue Eng. Regen. Med. 2007, 1, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Huey, D.J.; Hu, J.C.; Athanasiou, K.A. Unlike bone, cartilage regeneration remains elusive. Science 2012, 338, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Mackay, A.M.; Beck, S.C.; Murphy, J.M.; Barry, F.P.; Chichester, C.O.; Pittenger, M.F. Chondrogenic differentiation of cultured human mesenchymal stem cells from marrow. Tissue Eng. 1998, 4, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Hayashi, Y.; Schlieve, C.R.; Ikeya, M.; Kim, H.; Nguyen, T.D.; Sami, S.; Baba, S.; Barruet, E.; Nasu, A.; et al. Induced pluripotent stem cells from patients with human fibrodysplasia ossificans progressiva show increased mineralization and cartilage formation. Orphanet J. Rare Dis. 2013, 8, 190. [Google Scholar] [CrossRef] [PubMed]
- De Kroon, L.M.; Narcisi, R.; Blaney Davidson, E.N.; Cleary, M.A.; van Beuningen, H.M.; Koevoet, W.J.; van Osch, G.J.; van der Kraan, P.M. Activin Receptor-Like Kinase Receptors ALK5 and ALK1 Are Both Required for TGFbeta-Induced Chondrogenic Differentiation of Human Bone Marrow-Derived Mesenchymal Stem Cells. PLoS ONE 2015, 10, e0146124. [Google Scholar] [CrossRef] [PubMed]
- Lolli, A.; Narcisi, R.; Lambertini, E.; Penolazzi, L.; Angelozzi, M.; Kops, N.; Gasparini, S.; van Osch, G.J.; Piva, R. Silencing of Antichondrogenic MicroRNA-221 in Human Mesenchymal Stem Cells Promotes Cartilage Repair In Vivo. Stem. Cells 2016, 34, 1801–1811. [Google Scholar] [CrossRef] [PubMed]
- Lolli, A.; Narcisi, R.; Lambertini, E.; Penolazzi, L.; Angelozzi, M.; Kops, N.; Gasparini, S.; van Osch, G.J.; Piva, R. The effect of oxygen tension on human articular chondrocyte matrix synthesis: Integration of experimental and computational approaches. Biotechnol. Bioeng. 2014, 111, 1876–1885. [Google Scholar]
- Murdoch, A.D.; Hardingham, T.E.; Eyre, D.R.; Fernandes, R.J. The development of a mature collagen network in cartilage from human bone marrow stem cells in Transwell culture. Matrix Biol. 2016, 50, 6–26. [Google Scholar] [CrossRef] [PubMed]
- Van Beuningen, H.M.; Glansbeek, H.L.; van der Kraan, P.M.; van den Berg, W.B. Osteoarthritis-like changes in the murine knee joint resulting from intra-articular transforming growth factor-beta injections. Osteoarthr. Cartil. 2000, 8, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Farrell, E.; van der Jagt, O.P.; Koevoet, W.; Kops, N.; van Manen, C.J.; Hellingman, C.A.; Jahr, H.; O’Brien, F.J.; Verhaar, J.A.; Weinans, H.; et al. Chondrogenic priming of human bone marrow stromal cells: A better route to bone repair? Tissue Eng. Part C 2009, 15, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.B.; Fischer, M.; Zellner, J.; Berner, A.; Dienstknecht, T.; Prantl, L.; Kujat, R.; Nerlich, M.; Tuan, R.S.; Angele, P. Hypertrophy in mesenchymal stem cell chondrogenesis: Effect of TGF-beta isoforms and chondrogenic conditioning. Cells Tissues Organs 2010, 192, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Bauge, C.; Girard, N.; Lhuissier, E.; Bazille, C.; Boumediene, K. Regulation and Role of TGFbeta Signaling Pathway in Aging and Osteoarthritis Joints. Aging Dis. 2014, 5, 394–405. [Google Scholar] [PubMed]
- Kuroda, R.; Ishida, K.; Matsumoto, T.; Akisue, T.; Fujioka, H.; Mizuno, K.; Ohgushi, H.; Wakitani, S.; Kurosaka, M. Treatment of a full-thickness articular cartilage defect in the femoral condyle of an athlete with autologous bone-marrow stromal cells. Osteoarthr. Cartil. 2007, 15, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Orozco, L.; Munar, A.; Soler, R.; Alberca, M.; Soler, F.; Huguet, M.; Sentis, J.; Sanchez, A.; Garcia-Sancho, J. Treatment of knee osteoarthritis with autologous mesenchymal stem cells: Two-year follow-up results. Transplantation 2014, 97, e66–e68. [Google Scholar] [CrossRef] [PubMed]
- Wolfstadt, J.I.; Cole, B.J.; Ogilvie-Harris, D.J.; Viswanathan, S.; Chahal, J. Current concepts: The role of mesenchymal stem cells in the management of knee osteoarthritis. Sports Health 2015, 7, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Koh, Y.G.; Kwon, O.R.; Kim, Y.S.; Choi, Y.J.; Tak, D.H. Adipose-Derived Mesenchymal Stem Cells With Microfracture Versus Microfracture Alone: 2-Year Follow-up of a Prospective Randomized Trial. Arthroscopy 2016, 32, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Pak, J.; Lee, J.H.; Park, K.S.; Jeong, B.C.; Lee, S.H. Regeneration of Cartilage in Human Knee Osteoarthritis with Autologous Adipose Tissue-Derived Stem Cells and Autologous Extracellular Matrix. Biores. Open Access 2016, 5, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.B.; Ha, C.W.; Lee, C.H.; Yoon, Y.C.; Park, Y.G. Cartilage Regeneration in Osteoarthritic Patients by a Composite of Allogeneic Umbilical Cord Blood-Derived Mesenchymal Stem Cells and Hyaluronate Hydrogel: Results From a Clinical Trial for Safety and Proof-of-Concept With 7 Years of Extended Follow-Up. Stem. Cells Transl. Med. 2016, 6, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, M.; Cosenza, S.; Maumus, M.; Jorgensen, C.; Noel, D. Therapeutic application of mesenchymal stem cells in osteoarthritis. Exp. Opin. Biol. Ther. 2016, 16, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Koh, Y.G.; Choi, Y.J.; Kwon, O.R.; Kim, Y.S. Second-Look Arthroscopic Evaluation of Cartilage Lesions After Mesenchymal Stem Cell Implantation in Osteoarthritic Knees. Am. J. Sports Med. 2014, 42, 1628–1637. [Google Scholar] [CrossRef] [PubMed]
- Davatchi, F.; Sadeghi Abdollahi, B.; Mohyeddin, M.; Nikbin, B. Mesenchymal stem cell therapy for knee osteoarthritis: 5 years follow-up of three patients. Int. J. Rheum. Dis. 2016, 19, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Liu, H.; Xie, Y.; Sang, L.; Liu, J.; Chen, B. Effect of mesenchymal stromal cells for articular cartilage degeneration treatment: A meta-analysis. Cytotherapy 2015, 17, 1342–1352. [Google Scholar] [CrossRef] [PubMed]
- Solchaga, L.A.; Penick, K.; Porter, J.D.; Goldberg, V.M.; Caplan, A.I.; Welter, J.F. FGF-2 enhances the mitotic and chondrogenic potentials of human adult bone marrow-derived mesenchymal stem cells. J. Cell Physiol. 2005, 203, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Sawada, R.; Fujiwara, Y.; Tsuchiya, T. FGF-2 increases osteogenic and chondrogenic differentiation potentials of human mesenchymal stem cells by inactivation of TGF-beta signaling. Cytotechnology 2008, 56, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Solchaga, L.A.; Penick, K.; Goldberg, V.M.; Caplan, A.I.; Welter, J.F. Fibroblast growth factor-2 enhances proliferation and delays loss of chondrogenic potential in human adult bone-marrow-derived mesenchymal stem cells. Tissue Eng. Part A 2010, 16, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- Correa, D.; Somoza, R.A.; Lin, P.; Greenberg, S.; Rom, E.; Duesler, L.; Welter, J.F.; Yayon, A.; Caplan, A.I. Sequential exposure to fibroblast growth factors (FGF) 2, 9 and 18 enhances hMSC chondrogenic differentiation. Osteoarthr. Cartil. 2015, 23, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Hellingman, C.A.; Koevoet, W.; Kops, N.; Farrell, E.; Jahr, H.; Liu, W.; Baatenburg de Jong, R.J.; Frenz, D.A.; van Osch, G.J. Fibroblast growth factor receptors in in vitro and in vivo chondrogenesis: Relating tissue engineering using adult mesenchymal stem cells to embryonic development. Tissue Eng. Part A 2010, 16, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.; Hennig, T.; Bock, R.; Steck, E.; Richter, W. Impact of growth factors and PTHrP on early and late chondrogenic differentiation of human mesenchymal stem cells. J. Cell Physiol. 2010, 223, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, G.; Banfi, A.; Mastrogiacomo, M.; Notaro, R.; Luzzatto, L.; Cancedda, R.; Quarto, R. Ex vivo enrichment of mesenchymal cell progenitors by fibroblast growth factor 2. Exp. Cell Res. 2003, 287, 98–105. [Google Scholar] [CrossRef]
- Turnbull, J.E.; Fernig, D.G.; Ke, Y.; Wilkinson, M.C.; Gallagher, J.T. Identification of the basic fibroblast growth factor binding sequence in fibroblast heparan sulfate. J. Biol. Chem. 1992, 267, 10337–10341. [Google Scholar] [PubMed]
- Lin, X.; Buff, E.M.; Perrimon, N.; Michelson, A.M. Heparan sulfate proteoglycans are essential for FGF receptor signaling during Drosophila embryonic development. Development 1999, 126, 3715–3723. [Google Scholar] [PubMed]
- Mohammadi, M.; Olsen, S.K.; Ibrahimi, O.A. Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev. 2005, 16, 107–137. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Ori, A.; Rudd, T.R.; Uniewicz, K.A.; Ahmed, Y.A.; Guimond, S.E.; Skidmore, M.A.; Siligardi, G.; Yates, E.A.; Fernig, D.G. Diversification of the structural determinants of fibroblast growth factor-heparin interactions: Implications for binding specificity. J. Biol. Chem. 2012, 287, 40061–40073. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sun, C.; Yates, E.A.; Jiang, C.; Wilkinson, M.C.; Fernig, D.G. Heparin binding preference and structures in the fibroblast growth factor family parallel their evolutionary diversification. Open Biol. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.M.; West, L.A.; Govindraj, P.; Zhang, X.; Ornitz, D.M.; Hassell, J.R. Heparan and chondroitin sulfate on growth plate perlecan mediate binding and delivery of FGF-2 to FGF receptors. Matrix Biol. 2007, 26, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Vincent, T.L.; McLean, C.J.; Full, L.E.; Peston, D.; Saklatvala, J. FGF-2 is bound to perlecan in the pericellular matrix of articular cartilage, where it acts as a chondrocyte mechanotransducer. Osteoarthr. Cartil. 2007, 15, 752–763. [Google Scholar] [CrossRef] [PubMed]
- Melrose, J.; Isaacs, M.D.; Smith, S.M.; Hughes, C.E.; Little, C.B.; Caterson, B.; Hayes, A.J. Chondroitin sulphate and heparan sulphate sulphation motifs and their proteoglycans are involved in articular cartilage formation during human foetal knee joint development. Histochem. Cell Biol. 2012, 138, 461–475. [Google Scholar] [PubMed]
- Wijesinghe, S.J.; Ling, L.; Murali, S.; Qing, Y.H.; Hinkley, S.F.; Carnachan, S.M.; Bell, T.J.; Swaminathan, K.; Hui, J.H.; van Wijnen, A.J.; et al. Affinity Selection of FGF2-Binding Heparan Sulfates for Ex Vivo Expansion of Human Mesenchymal Stem Cells. J. Cell Physiol. 2016, 232, 566–575. [Google Scholar] [PubMed]
- Davidson, D.; Blanc, A.; Filion, D.; Wang, H.; Plut, P.; Pfeffer, G.; Buschmann, M.D.; Henderson, J.E. Fibroblast growth factor (FGF) 18 signals through FGF receptor 3 to promote chondrogenesis. J. Biol. Chem. 2005, 280, 20509–20515. [Google Scholar] [PubMed]
- Sekiya, I.; Vuoristo, J.T.; Larson, B.L.; Prockop, D.J. In vitro cartilage formation by human adult stem cells from bone marrow stroma defines the sequence of cellular and molecular events during chondrogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 4397–4402. [Google Scholar] [PubMed]
- McCaffrey, T.A.; Falcone, D.J.; Vicente, D.; Du, B.; Consigli, S.; Borth, W. Protection of transforming growth factor-beta 1 activity by heparin and fucoidan. J. Cell Physiol. 1994, 159, 51–59. [Google Scholar] [PubMed]
- Lyon, M.; Rushton, G.; Gallagher, J.T. The interaction of the transforming growth factor-betas with heparin/heparan sulfate is isoform-specific. J. Biol. Chem. 1997, 272, 18000–18006. [Google Scholar] [PubMed]
- Lee, M.J. Heparin inhibits activation of latent transforming growth factor-beta1. Pharmacology 2013, 92, 238–244. [Google Scholar] [PubMed]
- Lee, J.; Wee, S.; Gunaratne, J.; Chua, R.J.; Smith, R.A.; Ling, L.; Fernig, D.G.; Swaminathan, K.; Nurcombe, V.; Cool, S.M. Structural determinants of heparin-transforming growth factor-beta1 interactions and their effects on signaling. Glycobiology 2015, 25, 1491–1504. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, A.; Romaris, M.; Rasmussen, L.M.; Heinegard, D.; Twardzik, D.R.; Border, W.A.; Ruoslahti, E. Interaction of the small interstitial proteoglycans biglycan, decorin and fibromodulin with transforming growth factor beta. Biochem. J. 1994, 302, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.C.; Hoang, B.; Thomas, J.T.; Vukicevic, S.; Luyten, F.P.; Ryba, N.J.; Kozak, C.A.; Reddi, A.H.; Moos, M., Jr. Cartilage-derived morphogenetic proteins. New members of the transforming growth factor-beta superfamily predominantly expressed in long bones during human embryonic development. J. Biol. Chem. 1994, 269, 28227–28234. [Google Scholar] [PubMed]
- Storm, E.E.; Kingsley, D.M. GDF5 coordinates bone and joint formation during digit development. Dev. Biol. 1999, 209, 11–27. [Google Scholar] [CrossRef] [PubMed]
- Masuya, H.; Nishida, K.; Furuichi, T.; Toki, H.; Nishimura, G.; Kawabata, H.; Yokoyama, H.; Yoshida, A.; Tominaga, S.; Nagano, J.; et al. A novel dominant-negative mutation in Gdf5 generated by ENU mutagenesis impairs joint formation and causes osteoarthritis in mice. Hum. Mol. Genet 2007, 16, 2366–2375. [Google Scholar] [CrossRef] [PubMed]
- Kwong, F.N.; Hoyland, J.A.; Evans, C.H.; Freemont, A.J. Regional and cellular localisation of BMPs and their inhibitors’ expression in human fractures. Int. Orthop. 2009, 33, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Garciadiego-Cazares, D.; Aguirre-Sanchez, H.I.; Abarca-Buis, R.F.; Kouri, J.B.; Velasquillo, C.; Ibarra, C. Regulation of alpha5 and alphaV Integrin Expression by GDF-5 and BMP-7 in Chondrocyte Differentiation and Osteoarthritis. PLoS ONE 2015, 10, e0127166. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.; Wan, Y.; Balian, G.; Laurencin, C.T.; Li, X. Adenovirus-mediated expression of growth and differentiation factor-5 promotes chondrogenesis of adipose stem cells. Growth Factors 2008, 26, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Yang, S.; Sun, Z.; Zhang, Y.; Xia, T.; Xu, W.; Ye, S. Human mesenchymal stem cells induced by growth differentiation factor 5: An improved self-assembly tissue engineering method for cartilage repair. Tissue Eng. Part C 2011, 17, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Shang, H.; Katz, A.; Li, X. A modified aggregate culture for chondrogenesis of human adipose-derived stem cells genetically modified with growth and differentiation factor 5. BioResour. Open Access 2013, 2, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Enochson, L.; Stenberg, J.; Brittberg, M.; Lindahl, A. GDF5 reduces MMP13 expression in human chondrocytes via DKK1 mediated canonical Wnt signaling inhibition. Osteoarthr. Cartil. 2014, 22, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Coleman, C.M.; Vaughan, E.E.; Browe, D.C.; Mooney, E.; Howard, L.; Barry, F. Growth differentiation factor-5 enhances in vitro mesenchymal stromal cell chondrogenesis and hypertrophy. Stem. Cells Dev. 2013, 22, 1968–1976. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.K.; Huey, D.J.; Hu, J.C.; Athanasiou, K.A. TGF-beta1, GDF-5, and BMP-2 stimulation induces chondrogenesis in expanded human articular chondrocytes and marrow-derived stromal cells. Stem. Cells 2015, 33, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, N.S.; Mancera, R.L. Prediction of heparin binding sites in bone morphogenetic proteins (BMPs). Biochim. Biophys. Acta 2012, 1824, 1374–1381. [Google Scholar] [CrossRef] [PubMed]
- Indrawattana, N.; Chen, G.; Tadokoro, M.; Shann, L.H.; Ohgushi, H.; Tateishi, T.; Tanaka, J.; Bunyaratvej, A. Growth factor combination for chondrogenic induction from human mesenchymal stem cell. Biochem. Biophys. Res. Commun. 2004, 320, 914–919. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, I.; Larson, B.L.; Vuoristo, J.T.; Reger, R.L.; Prockop, D.J. Comparison of effect of BMP-2, -4, and -6 on in vitro cartilage formation of human adult stem cells from bone marrow stroma. Cell Tissue Res. 2005, 320, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Steinert, A.F.; Proffen, B.; Kunz, M.; Hendrich, C.; Ghivizzani, S.C.; Noth, U.; Rethwilm, A.; Eulert, J.; Evans, C.H. Hypertrophy is induced during the in vitro chondrogenic differentiation of human mesenchymal stem cells by bone morphogenetic protein-2 and bone morphogenetic protein-4 gene transfer. Arthritis Res. Ther. 2009, 11, R148. [Google Scholar] [CrossRef] [PubMed]
- Caron, M.M.; Emans, P.J.; Cremers, A.; Surtel, D.A.; Coolsen, M.M.; van Rhijn, L.W.; Welting, T.J. Hypertrophic differentiation during chondrogenic differentiation of progenitor cells is stimulated by BMP-2 but suppressed by BMP-7. Osteoarthr. Cartil. 2013, 21, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Irie, A.; Habuchi, H.; Kimata, K.; Sanai, Y. Heparan sulfate is required for bone morphogenetic protein-7 signaling. Biochem. Biophys. Res. Commun. 2003, 308, 858–865. [Google Scholar] [CrossRef]
- Yang, H.S.; La, W.G.; Bhang, S.H.; Jeon, J.Y.; Lee, J.H.; Kim, B.S. Heparin-conjugated fibrin as an injectable system for sustained delivery of bone morphogenetic protein-2. Tissue Eng. Part A 2010, 16, 1225–1233. [Google Scholar] [CrossRef] [PubMed]
- Bhakta, G.; Rai, B.; Lim, Z.X.; Hui, J.H.; Stein, G.S.; van Wijnen, A.J.; Nurcombe, V.; Prestwich, G.D.; Cool, S.M. Hyaluronic acid-based hydrogels functionalized with heparin that support controlled release of bioactive BMP-2. Biomaterials 2012, 33, 6113–6122. [Google Scholar] [CrossRef] [PubMed]
- Bramono, D.S.; Murali, S.; Rai, B.; Ling, L.; Poh, W.T.; Lim, Z.X.; Stein, G.S.; Nurcombe, V.; van Wijnen, A.J.; Cool, S.M. Bone marrow-derived heparan sulfate potentiates the osteogenic activity of bone morphogenetic protein-2 (BMP-2). Bone 2012, 50, 954–964. [Google Scholar] [CrossRef] [PubMed]
- Kraushaar, D.C.; Rai, S.; Condac, E.; Nairn, A.; Zhang, S.; Yamaguchi, Y.; Moremen, K.; Dalton, S.; Wang, L. Heparan sulfate facilitates FGF and BMP signaling to drive mesoderm differentiation of mouse embryonic stem cells. J. Biol. Chem. 2012, 287, 22691–22700. [Google Scholar] [CrossRef] [PubMed]
- Koo, K.H.; Lee, J.M.; Ahn, J.M.; Kim, B.S.; La, W.G.; Kim, C.S.; Im, G.I. Controlled delivery of low-dose bone morphogenetic protein-2 using heparin-conjugated fibrin in the posterolateral lumbar fusion of rabbits. Artif. Organs 2013, 37, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Murali, S.; Rai, B.; Dombrowski, C.; Lee, J.L.; Lim, Z.X.; Bramono, D.S.; Ling, L.; Bell, T.; Hinkley, S.; Nathan, S.S.; et al. Affinity-selected heparan sulfate for bone repair. Biomaterials 2013, 34, 5594–5605. [Google Scholar] [CrossRef] [PubMed]
- Hettiaratchi, M.H.; Miller, T.; Temenoff, J.S.; Guldberg, R.E.; McDevitt, T.C. Heparin microparticle effects on presentation and bioactivity of bone morphogenetic protein-2. Biomaterials 2014, 35, 7228–7238. [Google Scholar] [CrossRef] [PubMed]
- Rai, B.; Chatterjea, A.; Lim, Z.X.; Tan, T.C.; Sawyer, A.A.; Hosaka, Y.Z.; Murali, S.; Lee, J.J.; Fenwick, S.A.; Hui, J.H.; et al. Repair of segmental ulna defects using a beta-TCP implant in combination with a heparan sulfate glycosaminoglycan variant. Acta Biomater. 2015, 28, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Church, V.; Nohno, T.; Linker, C.; Marcelle, C.; Francis-West, P. Wnt regulation of chondrocyte differentiation. J. Cell Sci. 2002, 115, 4809–4818. [Google Scholar] [CrossRef] [PubMed]
- Bradley, E.W.; Drissi, M.H. WNT5A regulates chondrocyte differentiation through differential use of the CaN/NFAT and IKK/NF-kappaB pathways. Mol. Endocrinol. 2010, 24, 1581–1593. [Google Scholar] [CrossRef] [PubMed]
- Narcisi, R.; Cleary, M.A.; Brama, P.A.; Hoogduijn, M.J.; Tuysuz, N.; ten Berge, D.; van Osch, G.J. Long-term expansion, enhanced chondrogenic potential, and suppression of endochondral ossification of adult human MSCs via WNT signaling modulation. Stem. Cell. Reports 2015, 4, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Studer, D.; Millan, C.; Ozturk, E.; Maniura-Weber, K.; Zenobi-Wong, M. Molecular and biophysical mechanisms regulating hypertrophic differentiation in chondrocytes and mesenchymal stem cells. Eur. Cell Mater. 2012, 24, 118–135, discussion 135. [Google Scholar] [CrossRef] [PubMed]
- Dhoot, G.K.; Gustafsson, M.K.; Ai, X.; Sun, W.; Standiford, D.M.; Emerson, C.P., Jr. Regulation of Wnt signaling and embryo patterning by an extracellular sulfatase. Science 2001, 293, 1663–1666. [Google Scholar] [CrossRef] [PubMed]
- Ai, X.; Do, A.T.; Lozynska, O.; Kusche-Gullberg, M.; Lindahl, U.; Emerson, C.P., Jr. QSulf1 remodels the 6-O sulfation states of cell surface heparan sulfate proteoglycans to promote Wnt signaling. J. Cell Biol. 2003, 162, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Song, H.H.; Shi, W.; Xiang, Y.Y.; Filmus, J. The loss of glypican-3 induces alterations in Wnt signaling. J. Biol. Chem. 2005, 280, 2116–2125. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhang, E.; Yang, M.; Lu, L. Overexpression of Wnt11 promotes chondrogenic differentiation of bone marrow-derived mesenchymal stem cells in synergism with TGF-beta. Mol. Cell Biochem. 2014, 390, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Enomoto-Iwamoto, M.; Kitagaki, J.; Koyama, E.; Tamamura, Y.; Wu, C.; Kanatani, N.; Koike, T.; Okada, H.; Komori, T.; Yoneda, T.; et al. The Wnt antagonist Frzb-1 regulates chondrocyte maturation and long bone development during limb skeletogenesis. Dev. Biol. 2002, 251, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, C.; Tabin, C.J. Wnt-14 plays a pivotal role in inducing synovial joint formation in the developing appendicular skeleton. Cell 2001, 104, 341–351. [Google Scholar] [CrossRef]
- Frisch, J.; Venkatesan, J.K.; Rey-Rico, A.; Schmitt, G.; Madry, H.; Cucchiarini, M. Influence of insulin-like growth factor I overexpression via recombinant adeno-associated vector gene transfer upon the biological activities and differentiation potential of human bone marrow-derived mesenchymal stem cells. Stem. Cell Res. Ther. 2014, 5, 103. [Google Scholar] [CrossRef] [PubMed]
- Moller, A.V.; Jorgensen, S.P.; Chen, J.W.; Larnkjaer, A.; Ledet, T.; Flyvbjerg, A.; Frystyk, J. Glycosaminoglycans increase levels of free and bioactive IGF-I in vitro. Eur. J. Endocrinol. 2006, 155, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Kim, H.J.; Im, G.I. PTHrP promotes chondrogenesis and suppresses hypertrophy from both bone marrow-derived and adipose tissue-derived MSCs. Biochem. Biophys. Res. Commun. 2008, 373, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Im, G.I. PTHrP isoforms have differing effect on chondrogenic differentiation and hypertrophy of mesenchymal stem cells. Biochem. Biophys. Res. Commun. 2012, 421, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Zak, B.M.; Crawford, B.E.; Esko, J.D. Hereditary multiple exostoses and heparan sulfate polymerization. Biochim. Biophys. Acta 2002, 1573, 346–355. [Google Scholar] [CrossRef]
- Koziel, L.; Kunath, M.; Kelly, O.G.; Vortkamp, A. Ext1-dependent heparan sulfate regulates the range of Ihh signaling during endochondral ossification. Dev. Cell 2004, 6, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Newfeld, S.J.; Wisotzkey, R.G.; Kumar, S. Molecular evolution of a developmental pathway: Phylogenetic analyses of transforming growth factor-beta family ligands, receptors and Smad signal transducers. Genetics 1999, 152, 783–795. [Google Scholar] [PubMed]
- Moses, H.L.; Roberts, A.B.; Derynck, R. The Discovery and Early Days of TGF-beta: A Historical Perspective. Cold Spring Harb Perspect Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Constam, D.B.; Robertson, E.J. Regulation of bone morphogenetic protein activity by pro domains and proprotein convertases. J. Cell Biol. 1999, 144, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Griffith, D.L.; Keck, P.C.; Sampath, T.K.; Rueger, D.C.; Carlson, W.D. Three-dimensional structure of recombinant human osteogenic protein 1: Structural paradigm for the transforming growth factor beta superfamily. Proc. Natl. Acad. Sci. USA 1996, 93, 878–883. [Google Scholar] [CrossRef] [PubMed]
- Mittl, P.R.; Priestle, J.P.; Cox, D.A.; McMaster, G.; Cerletti, N.; Grutter, M.G. The crystal structure of TGF-beta 3 and comparison to TGF-beta 2: Implications for receptor binding. Protein Sci. 1996, 5, 1261–1271. [Google Scholar] [CrossRef] [PubMed]
- McPherron, A.C.; Lee, S.J. GDF-3 and GDF-9: Two new members of the transforming growth factor-beta superfamily containing a novel pattern of cysteines. J. Biol. Chem. 1993, 268, 3444–3449. [Google Scholar] [PubMed]
- Corradini, E.; Babitt, J.L.; Lin, H.Y. The RGM/DRAGON family of BMP co-receptors. Cytokine Growth Factor Rev. 2009, 20, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, M.; Ota, M.; Rifkin, D.B. Matrix control of transforming growth factor-beta function. J. Biochem. 2012, 152, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Taipale, J.; Miyazono, K.; Heldin, C.H.; Keski-Oja, J. Latent transforming growth factor-beta 1 associates to fibroblast extracellular matrix via latent TGF-beta binding protein. J. Cell Biol. 1994, 124, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Barcellos-Hoff, M.H.; Dix, T.A. Redox-mediated activation of latent transforming growth factor-beta 1. Mol. Endocrinol. 1996, 10, 1077–1083. [Google Scholar] [PubMed]
- Munger, J.S.; Harpel, J.G.; Gleizes, P.E.; Mazzieri, R.; Nunes, I.; Rifkin, D.B. Latent transforming growth factor-beta: Structural features and mechanisms of activation. Kidney Int. 1997, 51, 1376–1382. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Stamenkovic, I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000, 14, 163–176. [Google Scholar] [PubMed]
- Anderson, S.B.; Goldberg, A.L.; Whitman, M. Identification of a novel pool of extracellular pro-myostatin in skeletal muscle. J. Biol. Chem. 2008, 283, 7027–7035. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.A.; Al-Musawi, S.L.; Walton, K.L. Prodomains regulate the synthesis, extracellular localisation and activity of TGF-beta superfamily ligands. Growth Factors 2011, 29, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Akhurst, R.J.; Hata, A. Targeting the TGFbeta signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.; Attisano, L. The TGFbeta superfamily signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 47–63. [Google Scholar] [CrossRef] [PubMed]
- Zhai, G.; Dore, J.; Rahman, P. TGF-beta signal transduction pathways and osteoarthritis. Rheumatol. Int. 2015, 35, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Hanyu, A.; Ishidou, Y.; Ebisawa, T.; Shimanuki, T.; Imamura, T.; Miyazono, K. The N domain of Smad7 is essential for specific inhibition of transforming growth factor-beta signaling. J. Cell Biol. 2001, 155, 1017–1027. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Miyauchi, A.; Goto, J.; Takagi, Y.; Okuizumi, H.; Kanematsu, M.; Hase, M.; Takai, H.; Harada, A.; Ikeda, K. Association of a polymorphism of the transforming growth factor-beta1 gene with genetic susceptibility to osteoporosis in postmenopausal Japanese women. J. Bone Miner Res. 1998, 13, 1569–1576. [Google Scholar] [CrossRef] [PubMed]
- Kizawa, H.; Kou, I.; Iida, A.; Sudo, A.; Miyamoto, Y.; Fukuda, A.; Mabuchi, A.; Kotani, A.; Kawakami, A.; Yamamoto, S.; et al. An aspartic acid repeat polymorphism in asporin inhibits chondrogenesis and increases susceptibility to osteoarthritis. Nat. Genet 2005, 37, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Blaney Davidson, E.N.; Scharstuhl, A.; Vitters, E.L.; van der Kraan, P.M.; van den Berg, W.B. Reduced transforming growth factor-beta signaling in cartilage of old mice: Role in impaired repair capacity. Arthritis Res. Ther. 2005, 7, R1338–R1347. [Google Scholar] [CrossRef] [PubMed]
- Blaney Davidson, E.N.; Vitters, E.L.; van der Kraan, P.M.; van den Berg, W.B. Expression of transforming growth factor-beta (TGFbeta) and the TGFbeta signalling molecule SMAD-2P in spontaneous and instability-induced osteoarthritis: Role in cartilage degradation, chondrogenesis and osteophyte formation. Ann. Rheum. Dis. 2006, 65, 1414–1421. [Google Scholar] [CrossRef] [PubMed]
- Narcisi, R.; Quarto, R.; Ulivi, V.; Muraglia, A.; Molfetta, L.; Giannoni, P. TGF beta-1 administration during ex vivo expansion of human articular chondrocytes in a serum-free medium redirects the cell phenotype toward hypertrophy. J. Cell Physiol. 2012, 227, 3282–3290. [Google Scholar] [CrossRef] [PubMed]
- Cals, F.L.; Hellingman, C.A.; Koevoet, W.; Baatenburg de Jong, R.J.; van Osch, G.J. Effects of transforming growth factor-beta subtypes on in vitro cartilage production and mineralization of human bone marrow stromal-derived mesenchymal stem cells. J. Tissue Eng. Regen Med. 2012, 6, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Campos-Xavier, B.; Saraiva, J.M.; Savarirayan, R.; Verloes, A.; Feingold, J.; Faivre, L.; Munnich, A.; Le Merrer, M.; Cormier-Daire, V. Phenotypic variability at the TGF-beta1 locus in Camurati-Engelmann disease. Hum. Genet 2001, 109, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Janssens, K.; ten Dijke, P.; Ralston, S.H.; Bergmann, C.; Van Hul, W. Transforming growth factor-beta 1 mutations in Camurati-Engelmann disease lead to increased signaling by altering either activation or secretion of the mutant protein. J. Biol. Chem. 2003, 278, 7718–7724. [Google Scholar] [CrossRef] [PubMed]
- Van Beuningen, H.M.; van der Kraan, P.M.; Arntz, O.J.; van den Berg, W.B. Transforming growth factor-beta 1 stimulates articular chondrocyte proteoglycan synthesis and induces osteophyte formation in the murine knee joint. Lab. Invest. 1994, 71, 279–290. [Google Scholar] [PubMed]
- Somoza, R.A.; Welter, J.F.; Correa, D.; Caplan, A.I. Chondrogenic differentiation of mesenchymal stem cells: Challenges and unfulfilled expectations. Tissue Eng. Part B 2014, 20, 596–608. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Wei, A.; Whittaker, S.; Williams, L.A.; Tao, H.; Ma, D.D.; Diwan, A.D. The role of BMP-7 in chondrogenic and osteogenic differentiation of human bone marrow multipotent mesenchymal stromal cells in vitro. J. Cell Biochem. 2010, 109, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.T.; Li, W.J. Chondrogenesis of Embryonic Stem Cell-Derived Mesenchymal Stem Cells Induced by TGFbeta1 and BMP7 through Increased TGFbeta Receptor Expression and Endogenous TGFbeta1 Production. J. Cell Biochem. 2017, 118, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Handorf, A.M.; Li, W.J. Induction of mesenchymal stem cell chondrogenesis through sequential administration of growth factors within specific temporal windows. J. Cell Physiol. 2014, 229, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Denker, A.E.; Haas, A.R.; Nicoll, S.B.; Tuan, R.S. Chondrogenic differentiation of murine C3H10T1/2 multipotential mesenchymal cells: I. Stimulation by bone morphogenetic protein-2 in high-density micromass cultures. Differentiation 1999, 64, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Lyons, K.M.; McMahon, A.P.; Kronenberg, H.M. BMP signaling stimulates cellular differentiation at multiple steps during cartilage development. Proc. Natl. Acad. Sci. USA 2005, 102, 18023–18027. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, A.; Tsuji, K.; Cox, K.; Harfe, B.D.; Rosen, V.; Tabin, C.J. Genetic analysis of the roles of BMP2, BMP4, and BMP7 in limb patterning and skeletogenesis. PLoS Genet. 2006, 2, e216. [Google Scholar] [CrossRef] [PubMed]
- Bian, Q.; Jia, K.; Liu, S.F.; Shu, B.; Liang, Q.Q.; Zhou, C.J.; Zhou, Q.; Wang, Y.J. Inhibitory effect of YQHYRJ recipe on osteoblast differentiation induced by BMP-2 in fibroblasts from posterior longitudinal ligament of mice. Pharmazie 2011, 66, 784–790. [Google Scholar] [PubMed]
- Sekiya, I.; Tang, T.; Hayashi, M.; Morito, T.; Ju, Y.J.; Mochizuki, T.; Muneta, T. Periodic knee injections of BMP-7 delay cartilage degeneration induced by excessive running in rats. J. Orthop. Res. 2009, 27, 1088–1092. [Google Scholar] [CrossRef] [PubMed]
- Hotten, G.; Neidhardt, H.; Jacobowsky, B.; Pohl, J. Cloning and expression of recombinant human growth/differentiation factor 5. Biochem. Biophys. Res. Commun. 1994, 204, 646–652. [Google Scholar] [CrossRef] [PubMed]
- McDonald, N.Q.; Hendrickson, W.A. A structural superfamily of growth factors containing a cystine knot motif. Cell 1993, 73, 421–424. [Google Scholar] [CrossRef]
- Luyten, F.P. Cartilage-derived morphogenetic protein-1. Int. J. Biochem. Cell Biol. 1997, 29, 1241–1244. [Google Scholar] [CrossRef]
- Thieme, T.; Patzschke, R.; Job, F.; Liebold, J.; Seemann, P.; Lilie, H.; Balbach, J.; Schwarz, E. Biophysical and structural characterization of a folded core domain within the proregion of growth and differentiation factor-5. FEBS J. 2014, 281, 4866–4877. [Google Scholar] [CrossRef] [PubMed]
- Storm, E.E.; Huynh, T.V.; Copeland, N.G.; Jenkins, N.A.; Kingsley, D.M.; Lee, S.J. Limb alterations in brachypodism mice due to mutations in a new member of the TGF beta-superfamily. Nature 1994, 368, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Hotten, G.C.; Matsumoto, T.; Kimura, M.; Bechtold, R.F.; Kron, R.; Ohara, T.; Tanaka, H.; Satoh, Y.; Okazaki, M.; Shirai, T.; et al. Recombinant human growth/differentiation factor 5 stimulates mesenchyme aggregation and chondrogenesis responsible for the skeletal development of limbs. Growth Factors 1996, 13, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Buxton, P.; Edwards, C.; Archer, C.W.; Francis-West, P. Growth/differentiation factor-5 (GDF-5) and skeletal development. J. Bone Joint Surg. Am. 2001, 83-A (Suppl. 1), S23–S30. [Google Scholar] [CrossRef]
- Shwartz, Y.; Viukov, S.; Krief, S.; Zelzer, E. Joint Development Involves a Continuous Influx of Gdf5-Positive Cells. Cell Rep. 2016, 15, 2577–2587. [Google Scholar] [CrossRef] [PubMed]
- Storm, E.E.; Kingsley, D.M. Joint patterning defects caused by single and double mutations in members of the bone morphogenetic protein (BMP) family. Development 1996, 122, 3969–3979. [Google Scholar] [PubMed]
- Settle, S.H., Jr.; Rountree, R.B.; Sinha, A.; Thacker, A.; Higgins, K.; Kingsley, D.M. Multiple joint and skeletal patterning defects caused by single and double mutations in the mouse Gdf6 and Gdf5 genes. Dev. Biol. 2003, 254, 116–130. [Google Scholar] [CrossRef]
- Thomas, J.T.; Kilpatrick, M.W.; Lin, K.; Erlacher, L.; Lembessis, P.; Costa, T.; Tsipouras, P.; Luyten, F.P. Disruption of human limb morphogenesis by a dominant negative mutation in CDMP1. Nat. Genet. 1997, 17, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Dawson, K.; Seeman, P.; Sebald, E.; King, L.; Edwards, M.; Williams, J., 3rd; Mundlos, S.; Krakow, D. GDF5 is a second locus for multiple-synostosis syndrome. Am. J. Hum. Genet. 2006, 78, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, Y.; Mabuchi, A.; Shi, D.; Kubo, T.; Takatori, Y.; Saito, S.; Fujioka, M.; Sudo, A.; Uchida, A.; Yamamoto, S.; et al. A functional polymorphism in the 5′ UTR of GDF5 is associated with susceptibility to osteoarthritis. Nat. Genet. 2007, 39, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Degenkolbe, E.; Konig, J.; Zimmer, J.; Walther, M.; Reissner, C.; Nickel, J.; Ploger, F.; Raspopovic, J.; Sharpe, J.; Dathe, K.; et al. A GDF5 point mutation strikes twice—Causing BDA1 and SYNS2. PLoS Genet. 2013, 9, e1003846. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Garcia, M.; Garcia-Canto, E.; Fenollar-Cortes, M.; Aytes, A.P.; Trujillo-Tiebas, M.J. Characterization of an acromesomelic dysplasia, Grebe type case: Novel mutation affecting the recognition motif at the processing site of GDF5. J. Bone Miner Metab. 2016, 34, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Xiao, Z.; Pan, Y.; Hu, J.; Pohl, J.; Wen, J.; Li, L. Cartilage-derived morphogenetic protein-1 promotes the differentiation of mesenchymal stem cells into chondrocytes. Biochem. Biophys. Res. Commun. 2004, 325, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Katayama, R.; Wakitani, S.; Tsumaki, N.; Morita, Y.; Matsushita, I.; Gejo, R.; Kimura, T. Repair of articular cartilage defects in rabbits using CDMP1 gene-transfected autologous mesenchymal cells derived from bone marrow. Rheumatology (Oxford) 2004, 43, 980–985. [Google Scholar] [CrossRef] [PubMed]
- Ayerst, B.I.; Smith, R.A.; Nurcombe, V.; Day, A.J.; Merry, C.L.; Cool, S.M. Growth Differentiation Factor 5-Mediated Enhancement of Chondrocyte Phenotype Is Inhibited by Heparin: Implications for the Use of Heparin in the Clinic and in Tissue Engineering Applications. Tissue Eng. Part A 2017, 23, 275–292. [Google Scholar] [CrossRef] [PubMed]
- DeLise, A.M.; Fischer, L.; Tuan, R.S. Cellular interactions and signaling in cartilage development. Osteoarthr. Cartil. 2000, 8, 309–334. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Zhou, G.; Morello, R.; Chen, Y.; Garcia-Rojas, X.; Lee, B. Type X collagen gene regulation by Runx2 contributes directly to its hypertrophic chondrocyte-specific expression in vivo. J. Cell Biol. 2003, 162, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.T.; Zhang, B.; Tian, Q.; Liu, Y.; Yang, S.H.; Shao, Z.W. Construction of self-assembled cartilage tissue from bone marrow mesenchymal stem cells induced by hypoxia combined with GDF-5. J. Huazhong Univ. Sci. Technolog. Med. Sci. 2013, 33, 700–706. [Google Scholar] [CrossRef] [PubMed]
- An, B.; Heo, H.-R.; Lee, S.; Park, J.-A.; Kim, K.-S.; Yang, J.; Hong, S.-H. Supplementation of Growth Differentiation Factor-5 Increases Proliferation and Size of Chondrogenic Pellets of Human Umbilical Cord-Derived Perivascular Stem Cells. Tissue Eng. Regen. Med. 2015, 12, 181–187. [Google Scholar] [CrossRef]
- Muraglia, A.; Cancedda, R.; Quarto, R. Clonal mesenchymal progenitors from human bone marrow differentiate in vitro according to a hierarchical model. J. Cell Sci. 2000, 113, 1161–1166. [Google Scholar] [PubMed]
- Roelofs, A.J.; Rocke, J.P.; de Bari, C. Cell-based approaches to joint surface repair: A research perspective. Osteoarthr. Cartil. 2013, 21, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Baraniak, P.R.; McDevitt, T.C. Stem cell paracrine actions and tissue regeneration. Regen. Med. 2010, 5, 121–143. [Google Scholar] [CrossRef] [PubMed]
- Anthony, D.F.; Shiels, P.G. Exploiting paracrine mechanisms of tissue regeneration to repair damaged organs. Transpl. Res. 2013, 2, 10. [Google Scholar] [CrossRef] [PubMed]
- Gnecchi, M.; Melo, L.G. Bone marrow-derived mesenchymal stem cells: Isolation, expansion, characterization, viral transduction, and production of conditioned medium. Methods Mol. Biol. 2009, 482, 281–294. [Google Scholar] [PubMed]
- Lee, R.H.; Pulin, A.A.; Seo, M.J.; Kota, D.J.; Ylostalo, J.; Larson, B.L.; Semprun-Prieto, L.; Delafontaine, P.; Prockop, D.J. Intravenous hMSCs improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti-inflammatory protein TSG-6. Cell Stem. Cell 2009, 5, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Ko, I.K.; Lee, S.J.; Atala, A.; Yoo, J.J. In situ tissue regeneration through host stem cell recruitment. Exp. Mol. Med. 2013, 45, e57. [Google Scholar] [CrossRef] [PubMed]
- Linero, I.; Chaparro, O. Paracrine effect of mesenchymal stem cells derived from human adipose tissue in bone regeneration. PLos ONE 2014, 9, e107001. [Google Scholar] [CrossRef] [PubMed]
- Hacker, S.; Mittermayr, R.; Nickl, S.; Haider, T.; Lebherz-Eichinger, D.; Beer, L.; Mitterbauer, A.; Leiss, H.; Zimmermann, M.; Schweiger, T.; et al. Paracrine Factors from Irradiated Peripheral Blood Mononuclear Cells Improve Skin Regeneration and Angiogenesis in a Porcine Burn Model. Sci. Rep. 2016, 6, 25168. [Google Scholar] [CrossRef] [PubMed]
- Martino, M.M.; Briquez, P.S.; Maruyama, K.; Hubbell, J.A. Extracellular matrix-inspired growth factor delivery systems for bone regeneration. Adv. Drug Deliv. Rev. 2015, 94, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Manning, M.C.; Patel, K.; Borchardt, R.T. Stability of protein pharmaceuticals. Pharm. Res. 1989, 6, 903–918. [Google Scholar] [CrossRef] [PubMed]
- Lauer, G.; Sollberg, S.; Cole, M.; Flamme, I.; Sturzebecher, J.; Mann, K.; Krieg, T.; Eming, S.A. Expression and proteolysis of vascular endothelial growth factor is increased in chronic wounds. J. Investig. Dermatol. 2000, 115, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Eppler, S.M.; Combs, D.L.; Henry, T.D.; Lopez, J.J.; Ellis, S.G.; Yi, J.H.; Annex, B.H.; McCluskey, E.R.; Zioncheck, T.F. A target-mediated model to describe the pharmacokinetics and hemodynamic effects of recombinant human vascular endothelial growth factor in humans. Clin. Pharmacol. Ther. 2002, 72, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Annex, B.H.; Laham, R.J.; Kleiman, N.; Henry, T.; Dauerman, H.; Udelson, J.E.; Gervino, E.V.; Pike, M.; Whitehouse, M.J.; et al. Pharmacological treatment of coronary artery disease with recombinant fibroblast growth factor-2: Double-blind, randomized, controlled clinical trial. Circulation 2002, 105, 788–793. [Google Scholar] [CrossRef] [PubMed]
- Shields, L.B.; Raque, G.H.; Glassman, S.D.; Campbell, M.; Vitaz, T.; Harpring, J.; Shields, C.B. Adverse effects associated with high-dose recombinant human bone morphogenetic protein-2 use in anterior cervical spine fusion. Spine 2006, 31, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Epstein, N.E. Complications due to the use of BMP/INFUSE in spine surgery: The evidence continues to mount. Surg. Neurol. Int. 2013, 4 (Suppl. 5), S343–S352. [Google Scholar] [CrossRef] [PubMed]
- Sreekumar, V.; Aspera-Werz, R.H.; Tendulkar, G.; Reumann, M.K.; Freude, T.; Breitkopf-Heinlein, K.; Dooley, S.; Pscherer, S.; Ochs, B.G.; Flesch, I.; et al. BMP9 a possible alternative drug for the recently withdrawn BMP7? New perspectives for (re-)implementation by personalized medicine. Arch. Toxicol. 2017, 91, 1353–1366. [Google Scholar] [CrossRef] [PubMed]
- Devine, J.G.; Dettori, J.R.; France, J.C.; Brodt, E.; McGuire, R.A. The use of rhBMP in spine surgery: Is there a cancer risk? Evid. Based Spine Care J. 2012, 3, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Carragee, E.J.; Chu, G.; Rohatgi, R.; Hurwitz, E.L.; Weiner, B.K.; Yoon, S.T.; Comer, G.; Kopjar, B. Cancer risk after use of recombinant bone morphogenetic protein-2 for spinal arthrodesis. J. Bone Joint Surg. Am. 2013, 95, 1537–1545. [Google Scholar] [CrossRef] [PubMed]
- Epstein, N.E. Basic science and spine literature document bone morphogenetic protein increases cancer risk. Surg. Neurol. Int. 2014, 5 (Suppl. 15), S552–S560. [Google Scholar] [CrossRef] [PubMed]
- Dettori, J.R.; Chapman, J.R.; DeVine, J.G.; McGuire, R.A.; Norvell, D.C.; Weiss, N.S. The Risk of Cancer With the Use of Recombinant Human Bone Morphogenetic Protein in Spine Fusion. Spine 2016, 41, 1317–1324. [Google Scholar] [CrossRef] [PubMed]
- Blanquaert, F.; Saffar, J.L.; Colombier, M.L.; Carpentier, G.; Barritault, D.; Caruelle, J.P. Heparan-like molecules induce the repair of skull defects. Bone 1995, 17, 499–506. [Google Scholar] [CrossRef]
- Uludag, H.; D'Augusta, D.; Palmer, R.; Timony, G.; Wozney, J. Characterization of rhBMP-2 pharmacokinetics implanted with biomaterial carriers in the rat ectopic model. J. Biomed. Mater. Res. 1999, 46, 193–202. [Google Scholar] [CrossRef]
- Lafont, J.; Blanquaert, F.; Colombier, M.L.; Barritault, D.; Carueelle, J.P.; Saffar, J.L. Kinetic study of early regenerative effects of RGTA11, a heparan sulfate mimetic, in rat craniotomy defects. Calcif. Tissue Int. 2004, 75, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Poon, S.; Murali, S.; Koo, C.Y.; Bell, T.J.; Hinkley, S.F.; Yeong, H.; Bhakoo, K.; Nurcombe, V.; Cool, S.M. Engineering a vascular endothelial growth factor 165-binding heparan sulfate for vascular therapy. Biomaterials 2014, 35, 6776–6786. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Fung, K.W.; Rodriguez, E.; Patel, R.; Gor, J.; Mulloy, B.; Perkins, S.J. The solution structure of heparan sulfate differs from that of heparin: Implications for function. J. Biol. Chem. 2013, 288, 27737–27751. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L. Glycosaminoglycan (GAG) biosynthesis and GAG-binding proteins. Prog. Mol. Biol. Transl. Sci. 2010, 93, 1–17. [Google Scholar] [PubMed]
- Lindahl, U.; Kjellen, L. Pathophysiology of heparan sulphate: Many diseases, few drugs. J. Intern. Med. 2013, 273, 555–571. [Google Scholar] [CrossRef] [PubMed]
- Krusius, T.; Ruoslahti, E. Primary structure of an extracellular matrix proteoglycan core protein deduced from cloned cDNA. Proc. Natl. Acad. Sci. USA 1986, 83, 7683–7687. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.; Grodzinsky, A.J.; Patwari, P.; Sandy, J.; Plaas, A.; Ortiz, C. Individual cartilage aggrecan macromolecules and their constituent glycosaminoglycans visualized via atomic force microscopy. J. Struct. Biol. 2003, 143, 242–257. [Google Scholar] [CrossRef] [PubMed]
- Gibson, B.G.; Briggs, M.D. The aggrecanopathies; an evolving phenotypic spectrum of human genetic skeletal diseases. Orphanet J Rare Dis. 2016, 11, 86. [Google Scholar] [CrossRef] [PubMed]
- Roughley, P.J.; Mort, J.S. The role of aggrecan in normal and osteoarthritic cartilage. J. Exp. Orthop. 2014, 1, 8. [Google Scholar] [CrossRef] [PubMed]
- Kiani, C.; Chen, L.; Wu, Y.J.; Yee, A.J.; Yang, B.B. Structure and function of aggrecan. Cell Res. 2002, 12, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.S.; McLane, L.T.; Fogg, R.; Scrimgeour, J.; Temenoff, J.S.; Granqvist, A.; Curtis, J.E. Cell Surface Access Is Modulated by Tethered Bottlebrush Proteoglycans. Biophys. J. 2016, 110, 2739–2750. [Google Scholar] [CrossRef] [PubMed]
- SundarRaj, N.; Fite, D.; Ledbetter, S.; Chakravarti, S.; Hassell, J.R. Perlecan is a component of cartilage matrix and promotes chondrocyte attachment. J. Cell Sci. 1995, 108, 2663–2672. [Google Scholar] [PubMed]
- Gu, G.; Deutch, A.Y.; Franklin, J.; Levy, S.; Wallace, D.C.; Zhang, J. Profiling genes related to mitochondrial function in mice treated with N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Biochem. Biophys. Res. Commun. 2003, 308, 197–205. [Google Scholar] [CrossRef]
- Echtermeyer, F.; Bertrand, J.; Dreier, R.; Meinecke, I.; Neugebauer, K.; Fuerst, M.; Lee, Y.J.; Song, Y.W.; Herzog, C.; Theilmeier, G.; et al. Syndecan-4 regulates ADAMTS-5 activation and cartilage breakdown in osteoarthritis. Nat. Med. 2009, 15, 1072–1076. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, R.E.; Defrate, L.E.; Guilak, F. A biomechanical role for perlecan in the pericellular matrix of articular cartilage. Matrix Biol. 2012, 31, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ly, M.; Linhardt, R.J. Proteoglycan sequence. Mol. Biosyst. 2012, 8, 1613–1625. [Google Scholar] [CrossRef] [PubMed]
- Caterson, B. Fell-Muir Lecture: Chondroitin sulphate glycosaminoglycans: Fun for some and confusion for others. Int. J. Exp. Pathol. 2012, 93, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yayon, A.; Klagsbrun, M.; Esko, J.D.; Leder, P.; Ornitz, D.M. Cell surface, heparin-like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor. Cell 1991, 64, 841–848. [Google Scholar] [CrossRef]
- Yayon, A.; Klagsbrun, M.; Esko, J.D.; Leder, P.; Ornitz, D.M. Heparin is required for cell-free binding of basic fibroblast growth factor to a soluble receptor and for mitogenesis in whole cells. Mol. Cell Biol. 1992, 12, 240–247. [Google Scholar]
- Schaefer, T.; Roux, M.; Stuhlsatz, H.W.; Herken, R.; Coulomb, B.; Krieg, T.; Smola, H. Glycosaminoglycans modulate cell-matrix interactions of human fibroblasts and endothelial cells in vitro. J. Cell Sci. 1996, 109, 479–488. [Google Scholar] [PubMed]
- Ji, Z.S.; Pitas, R.E.; Mahley, R.W. Differential cellular accumulation/retention of apolipoprotein E mediated by cell surface heparan sulfate proteoglycans. Apolipoproteins E3 and E2 greater than e4. J. Biol. Chem. 1998, 273, 13452–13460. [Google Scholar] [CrossRef] [PubMed]
- Linhardt, R.J.; Toida, T. Role of glycosaminoglycans in cellular communication. Acc. Chem. Res. 2004, 37, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Sadir, R.; Imberty, A.; Baleux, F.; Lortat-Jacob, H. Heparan sulfate/heparin oligosaccharides protect stromal cell-derived factor-1 (SDF-1)/CXCL12 against proteolysis induced by CD26/dipeptidyl peptidase IV. J. Biol. Chem. 2004, 279, 43854–43860. [Google Scholar] [CrossRef] [PubMed]
- Vives, R.R.; Lortat-Jacob, H.; Chroboczek, J.; Fender, P. Heparan sulfate proteoglycan mediates the selective attachment and internalization of serotype 3 human adenovirus dodecahedron. Virology 2004, 321, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Capurro, M.I.; Xu, P.; Shi, W.; Li, F.; Jia, A.; Filmus, J. Glypican-3 inhibits Hedgehog signaling during development by competing with patched for Hedgehog binding. Dev. Cell 2008, 14, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.N.; Pinali, C.; Young, R.D.; Meek, K.M.; Quantock, A.J.; Knupp, C. Structural interactions between collagen and proteoglycans are elucidated by three-dimensional electron tomography of bovine cornea. Structure 2010, 18, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Ortmann, C.; Pickhinke, U.; Exner, S.; Ohlig, S.; Lawrence, R.; Jboor, H.; Dreier, R.; Grobe, K. Sonic hedgehog processing and release are regulated by glypican heparan sulfate proteoglycans. J. Cell Sci. 2015, 128, 4462. [Google Scholar] [CrossRef] [PubMed]
- Shriver, Z.; Capila, I.; Venkataraman, G.; Sasisekharan, R. Heparin and heparan sulfate: Analyzing structure and microheterogeneity. Handb. Exp. Pharmacol. 2012, 207, 159–176. [Google Scholar]
- Mikami, T.; Kitagawa, H. Biosynthesis and function of chondroitin sulfate. Biochim. Biophys. Acta 2013, 1830, 4719–4733. [Google Scholar] [CrossRef] [PubMed]
- Poulain, F.E.; Yost, H.J. Heparan sulfate proteoglycans: A sugar code for vertebrate development? Development 2015, 142, 3456–3467. [Google Scholar] [CrossRef] [PubMed]
- Langford-Smith, A.; Keenan, T.D.; Clark, S.J.; Bishop, P.N.; Day, A.J. The role of complement in age-related macular degeneration: Heparan sulphate, a ZIP code for complement factor H? J. Innate Immun. 2014, 6, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Lamanna, W.C.; Kalus, I.; Padva, M.; Baldwin, R.J.; Merry, C.L.; Dierks, T. The heparanome—The enigma of encoding and decoding heparan sulfate sulfation. J. Biotechnol. 2007, 129, 290–307. [Google Scholar] [CrossRef] [PubMed]
- Pomin, V.H.; Mulloy, B. Current structural biology of the heparin interactome. Curr. Opin. Struct. Biol. 2015, 34, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Sasisekharan, R.; Venkataraman, G. Heparin and heparan sulfate: Biosynthesis, structure and function. Curr. Opin. Chem. Biol. 2000, 4, 626–631. [Google Scholar] [CrossRef]
- Kreuger, J.; et al. Interactions between heparan sulfate and proteins: The concept of specificity. J. Cell Biol. 2006, 174, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Whitelock, J.; Melrose, J. Heparan sulfate proteoglycans in healthy and diseased systems. Wiley Interdiscip. Rev. Syst. Biol. Med. 2011, 3, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Nugent, M.A.; Zaia, J.; Spencer, J.L. Heparan sulfate-protein binding specificity. Biochemistry (Moscow) 2013, 78, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.E.; Crawford, B.E.; Stavridis, M.; Ten Dam, G.; Wat, A.L.; Rushton, G.; Ward, C.M.; Wilson, V.; van Kuppevelt, T.H.; Esko, J.D.; et al. Essential alterations of heparan sulfate during the differentiation of embryonic stem cells to Sox1-enhanced green fluorescent protein-expressing neural progenitor cells. Stem. Cells 2007, 25, 1913–1923. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, R.J.; ten Dam, G.B.; van Kuppevelt, T.H.; Lacaud, G.; Gallagher, J.T.; Kouskoff, V.; Merry, C.L. A developmentally regulated heparan sulfate epitope defines a subpopulation with increased blood potential during mesodermal differentiation. Stem. Cells 2008, 26, 3108–3118. [Google Scholar] [CrossRef] [PubMed]
- Holley, R.J.; Pickford, C.E.; Rushton, G.; Lacaud, G.; Gallagher, J.T.; Kouskoff, V.; Merry, C.L. Influencing hematopoietic differentiation of mouse embryonic stem cells using soluble heparin and heparan sulfate saccharides. J. Biol. Chem. 2011, 286, 6241–6252. [Google Scholar] [CrossRef] [PubMed]
- Pickford, C.E.; Holley, R.J.; Rushton, G.; Stavridis, M.P.; Ward, C.M.; Merry, C.L. Specific glycosaminoglycans modulate neural specification of mouse embryonic stem cells. Stem. Cells 2011, 29, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Meade, K.A.; White, K.J.; Pickford, C.E.; Holley, R.J.; Marson, A.; Tillotson, D.; van Kuppevelt, T.H.; Whittle, J.D.; Day, A.J.; Merry, C.L. Immobilization of heparan sulfate on electrospun meshes to support embryonic stem cell culture and differentiation. J. Biol. Chem. 2013, 288, 5530–5538. [Google Scholar] [CrossRef] [PubMed]
- Nairn, A.V.; Aoki, K.; dela Rosa, M.; Porterfield, M.; Lim, J.M.; Kulik, M.; Pierce, J.M.; Wells, L.; Dalton, S.; Tiemeyer, M.; et al. Regulation of glycan structures in murine embryonic stem cells: Combined transcript profiling of glycan-related genes and glycan structural analysis. J. Biol. Chem. 2012, 287, 37835–37856. [Google Scholar] [CrossRef] [PubMed]
- Suchorska, W.M.; Lach, M.S.; Richter, M.; Kaczmarczyk, J.; Trzeciak, T. Bioimaging: An Useful Tool to Monitor Differentiation of Human Embryonic Stem Cells into Chondrocytes. Ann. Biomed. Eng. 2016, 44, 1845–1859. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Wei, G.; Shi, Z.; Dryer, L.; Esko, J.D.; Wells, D.E.; Matzuk, M.M. Disruption of gastrulation and heparan sulfate biosynthesis in EXT1-deficient mice. Dev. Biol. 2000, 224, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Stickens, D.; Zak, B.M.; Rougier, N.; Esko, J.D.; Werb, Z. Mice deficient in Ext2 lack heparan sulfate and develop exostoses. Development 2005, 132, 5055–5068. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Matsumoto, K.; Irie, F.; Fukushi, J.; Stallcup, W.B.; Yamaguchi, Y. Conditional ablation of the heparan sulfate-synthesizing enzyme Ext1 leads to dysregulation of bone morphogenic protein signaling and severe skeletal defects. J. Biol. Chem. 2010, 285, 19227–19234. [Google Scholar] [CrossRef] [PubMed]
- Vincent, T.; Hermansson, M.; Bolton, M.; Wait, R.; Saklatvala, J. Basic FGF mediates an immediate response of articular cartilage to mechanical injury. Proc. Natl. Acad. Sci. USA 2002, 99, 8259–8264. [Google Scholar] [CrossRef] [PubMed]
- Faiyaz ul Haque, M.; King, L.M.; Krakow, D.; Cantor, R.M.; Rusiniak, M.E.; Swank, R.T.; Superti-Furga, A.; Haque, S.; Abbas, H.; Ahmad, W.; Ahmad, M.; et al. Mutations in orthologous genes in human spondyloepimetaphyseal dysplasia and the brachymorphic mouse. Nat. Genet 1998, 20, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Otsuki, S.; Taniguchi, N.; Grogan, S.P.; D'Lima, D.; Kinoshita, M.; Lotz, M. Expression of novel extracellular sulfatases Sulf-1 and Sulf-2 in normal and osteoarthritic articular cartilage. Arthritis Res. Ther. 2008, 10, R61. [Google Scholar] [CrossRef] [PubMed]
- Otsuki, S.; Hanson, S.R.; Miyaki, S.; Grogan, S.P.; Kinoshita, M.; Asahara, H.; Wong, C.H.; Lotz, M.K. Extracellular sulfatases support cartilage homeostasis by regulating BMP and FGF signaling pathways. Proc. Natl. Acad. Sci. USA 2010, 107, 10202–10207. [Google Scholar] [CrossRef] [PubMed]
- San Antonio, J.D.; Winston, B.M.; Tuan, R.S. Regulation of chondrogenesis by heparan sulfate and structurally related glycosaminoglycans. Dev. Biol. 1987, 123, 17–24. [Google Scholar] [CrossRef]
- Fisher, M.C.; Li, Y.; Seghatoleslami, M.R.; Dealy, C.N.; Kosher, R.A. Heparan sulfate proteoglycans including syndecan-3 modulate BMP activity during limb cartilage differentiation. Matrix Biol. 2006, 25, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, Y.; Chen, C.; Lian, C.; Zhou, T.; Gao, B.; Wu, Z.; Xu, C. Exogenous Heparan Sulfate Enhances the TGF-beta3-Induced Chondrogenesis in Human Mesenchymal Stem Cells by Activating TGF-beta/Smad Signaling. Stem. Cells Int. 2016, 2016, 1520136. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.K.; Yang, W.; Kirn-Safran, C.B.; Farach-Carson, M.C.; Jia, X. Perlecan domain I-conjugated, hyaluronic acid-based hydrogel particles for enhanced chondrogenic differentiation via BMP-2 release. Biomaterials 2009, 30, 6964–6975. [Google Scholar] [CrossRef] [PubMed]
- Sadatsuki, R.; Kaneko, H.; Kinoshita, M.; Futami, I.; Nonaka, R.; Culley, K.L.; Otero, M.; Hada, S.; Goldring, M.B.; Yamada, Y.; et al. Perlecan is required for the chondrogenic differentiation of synovial mesenchymal cells through regulation of Sox9 gene expression. J. Orthop. Res. 2017, 35, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Turnbull, J.; Guimond, S. Extracellular matrix and cell signalling: The dynamic cooperation of integrin, proteoglycan and growth factor receptor. J. Endocrinol. 2011, 209, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Khoshgoftar, M.; Ito, K.; van Donkelaar, C.C. The influence of cell-matrix attachment and matrix development on the micromechanical environment of the chondrocyte in tissue-engineered cartilage. Tissue Eng. Part A 2014, 20, 3112–3121. [Google Scholar] [CrossRef] [PubMed]
- Hayes, A.J.; Shu, C.C.; Lord, M.S.; Little, C.B.; Whitelock, J.M.; Melrose, J. Pericellular colocalisation and interactive properties of type VI collagen and perlecan in the intervertebral disc. Eur. Cell Mater. 2016, 32, 40–57. [Google Scholar] [CrossRef] [PubMed]
- Schminke, B.; Frese, J.; Bode, C.; Goldring, M.B.; Miosge, N. Laminins and Nidogens in the Pericellular Matrix of Chondrocytes: Their Role in Osteoarthritis and Chondrogenic Differentiation. Am. J. Pathol. 2016, 186, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Li, Z.; Leng, Y.; Neu, C.P.; Calve, S. Knockdown of the pericellular matrix molecule perlecan lowers in situ cell and matrix stiffness in developing cartilage. Dev. Biol. 2016, 418, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, T.; Bayliss, M. Proteoglycans of articular cartilage: Changes in aging and in joint disease. Semin. Arthritis Rheum. 1990, 20 (Suppl. 1), 12–33. [Google Scholar] [CrossRef]
- Han, C.; Yan, D.; Belenkaya, T.Y.; Lin, X. Drosophila glypicans Dally and Dally-like shape the extracellular Wingless morphogen gradient in the wing disc. Development 2005, 132, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Ayerst, B.I.; Day, A.J.; Nurcombe, V.; Cool, S.M.; Merry, C.L. New strategies for cartilage regeneration exploiting selected glycosaminoglycans to enhance cell fate determination. Biochem. Soc. Trans. 2014, 42, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Coombe, D.R.; Kett, W.C. Heparan sulfate-protein interactions: Therapeutic potential through structure-function insights. Cell Mol. Life Sci. 2005, 62, 410–424. [Google Scholar] [CrossRef] [PubMed]
- Zaia, J. Glycosaminoglycan glycomics using mass spectrometry. Mol. Cell Proteomics 2013, 12, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Suflita, M.; Linhardt, R.J. Bioengineered heparins and heparan sulfates. Adv. Drug Deliv. Rev. 2016, 97, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Puvirajesinghe, T.M.; Turnbull, J.E. Glycoarray Technologies: Deciphering Interactions from Proteins to Live Cell Responses. Microarrays (Basel) 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.D.; Grande, D.A. The current state of scaffolds for musculoskeletal regenerative applications. Nat. Rev. Rheumatol. 2015, 11, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Yasui, N.; Ohsawa, S.; Ono, K. Chondrocytes embedded in collagen gels maintain cartilage phenotype during long-term cultures. Clin. Orthop. Relat. Res. 1984, 186, 231–239. [Google Scholar]
- Schulz, R.M.; Zscharnack, M.; Hanisch, I.; Geiling, M.; Hepp, P.; Bader, A. Cartilage tissue engineering by collagen matrix associated bone marrow derived mesenchymal stem cells. Biomed. Mater. Eng. 2008, 18 (Suppl. 1), S55–S70. [Google Scholar] [PubMed]
- Bertolo, A.; Arcolino, F.; Capossela, S.; Taddei, A.R.; Baur, M.; Potzel, T.; Stoyanov, J. Growth Factors Cross-Linked to Collagen Microcarriers Promote Expansion and Chondrogenic Differentiation of Human Mesenchymal Stem Cells. Tissue Eng. Part A 2015, 21, 2618–2628. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Portalatin, N.; Kilmer, C.E.; Panitch, A.; Liu, J.C. Characterization of Collagen Type I and II Blended Hydrogels for Articular Cartilage Tissue Engineering. Biomacromolecules 2016, 17, 3145–3152. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Portalatin, N.; Kilmer, C.E.; Panitch, A.; Liu, J.C. Knee joint preservation with combined neutralising high tibial osteotomy (HTO) and Matrix-induced Autologous Chondrocyte Implantation (MACI) in younger patients with medial knee osteoarthritis: A case series with prospective clinical and MRI follow-up over 5 years. Knee 2012, 19, 431–439. [Google Scholar]
- Ventura, A.; Memeo, A.; Borgo, E.; Terzaghi, C.; Legnani, C.; Albisetti, W. Repair of osteochondral lesions in the knee by chondrocyte implantation using the MACI(R) technique. Knee Surg. Sports Traumatol. Arthrosc. 2012, 20, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Deponti, D.; Di Giancamillo, A.; Gervaso, F.; Domenicucci, M.; Domeneghini, C.; Sannino, A.; Peretti, G.M. Collagen scaffold for cartilage tissue engineering: The benefit of fibrin glue and the proper culture time in an infant cartilage model. Tissue Eng. Part A 2014, 20, 1113–1126. [Google Scholar] [CrossRef] [PubMed]
- Kusano, T.; Jakob, R.P.; Gautier, E.; Magnussen, R.A.; Hoogewoud, H.; Jacobi, M. Treatment of isolated chondral and osteochondral defects in the knee by autologous matrix-induced chondrogenesis (AMIC). Knee Surg. Sports Traumatol. Arthrosc. 2012, 20, 2109–2115. [Google Scholar] [CrossRef] [PubMed]
- Gille, J.; Behrens, P.; Volpi, P.; de Girolamo, L.; Reiss, E.; Zoch, W.; Anders, S. Outcome of Autologous Matrix Induced Chondrogenesis (AMIC) in cartilage knee surgery: Data of the AMIC Registry. Arch. Orthop. Trauma Surg. 2013, 133, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Benthien, J.P.; Behrens, P. Nanofractured autologous matrix induced chondrogenesis (NAMIC(c))—Further development of collagen membrane aided chondrogenesis combined with subchondral needling: A technical note. Knee 2015, 22, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Levingstone, T.J.; Ramesh, A.; Brady, R.T.; Brama, P.A.; Kearney, C.; Gleeson, J.P.; O'Brien, F.J. Cell-free multi-layered collagen-based scaffolds demonstrate layer specific regeneration of functional osteochondral tissue in caprine joints. Biomaterials 2016, 87, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Levingstone, T.J.; Thompson, E.; Matsiko, A.; Schepens, A.; Gleeson, J.P.; O'Brien, F.J. Multi-layered collagen-based scaffolds for osteochondral defect repair in rabbits. Acta Biomater. 2016, 32, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Zhou, M.; Gough, J.; Glidle, A.; Yin, H. A novel culture system for modulating single cell geometry in 3D. Acta Biomater. 2015, 24, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Nehrer, S.; Domayer, S.; Dorotka, R.; Schatz, K.; Bindreiter, U.; Kotz, R. Three-year clinical outcome after chondrocyte transplantation using a hyaluronan matrix for cartilage repair. Eur. J. Radiol. 2006, 57, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Gobbi, A.; Kon, E.; Berruto, M.; Filardo, G.; Delcogliano, M.; Boldrini, L.; Bathan, L.; Marcacci, M. Patellofemoral full-thickness chondral defects treated with second-generation autologous chondrocyte implantation: Results at 5 years’ follow-up. Am. J. Sports Med. 2009, 37, 1083–1092. [Google Scholar] [CrossRef] [PubMed]
- Brix, M.O.; Stelzeneder, D.; Chiari, C.; Koller, U.; Nehrer, S.; Dorotka, R.; Windhager, R.; Domayer, S.E. Treatment of Full-Thickness Chondral Defects With Hyalograft C in the Knee: Long-term Results. Am. J. Sports Med. 2014, 42, 1426–1432. [Google Scholar] [CrossRef] [PubMed]
- Methot, S.; Changoor, A.; Tran-Khanh, N.; Hoemann, C.D.; Stanish, W.D.; Restrepo, A.; Shive, M.S.; Buschmann, M.D. Osteochondral Biopsy Analysis Demonstrates That BST-CarGel Treatment Improves Structural and Cellular Characteristics of Cartilage Repair Tissue Compared With Microfracture. Cartilage 2016, 7, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Merkle, H.P. Drug delivery’s quest for polymers: Where are the frontiers? Eur. J. Pharm. Biopharm. 2015, 97, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Holzwarth, J.M.; Ma, P.X. Functionalized synthetic biodegradable polymer scaffolds for tissue engineering. Macromol. Biosci. 2012, 12, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Temenoff, J.S.; Mikos, A.G. Review: Tissue engineering for regeneration of articular cartilage. Biomaterials 2000, 21, 431–440. [Google Scholar] [CrossRef]
- Gentile, P.; Chiono, V.; Carmagnola, I.; Hatton, P.V. An overview of poly(lactic-co-glycolic) acid (PLGA)-based biomaterials for bone tissue engineering. Int. J. Mol. Sci. 2014, 15, 3640–3659. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, Y.; Pan, Z.; Sun, H.; Wang, J.; Yu, D.; Zhu, S.; Dai, J.; Chen, Y.; Tian, N.; et al. The effects of lactate and acid on articular chondrocytes function: Implications for polymeric cartilage scaffold design. Acta Biomater. 2016, 42, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Gunatillake, P.A.; Adhikari, R. Biodegradable synthetic polymers for tissue engineering. Eur. Cell Mater. 2003, 5, 1–16, discussion 16. [Google Scholar] [CrossRef] [PubMed]
- Farah, S.; Anderson, D.G.; Langer, R. Physical and mechanical properties of PLA, and their functions in widespread applications—A comprehensive review. Adv. Drug Deliv. Rev. 2016, 107, 367–392. [Google Scholar] [CrossRef] [PubMed]
- Izal, I.; Aranda, P.; Sanz-Ramos, P.; Ripalda, P.; Mora, G.; Granero-Molto, F.; Deplaine, H.; Gomez-Ribelles, J.L.; Ferrer, G.G.; Acosta, V.; et al. Culture of human bone marrow-derived mesenchymal stem cells on of poly(L-lactic acid) scaffolds: Potential application for the tissue engineering of cartilage. Knee Surg. Sports Traumatol. Arthrosc. 2013, 21, 1737–1750. [Google Scholar] [CrossRef] [PubMed]
- Moran, J.M.; Pazzano, D.; Bonassar, L.J. Characterization of polylactic acid-polyglycolic acid composites for cartilage tissue engineering. Tissue Eng. 2003, 9, 63–70. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Feng, B.; Huang, C.; Wang, H.; Ge, Y.; Hu, R.; Yin, M.; Xu, Z.; Wang, W.; Fu, W.; et al. Electrospun gelatin/polycaprolactone nanofibrous membranes combined with a coculture of bone marrow stromal cells and chondrocytes for cartilage engineering. Int. J. Nanomedicine 2015, 10, 2089–2099. [Google Scholar] [PubMed]
- Olubamiji, A.D.; Izadifar, Z.; Si, J.L.; Cooper, D.M.; Eames, B.F.; Chen, D.X. Modulating mechanical behaviour of 3D-printed cartilage-mimetic PCL scaffolds: Influence of molecular weight and pore geometry. Biofabrication 2016, 8, 025020. [Google Scholar] [CrossRef] [PubMed]
- Deepthi, S.; Jayakumar, R. Prolonged release of TGF-beta from polyelectrolyte nanoparticle loaded macroporous chitin-poly(caprolactone) scaffold for chondrogenesis. Int. J. Biol. Macromol. 2016, 93, 1402–1409. [Google Scholar] [CrossRef] [PubMed]
- Casper, M.E.; Fitzsimmons, J.S.; Stone, J.J.; Meza, A.O.; Huang, Y.; Ruesink, T.J.; O’Driscoll, S.W.; Reinholz, G.G. Tissue engineering of cartilage using poly-epsilon-caprolactone nanofiber scaffolds seeded in vivo with periosteal cells. Osteoarthr. Cartil. 2010, 18, 981–991. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Ding, J. Poly(lactide-co-glycolide) porous scaffolds for tissue engineering and regenerative medicine. Interface Focus 2012, 2, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Haaparanta, A.M.; Jarvinen, E.; Cengiz, I.F.; Ella, V.; Kokkonen, H.T.; Kiviranta, I.; Kellomaki, M. Preparation and characterization of collagen/PLA, chitosan/PLA, and collagen/chitosan/PLA hybrid scaffolds for cartilage tissue engineering. J. Mater. Sci. Mater. Med. 2014, 25, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Chen, S.; Morsi, Y.; El-Hamshary, H.; El-Newhy, M.; Fan, C.; Mo, X. Superabsorbent 3D Scaffold Based on Electrospun Nanofibers for Cartilage Tissue Engineering. ACS Appl. Mater. Interfaces 2016, 8, 24415–24425. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Fan, D.; Zhao, L.; Yu, B.; Su, J.; Wei, J.; Shin, J.W. Degradability, cytocompatibility, and osteogenesis of porous scaffolds of nanobredigite and PCL-PEG-PCL composite. Int. J. Nanomedicine 2016, 11, 3545–3555. [Google Scholar] [PubMed]
- Lee, J.M.; Chae, T.; Sheikh, F.A.; Ju, H.W.; Moon, B.M.; Park, H.J.; Park, Y.R.; Park, C.H. Three dimensional poly(epsilon-caprolactone) and silk fibroin nanocomposite fibrous matrix for artificial dermis. Mater. Sci. Eng. C Mater. Biol. Appl. 2016, 68, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Sonomoto, K.; Yamaoka, K.; Kaneko, H.; Yamagata, K.; Sakata, K.; Zhang, X.; Kondo, M.; Zenke, Y.; Sabanai, K.; Nakayamada, S.; et al. Spontaneous Differentiation of Human Mesenchymal Stem Cells on Poly-Lactic-Co-Glycolic Acid Nano-Fiber Scaffold. PLoS ONE 2016, 11, e0153231. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ding, B.; Li, B. Biomimetic electrospun nanofibrous structures for tissue engineering. Mater. Today (Kidlington) 2013, 16, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Bonzani, I.C.; George, J.H.; Stevens, M.M. Novel materials for bone and cartilage regeneration. Curr. Opin. Chem. Biol. 2006, 10, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Holmes, B.; Castro, N.J.; Zhang, L.G.; Zussman, E. Electrospun fibrous scaffolds for bone and cartilage tissue generation: Recent progress and future developments. Tissue Eng. Part B 2012, 18, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Wise, J.K.; Yarin, A.L.; Megaridis, C.M.; Cho, M. Chondrogenic differentiation of human mesenchymal stem cells on oriented nanofibrous scaffolds: Engineering the superficial zone of articular cartilage. Tissue Eng. Part A 2009, 15, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Shores, L.; Guo, Q.; Aly, A.; Jeon, O.H.; Kim do, H.; Bernstein, N.; Bhattacharya, R.; Chae, J.J.; Yarema, K.J.; et al. Electrospun Microfiber Scaffolds with Anti-Inflammatory Tributanoylated N-Acetyl-d-Glucosamine Promote Cartilage Regeneration. Tissue Eng. Part A 2016, 22, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Tzezana, R.; Zussman, E.; Levenberg, S. A layered ultra-porous scaffold for tissue engineering, created via a hydrospinning method. Tissue Eng. Part C 2008, 14, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Rnjak-Kovacina, J.; Weiss, A.S. Increasing the pore size of electrospun scaffolds. Tissue Eng. Part B 2011, 17, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Pham, Q.P.; Sharma, U.; Mikos, A.G. Electrospun poly(epsilon-caprolactone) microfiber and multilayer nanofiber/microfiber scaffolds: Characterization of scaffolds and measurement of cellular infiltration. Biomacromolecules 2006, 7, 2796–2805. [Google Scholar] [CrossRef] [PubMed]
- Ekaputra, A.K.; Prestwich, G.D.; Cool, S.M.; Hutmacher, D.W. Combining electrospun scaffolds with electrosprayed hydrogels leads to three-dimensional cellularization of hybrid constructs. Biomacromolecules 2008, 9, 2097–2103. [Google Scholar] [CrossRef] [PubMed]
- Soliman, S.; Pagliari, S.; Rinaldi, A.; Forte, G.; Fiaccavento, R.; Pagliari, F.; Franzese, O.; Minieri, M.; Di Nardo, P.; Licoccia, S.; et al. Multiscale three-dimensional scaffolds for soft tissue engineering via multimodal electrospinning. Acta Biomater. 2010, 6, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Takanari, K.; Hong, Y.; Hashizume, R.; Huber, A.; Amoroso, N.J.; D'Amore, A.; Badylak, S.F.; Wagner, W.R. Abdominal wall reconstruction by a regionally distinct biocomposite of extracellular matrix digest and a biodegradable elastomer. J. Tissue Eng. Regen. Med. 2016, 10, 748–761. [Google Scholar] [CrossRef] [PubMed]
- Coburn, J.; Gibson, M.; Bandalini, P.A.; Laird, C.; Mao, H.Q.; Moroni, L.; Seliktar, D.; Elisseeff, J. Biomimetics of the Extracellular Matrix: An Integrated Three-Dimensional Fiber-Hydrogel Composite for Cartilage Tissue Engineering. Smart Struct. Syst. 2011, 7, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.L.; Mauck, R.L.; Burdick, J.A. Hydrogel design for cartilage tissue engineering: A case study with hyaluronic acid. Biomaterials 2011, 32, 8771–8782. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; He, Z.Y.; Wei, X.W.; Wei, Y.Q. Recent advances of biomaterials in biotherapy. Regen. Biomater. 2016, 3, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.T.; Cool, S.M.; Hui, J.H.; Hutmacher, D.W. The influence of fibrin based hydrogels on the chondrogenic differentiation of human bone marrow stromal cells. Biomaterials 2010, 31, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; Ozturk, E.; Arlov, O.; Gatenholm, P.; Zenobi-Wong, M. Alginate Sulfate-Nanocellulose Bioinks for Cartilage Bioprinting Applications. Ann. Biomed. Eng. 2017, 45, 210–223. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, M.; Azadi, A.; Rafiei, P. Hydrogel nanoparticles in drug delivery. Adv. Drug Deliv. Rev. 2008, 60, 1638–1649. [Google Scholar] [CrossRef] [PubMed]
- Koutsopoulos, S.; Unsworth, L.D.; Nagai, Y.; Zhang, S. Controlled release of functional proteins through designer self-assembling peptide nanofiber hydrogel scaffold. Proc. Natl. Acad. Sci. USA 2009, 106, 4623–4628. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cheetham, A.G.; Angacian, G.; Su, H.; Xie, L.; Cui, H. Peptide-drug conjugates as effective prodrug strategies for targeted delivery. Adv. Drug. Deliv. Rev. 2016. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cheetham, A.G.; Angacian, G.; Su, H.; Xie, L.; Cui, H. Self-assembled peptide-based hydrogels as scaffolds for anchorage-dependent cells. Biomaterials 2009, 30, 2523–2530. [Google Scholar]
- Zhou, M.; Ulijn, R.V.; Gough, J.E. Extracellular matrix formation in self-assembled minimalistic bioactive hydrogels based on aromatic peptide amphiphiles. J. Tissue Eng. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Saiani, A.; Frielinghaus, H. Self-assembly and gelation properties of a-helix versus b-sheet forming peptides. Soft Matter 2009, 5, 193–202. [Google Scholar] [CrossRef]
- Zhou, M.; Smith, A.M.; Das, A.K.; Hodson, N.W.; Collins, R.F.; Ulijn, R.V.; Gough, J.E. Self-assembled octapeptide scaffolds for in vitro chondrocyte culture. Acta Biomater. 2013, 9, 4609–4617. [Google Scholar]
- King, P.J.; Giovanna Lizio, M.; Booth, A.; Collins, R.F.; Gough, J.E.; Miller, A.F.; Webb, S.J. A modular self-assembly approach to functionalised beta-sheet peptide hydrogel biomaterials. Soft Matter 2016, 12, 1915–1923. [Google Scholar] [CrossRef] [PubMed]
- Coxon, T.P.; Fallows, T.W.; Gough, J.E.; Webb, S.J. A versatile approach towards multivalent saccharide displays on magnetic nanoparticles and phospholipid vesicles. Org. Biomol. Chem. 2015, 13, 10751–10761. [Google Scholar] [CrossRef] [PubMed]
- Freudenberg, U.; Liang, Y.; Kiick, K.L.; Werner, C. Glycosaminoglycan-Based Biohybrid Hydrogels: A Sweet and Smart Choice for Multifunctional Biomaterials. Adv. Mater. 2016, 28, 8861–8891. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Erickson, I.E.; Choudhury, M.; Pleshko, N.; Mauck, R.L. Transient exposure to TGF-beta3 improves the functional chondrogenesis of MSC-laden hyaluronic acid hydrogels. J. Mech. Behav. Biomed. Mater. 2012, 11, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Getgood, A.; Henson, F.; Skelton, C.; Herrera, E.; Brooks, R.; Fortier, L.A.; Rushton, N. The Augmentation of a Collagen/Glycosaminoglycan Biphasic Osteochondral Scaffold with Platelet-Rich Plasma and Concentrated Bone Marrow Aspirate for Osteochondral Defect Repair in Sheep: A Pilot Study. Cartilage 2012, 3, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Kang, I.K.; Huh, M.W.; Yoon, S.C. Surface characterization and in vitro blood compatibility of poly(ethylene terephthalate) immobilized with insulin and/or heparin using plasma glow discharge. Biomaterials 2000, 21, 121–130. [Google Scholar] [CrossRef]
- Ji, Y.; Ghosh, K.; Shu, X.Z.; Li, B.; Sokolov, J.C.; Prestwich, G.D.; Clark, R.A.; Rafailovich, M.H. Electrospun three-dimensional hyaluronic acid nanofibrous scaffolds. Biomaterials 2006, 27, 3782–3792. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Chen, X.; Pan, Y.; Cui, Y.; Zhou, X.; Kong, D.; Zhao, Q. Enhanced vascularization in hybrid PCL/gelatin fibrous scaffolds with sustained release of VEGF. Biomed. Res. Int. 2015, 2015, 865076. [Google Scholar] [CrossRef] [PubMed]
- Hua, Q.; Knudson, C.B.; Knudson, W. Internalization of hyaluronan by chondrocytes occurs via receptor-mediated endocytosis. J. Cell Sci. 1993, 106, 365–375. [Google Scholar] [PubMed]
- Sorensen, V.; Nilsen, T.; Wiedlocha, A. Functional diversity of FGF-2 isoforms by intracellular sorting. Bioessays 2006, 28, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Payne, C.K.; Jones, S.A.; Chen, C.; Zhuang, X. Internalization and trafficking of cell surface proteoglycans and proteoglycan-binding ligands. Traffic 2007, 8, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, D.J.; Whittle, J.D.; Milner, C.M.; Clark, S.J.; Mulloy, B.; Buttle, D.J.; Jones, G.C.; Day, A.J.; Short, R.D. A method for the non-covalent immobilization of heparin to surfaces. Anal. Biochem. 2004, 330, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Keselowsky, B.G.; Collard, D.M.; Garcia, A.J. Surface chemistry modulates fibronectin conformation and directs integrin binding and specificity to control cell adhesion. J. Biomed. Mater. Res. A 2003, 66, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.E.; Buttle, D.J.; Short, R.D.; McArthur, S.L.; Steele, D.A.; Whittle, J.D. Glycosaminoglycan (GAG) binding surfaces for characterizing GAG-protein interactions. Biomaterials 2012, 33, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Rinsch, C.L.; Chen, X.; Panchalingam, V.; Eberhart, R.C.; Wang, J.H.; Timmons, R.B. Pulsed radio frequency plasma polymerization of allyl alcohol: Controlled deposition of surface hydroxyl groups. Langmuir 1996, 12, 2995–3002. [Google Scholar] [CrossRef]
- Marson, A.; Robinson, D.E.; Brookes, P.N.; Mulloy, B.; Wiles, M.; Clark, S.J.; Fielder, H.L.; Collinson, L.J.; Cain, S.A.; Kielty, C.M.; et al. Development of a microtiter plate-based glycosaminoglycan array for the investigation of glycosaminoglycan-protein interactions. Glycobiology 2009, 19, 1537–1546. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.E.; Marson, A.; Short, R.D.; Buttle, D.J.; Day, A.J.; Parry, K.L.; Wiles, M.; Highfield, P.; Mistry, A.; Whittle, J.D. Surface gradient of functional heparin. Adv. Mater. 2008, 20, 1166–1169. [Google Scholar] [CrossRef]
- Yang, Z.; Tu, Q.; Wang, J.; Huang, N. The role of heparin binding surfaces in the direction of endothelial and smooth muscle cell fate and re-endothelialization. Biomaterials 2012, 33, 6615–6625. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Hong, B.; Lee, J.; Kim, S.E.; Kang, S.S.; Kim, Y.H.; Tae, G. Composite system of PLCL scaffold and heparin-based hydrogel for regeneration of partial-thickness cartilage defects. Biomacromolecules 2012, 13, 2287–2298. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; An, Q.; Li, D.; Wu, T.; Chen, W.; Sun, B.; El-Hamshary, H.; Al-Deyab, S.S.; Zhu, W.; Mo, X. Heparin and Vascular Endothelial Growth Factor Loaded Poly(L-lactide-co-caprolactone) Nanofiber Covered Stent-Graft for Aneurysm Treatment. J. Biomed. Nanotechnol. 2015, 11, 1947–1960. [Google Scholar] [CrossRef] [PubMed]
- Sackler, J.P.; Liu, L. Heparin-induced osteoporosis. Br. J. Radiol. 1973, 46, 548–550. [Google Scholar] [CrossRef] [PubMed]
- Bounameaux, H.; Schneider, P.A.; Mossaz, A.; Suter, P.; Vasey, H. Severe vasospastic reactions (ergotism) during prophylactic administration of heparin-dihydroergotamine. Vasa 1987, 16, 370–372. [Google Scholar] [PubMed]
- Mazziotti, G.; Canalis, E.; Giustina, A. Drug-induced osteoporosis: Mechanisms and clinical implications. Am. J. Med. 2010, 123, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Bambrah, R.K.; Pham, D.C.; Zaiden, R.; Vu, H.; Tai, S. Heparin-induced thrombocytopenia. Clin. Adv. Hematol. Oncol. 2011, 9, 594–599. [Google Scholar] [PubMed]
- Ling, L.; Camilleri, E.T.; Helledie, T.; Samsonraj, R.M.; Titmarsh, D.M.; Chua, R.J.; Dreesen, O.; Dombrowski, C.; Rider, D.A.; Galindo, M.; et al. Effect of heparin on the biological properties and molecular signature of human mesenchymal stem cells. Gene 2016, 576, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Ling, L.; Camilleri, E.T.; Helledie, T.; Samsonraj, R.M.; Titmarsh, D.M.; Chua, R.J.; Dreesen, O.; Dombrowski, C.; Rider, D.A.; Galindo, M.; et al. Oversulfated chondroitin sulfate is a contaminant in heparin associated with adverse clinical events. Nat. Biotechnol. 2008, 26, 669–675. [Google Scholar]
- Mulloy, B.; Wu, N.; Gyapon-Quast, F.; Lin, L.; Zhang, F.; Pickering, M.C.; Linhardt, R.J.; Feizi, T.; Chai, W. Abnormally High Content of Free Glucosamine Residues Identified in a Preparation of Commercially Available Porcine Intestinal Heparan Sulfate. Anal. Chem. 2016, 88, 6648–6652. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Linhardt, R.J. Chemoenzymatic synthesis of heparan sulfate and heparin. Nat. Prod. Rep. 2014, 31, 1676–1685. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Xu, Y.; Chen, M.; Weiwer, M.; Zhou, X.; Bridges, A.S.; DeAngelis, P.L.; Zhang, Q.; Linhardt, R.J.; Liu, J. Chemoenzymatic design of heparan sulfate oligosaccharides. J. Biol. Chem. 2010, 285, 34240–34249. [Google Scholar] [CrossRef] [PubMed]
- Peterson, S.; Frick, A.; Liu, J. Design of biologically active heparan sulfate and heparin using an enzyme-based approach. Nat. Prod. Rep. 2009, 26, 610–627. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, Y.; Yu, H.; Sugiarto, G.; Thon, V.; Hwang, J.; Ding, L.; Hie, L.; Chen, X. Tailored design and synthesis of heparan sulfate oligosaccharide analogues using sequential one-pot multienzyme systems. Angew. Chem. Int. Ed. Engl. 2013, 52, 11852–11856. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Cai, C.; Chandarajoti, K.; Hsieh, P.H.; Li, L.; Pham, T.Q.; Sparkenbaugh, E.M.; Sheng, J.; Key, N.S.; Pawlinski, R.; et al. Homogeneous low-molecular-weight heparins with reversible anticoagulant activity. Nat. Chem. Biol. 2014, 10, 248–250. [Google Scholar] [CrossRef] [PubMed]
- Ori, A.; Wilkinson, M.C.; Fernig, D.G. A systems biology approach for the investigation of the heparin/heparan sulfate interactome. J. Biol. Chem. 2011, 286, 19892–19904. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Kim, S.E.; Kang, S.S.; Kim, Y.H.; Tae, G. The use of de-differentiated chondrocytes delivered by a heparin-based hydrogel to regenerate cartilage in partial-thickness defects. Biomaterials 2011, 32, 7883–7896. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.C.; Tsai, Y.T. Heparin-conjugated scaffolds with pore structure of inverted colloidal crystals for cartilage regeneration. Coll. Surf. B Biointerfaces 2011, 82, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Muinos, T.; Recha-Sancho, L.; Lopez-Chicon, P.; Castells-Sala, C.; Mata, A.; Semino, C.E. Bimolecular based heparin and self-assembling hydrogel for tissue engineering applications. Acta Biomater. 2015, 16, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Deng, L.Z.; Sun, H.P.; Xu, J.Y.; Li, Y.M.; Xie, X.; Zhang, L.M.; Deng, F.L. Sustained dual release of placental growth factor-2 and bone morphogenic protein-2 from heparin-based nanocomplexes for direct osteogenesis. Int. J. Nanomedicine 2016, 11, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Recha-Sancho, L.; Semino, C.E. Heparin based self-assembling peptide scaffold reestablish chondrogenic phenotype of expanded de-differentiated human chondrocytes. J. Biomed. Mater. Res. A 2016, 104, 1694–1706. [Google Scholar] [CrossRef] [PubMed]
- Zwingenberger, S.; Langanke, R.; Vater, C.; Lee, G.; Niederlohmann, E.; Sensenschmidt, M.; Jacobi, A.; Bernhardt, R.; Muders, M.; Rammelt, S.; et al. The effect of SDF-1alpha on low dose BMP-2 mediated bone regeneration by release from heparinized mineralized collagen type I matrix scaffolds in a murine critical size bone defect model. J. Biomed. Mater. Res. A 2016, 104, 2126–2134. [Google Scholar] [CrossRef] [PubMed]
- Salek-Ardakani, S.; Arrand, J.R.; Shaw, D.; Mackett, M. Heparin and heparan sulfate bind interleukin-10 and modulate its activity. Blood 2000, 96, 1879–1888. [Google Scholar] [PubMed]
- Kanzaki, S.; Takahashi, T.; Kanno, T.; Ariyoshi, W.; Shinmyouzu, K.; Tujisawa, T.; Nishihara, T. Heparin inhibits BMP-2 osteogenic bioactivity by binding to both BMP-2 and BMP receptor. J. Cell Physiol. 2008, 216, 844–850. [Google Scholar] [CrossRef] [PubMed]
- Hirsh, J.; Anand, S.S.; Halperin, J.L.; Fuster, V. AHA Scientific Statement: Guide to anticoagulant therapy: Heparin: A statement for healthcare professionals from the American Heart Association. Arterioscler. Thromb. Vasc. Biol. 2001, 21, E9. [Google Scholar] [CrossRef] [PubMed]
- Kan, A.; Ikeda, T.; Fukai, A.; Nakagawa, T.; Nakamura, K.; Chung, U.I.; Kawaguchi, H.; Tabin, C.J. SOX11 contributes to the regulation of GDF5 in joint maintenance. BMC Dev. Biol. 2013, 13, 4. [Google Scholar] [CrossRef] [PubMed]
- Fujise, M.; Takeo, S.; Kamimura, K.; Matsuo, T.; Aigaki, T.; Izumi, S.; Nakato, H. Dally regulates Dpp morphogen gradient formation in the Drosophila wing. Development 2003, 130, 1515–1522. [Google Scholar] [CrossRef] [PubMed]
- Bornemann, D.J.; Duncan, J.E.; Staatz, W.; Selleck, S.; Warrior, R. Abrogation of heparan sulfate synthesis in Drosophila disrupts the Wingless, Hedgehog and Decapentaplegic signaling pathways. Development 2004, 131, 1927–1938. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M. FGFs, heparan sulfate and FGFRs: Complex interactions essential for development. Bioessays 2000, 22, 108–112. [Google Scholar] [CrossRef]
- Pellegrini, L. Role of heparan sulfate in fibroblast growth factor signalling: A structural view. Curr. Opin. Struct. Biol. 2001, 11, 629–634. [Google Scholar] [CrossRef]
- Kuo, W.J.; Digman, M.A.; Lander, A.D. Heparan sulfate acts as a bone morphogenetic protein coreceptor by facilitating ligand-induced receptor hetero-oligomerization. Mol. Biol. Cell. 2010, 21, 4028–4041. [Google Scholar] [CrossRef] [PubMed]
- Irimura, T.; Nakajima, M.; Nicolson, G.L. Chemically modified heparins as inhibitors of heparan sulfate specific endo-beta-glucuronidase (heparanase) of metastatic melanoma cells. Biochemistry 1986, 25, 5322–5328. [Google Scholar] [CrossRef] [PubMed]
- Bar-Ner, M.; Eldor, A.; Wasserman, L.; Matzner, Y.; Cohen, I.R.; Fuks, Z.; Vlodavsky, I. Inhibition of heparanase-mediated degradation of extracellular matrix heparan sulfate by non-anticoagulant heparin species. Blood 1987, 70, 551–557. [Google Scholar] [PubMed]
- Parish, C.R.; Coombe, D.R.; Jakobsen, K.B.; Bennett, F.A.; Underwood, P.A. Evidence that sulphated polysaccharides inhibit tumour metastasis by blocking tumour-cell-derived heparanases. Int. J. Cancer 1987, 40, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Huegel, J.; Enomoto-Iwamoto, M.; Sgariglia, F.; Koyama, E.; Pacifici, M. Heparanase stimulates chondrogenesis and is up-regulated in human ectopic cartilage: A mechanism possibly involved in hereditary multiple exostoses. Am. J. Pathol. 2015, 185, 1676–1685. [Google Scholar] [CrossRef] [PubMed]
- Cardin, A.D.; Weintraub, H.J. Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis 1989, 9, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Ori, A.; Free, P.; Courty, J.; Wilkinson, M.C.; Fernig, D.G. Identification of heparin-binding sites in proteins by selective labeling. Mol. Cell Proteom. 2009, 8, 2256–2265. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.C.; Mulloy, B.; Magee, A.I.; Couchman, J.R. Two distinct sites in sonic Hedgehog combine for heparan sulfate interactions and cell signaling functions. J. Biol. Chem. 2011, 286, 44391–44402. [Google Scholar] [CrossRef] [PubMed]
- Uniewicz, K.A.; Ori, A.; Ahmed, Y.A.; Yates, E.A.; Fernig, D.G. Characterisation of the interaction of neuropilin-1 with heparin and a heparan sulfate mimetic library of heparin-derived sugars. PeerJ 2014, 2, e461. [Google Scholar] [CrossRef] [PubMed]
- Tatsinkam, A.J.; Mulloy, B.; Rider, C.C. Mapping the heparin-binding site of the BMP antagonist gremlin by site-directed mutagenesis based on predictive modelling. Biochem. J. 2015, 470, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Esko, J.D.; Kimata, K.; Lindahl, U. Proteoglycans and Sulfated Glycosaminoglycans. In Essentials of Glycobiology; Varki, A., Ed.; Cold Spring Harbor: New York, NY, USA, 2009. [Google Scholar]
- Powell, A.K.; Yates, E.A.; Fernig, D.G.; Turnbull, J.E. Interactions of heparin/heparan sulfate with proteins: Appraisal of structural factors and experimental approaches. Glycobiology 2004, 14, 17R–30R. [Google Scholar] [CrossRef] [PubMed]
- Mosier, P.D.; Krishnasamy, C.; Kellogg, G.E.; Desai, U.R. On the specificity of heparin/heparan sulfate binding to proteins. Anion-binding sites on antithrombin and thrombin are fundamentally different. PLoS ONE 2012, 7, e48632. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, J. Fell-Muir Lecture: Heparan sulphate and the art of cell regulation: A polymer chain conducts the protein orchestra. Int. J. Exp. Pathol. 2015, 96, 203–231. [Google Scholar] [CrossRef] [PubMed]
- Olson, S.T.; Halvorson, H.R.; Bjork, I. Quantitative characterization of the thrombin-heparin interaction. Discrimination between specific and nonspecific binding models. J. Biol. Chem. 1991, 266, 6342–6352. [Google Scholar] [PubMed]
- Xu, D.; Esko, J.D. Demystifying heparan sulfate-protein interactions. Annu. Rev. Biochem. 2014, 83, 129–157. [Google Scholar] [CrossRef] [PubMed]
- Petitou, M.; van Boeckel, C.A. A synthetic antithrombin III binding pentasaccharide is now a drug! What comes next? Angew. Chem. Int. Ed. Engl. 2004, 43, 3118–3133. [Google Scholar] [CrossRef] [PubMed]
- Atha, D.H.; Lormeau, J.C.; Petitou, M.; Rosenberg, R.D.; Choay, J. Contribution of monosaccharide residues in heparin binding to antithrombin III. Biochemistry 1985, 24, 6723–6729. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wang, Z.; Liu, R.; Bridges, A.S.; Huang, X.; Liu, J. Directing the biological activities of heparan sulfate oligosaccharides using a chemoenzymatic approach. Glycobiology 2012, 22, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Lortat-Jacob, H.; Turnbull, J.E.; Grimaud, J.A. Molecular organization of the interferon gamma-binding domain in heparan sulphate. Biochem. J. 1995, 310, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Merry, C.L.; Bullock, S.L.; Swan, D.C.; Backen, A.C.; Lyon, M.; Beddington, R.S.; Wilson, V.A.; Gallagher, J.T. The molecular phenotype of heparan sulfate in the Hs2st-/- mutant mouse. J. Biol. Chem. 2001, 276, 35429–35434. [Google Scholar] [CrossRef] [PubMed]
- Skidmore, M.A.; Guimond, S.E.; Rudd, T.R.; Fernig, D.G.; Turnbull, J.E.; Yates, E.A. The activities of heparan sulfate and its analogue heparin are dictated by biosynthesis, sequence, and conformation. Connect. Tissue Res. 2008, 49, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Catlow, K.R.; Deakin, J.A.; Wei, Z.; Delehedde, M.; Fernig, D.G.; Gherardi, E.; Gallagher, J.T.; Pavao, M.S.; Lyon, M. Interactions of hepatocyte growth factor/scatter factor with various glycosaminoglycans reveal an important interplay between the presence of iduronate and sulfate density. J. Biol. Chem. 2008, 283, 5235–5248. [Google Scholar] [CrossRef] [PubMed]
- Raman, R.; Sasisekharan, V.; Sasisekharan, R. Structural insights into biological roles of protein-glycosaminoglycan interactions. Chem. Biol. 2005, 12, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Capila, I.; Linhardt, R.J. Heparin-protein interactions. Angew. Chem. Int. Ed. Engl. 2002, 41, 391–412. [Google Scholar] [CrossRef]
- Thompson, L.D.; Pantoliano, M.W.; Springer, B.A. Energetic characterization of the basic fibroblast growth factor-heparin interaction: Identification of the heparin binding domain. Biochemistry 1994, 33, 3831–3840. [Google Scholar] [CrossRef] [PubMed]
- Asensio, J.L.; Arda, A.; Canada, F.J.; Jimenez-Barbero, J. Carbohydrate-aromatic interactions. Acc. Chem. Res. 2013, 46, 946–954. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Desai, U.R. A Simple Method for Discovering Druggable, Specific Glycosaminoglycan-Protein Systems. Elucidation of Key Principles from Heparin/Heparan Sulfate-Binding Proteins. PLoS ONE 2015, 10, e0141127. [Google Scholar] [CrossRef] [PubMed]
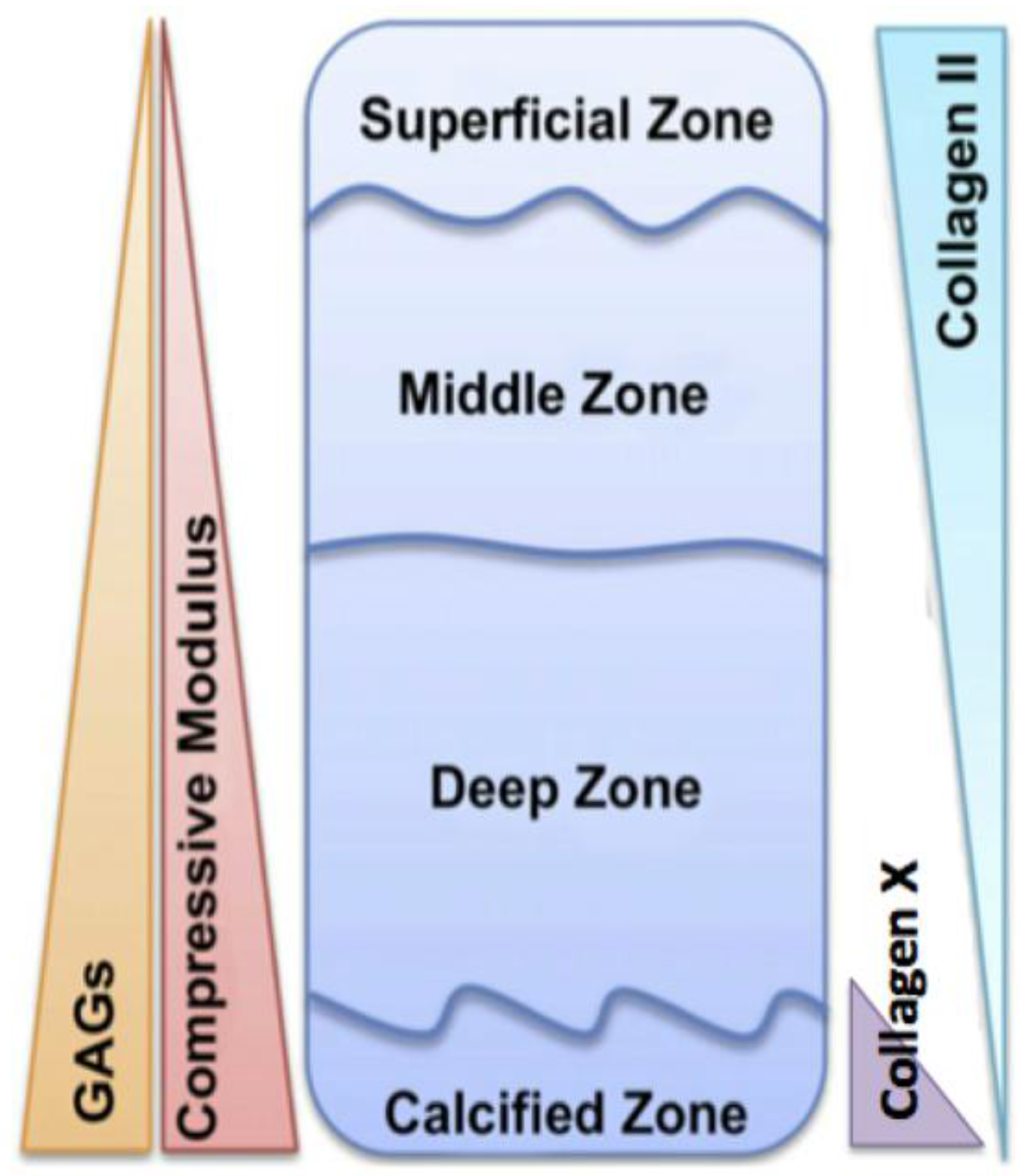
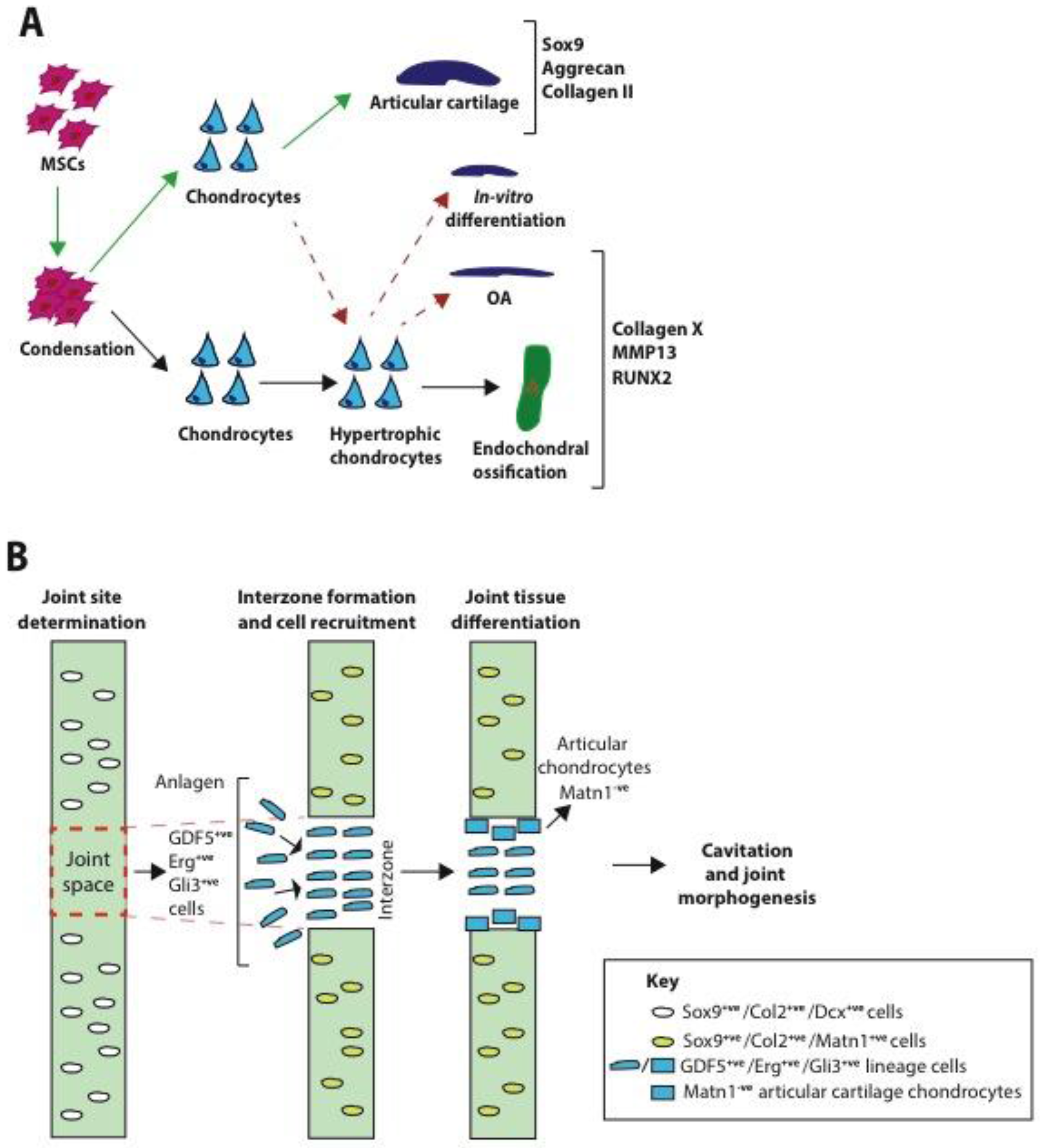
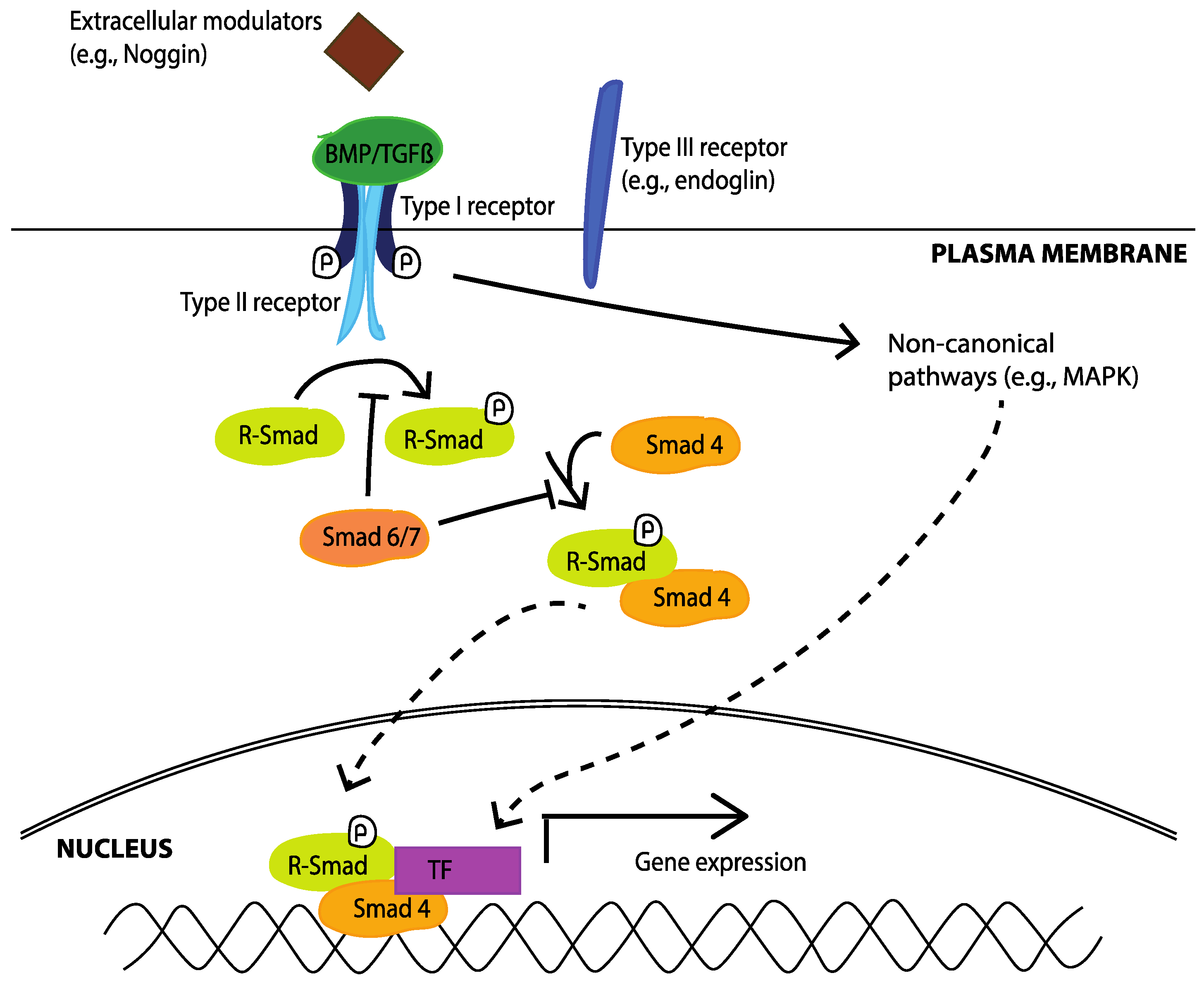
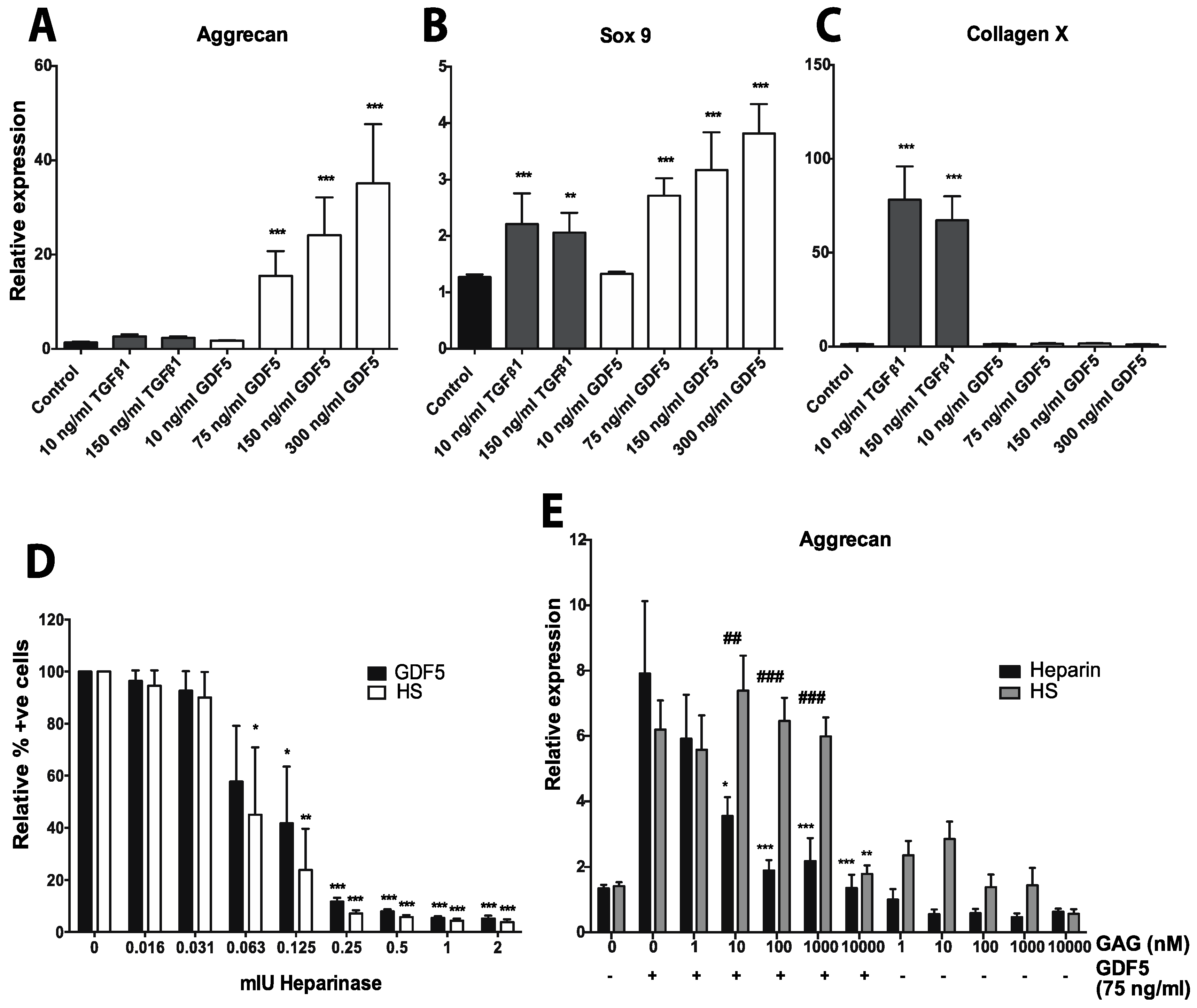
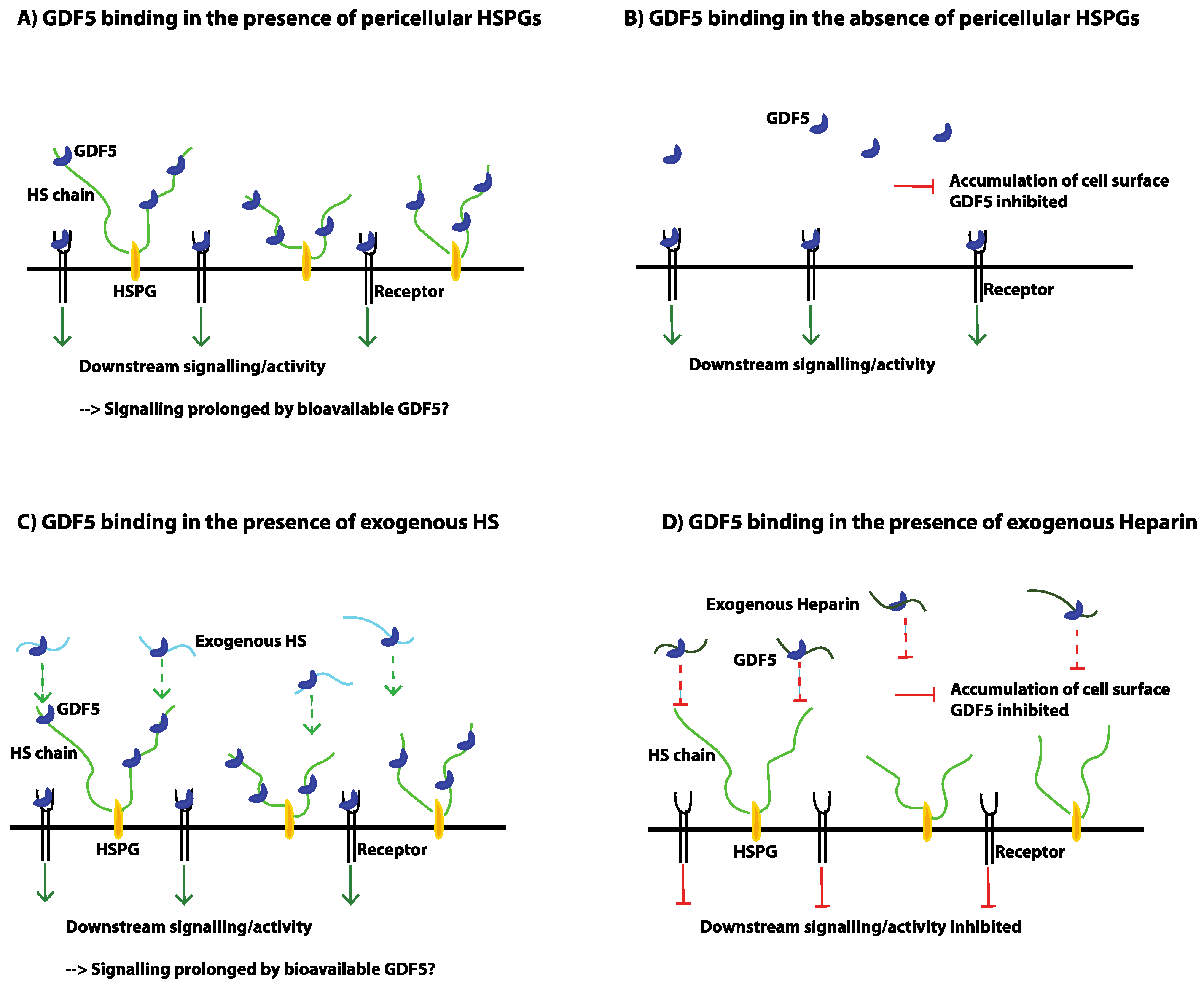

{kind=link}
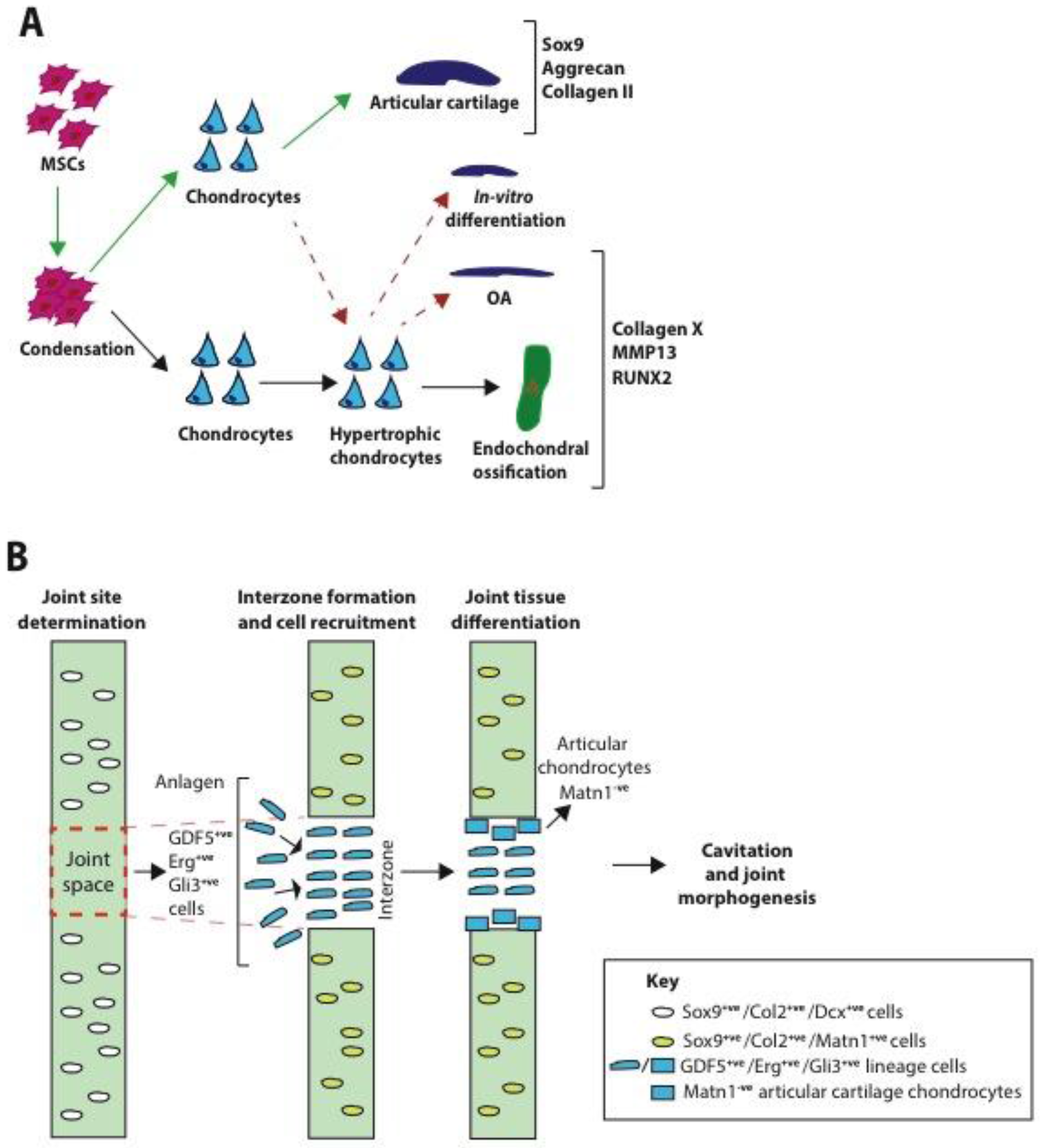{kind=link}
{kind=link}
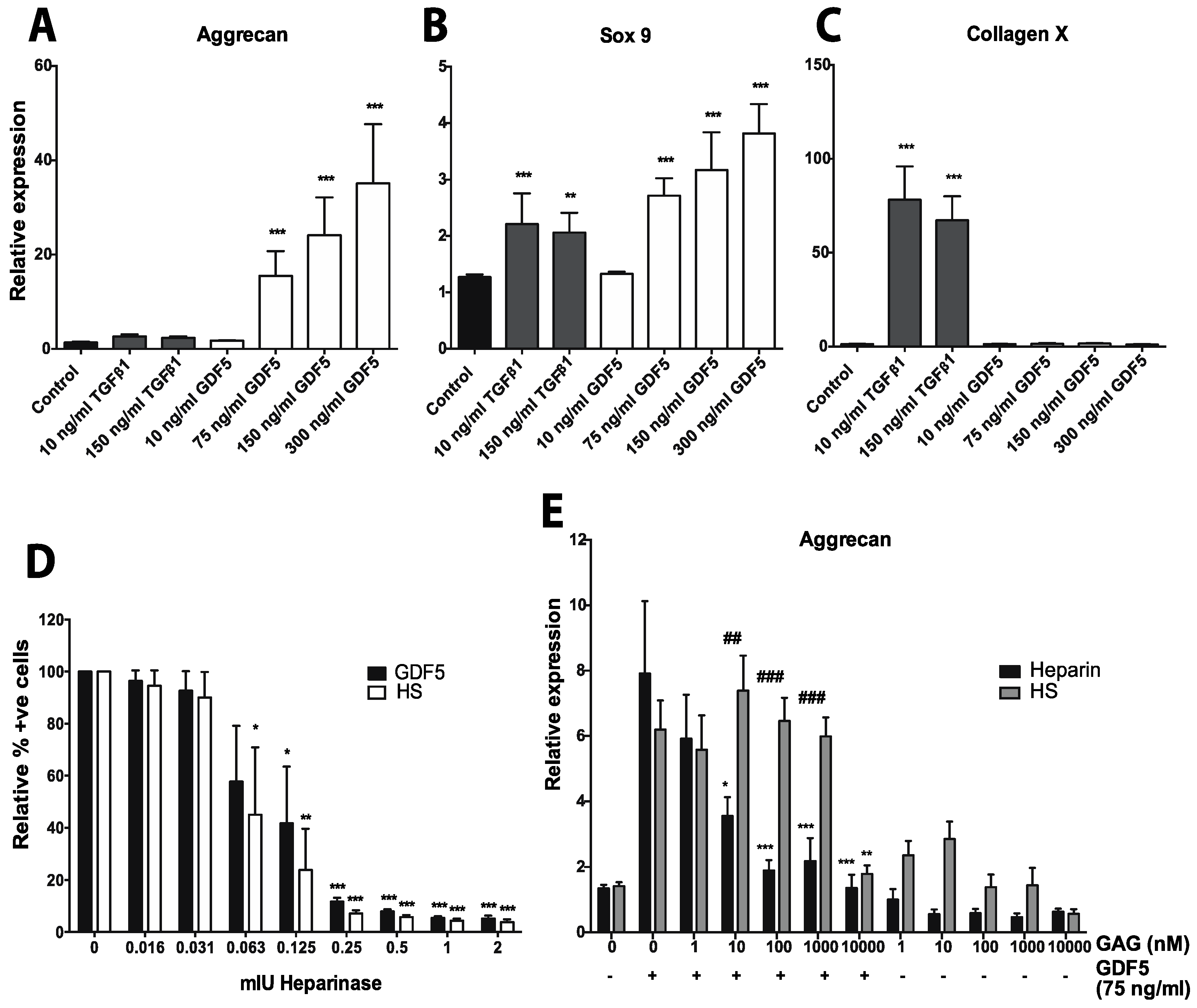{kind=link}
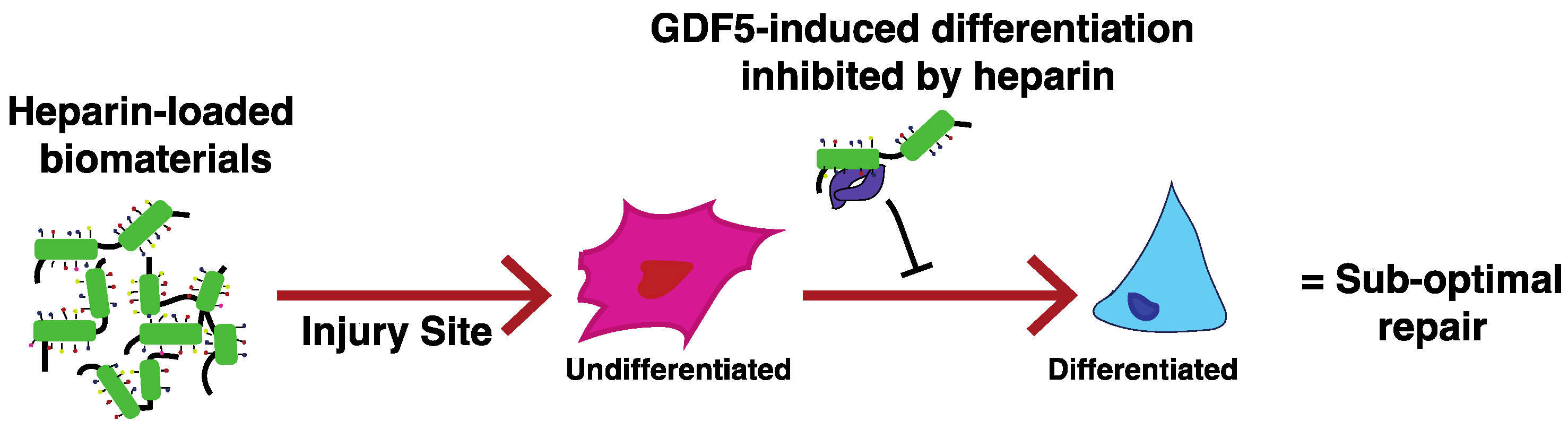{kind=link}
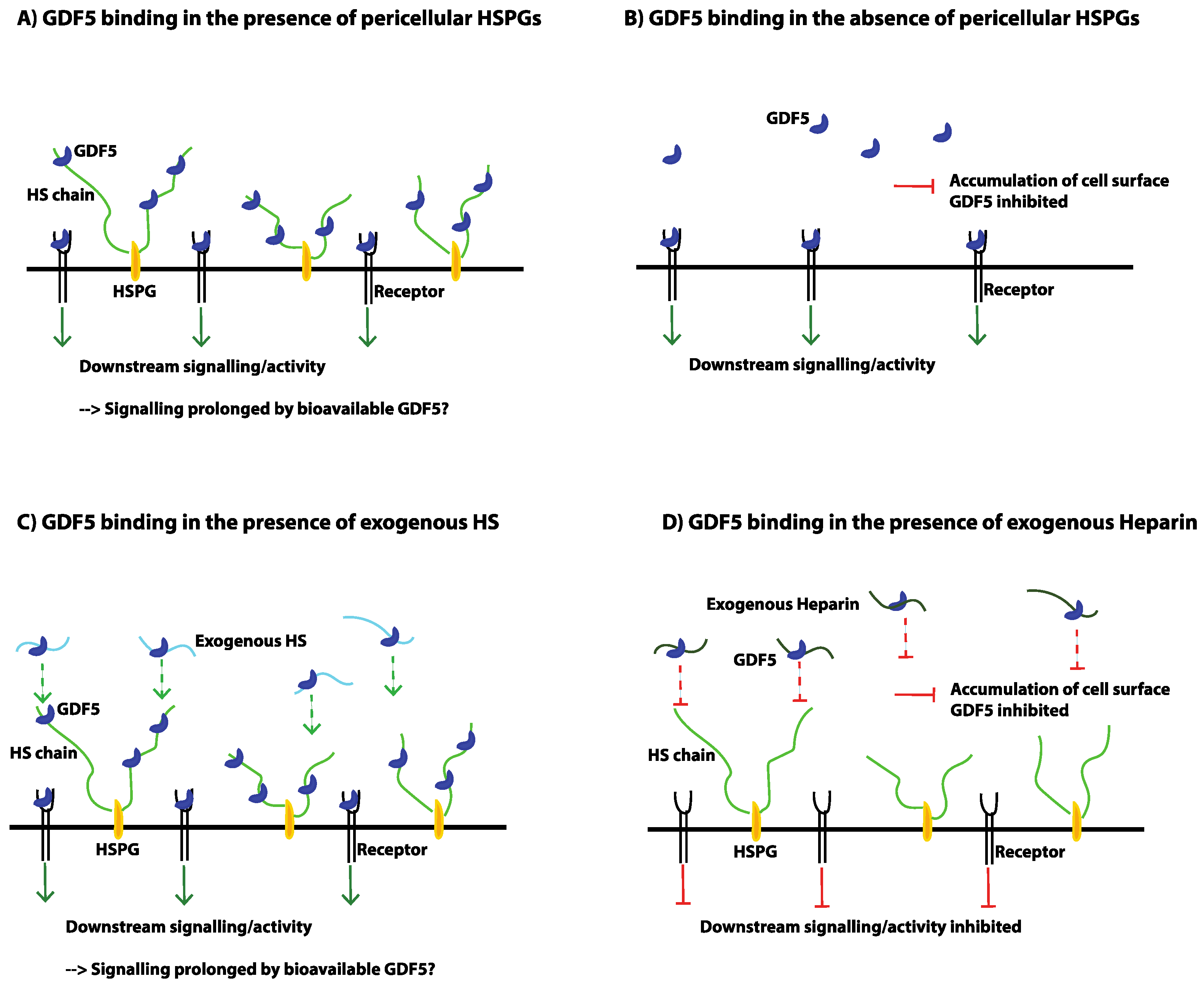{kind=link}
| Molecule Family | Molecule | Proposed Function during Chondrogenesis | Reference | Associated PGs/GAGs | Reference |
|---|---|---|---|---|---|
| FGF 1 | FGF2 | Enhances proliferation and chondrogenic potential during expansion. | [59,148,149,150,151] | Role of HSPGs in FGF-receptor binding has been extensively studied; HSPGs play an important role in FGF-receptor signalling by facilitating ligand-receptor oligomerisation. | [155,156,157,158,159] |
| Negative effect on matrix deposition and differentiation. | [151,152,153] | ||||
| Addition during expansion primes cells for hypertrophy. | [149,151] | ||||
| Prolongs lifespan of MSCs. | [154] | ||||
| FGF9 | Increases matrix production early on, but then promotes matrix resorption and hypertrophy. | [152] | CS sulfation patterns have also been implicated in articular cartilage formation and expression has been co-localised with FGF2. Perlecan can only deliver FGF2 to its receptors after its CS chains have been removed. | [160,161,162] | |
| However, also reported to promote matrix production and delay terminal hypertrophy. | [151] | ||||
| FGF18 | Suppresses proliferation and promotes matrix production. | [151,164] | Exogenous HS can be used to improve hMSC expansion. | [163] | |
| Delays terminal hypertrophy. | [151] | ||||
| TGFβ | TGFβ1/3 | Promotes chondrogenic differentiation of MSCs. Considered a main chondrogenic inducer of MSCs, however, leads to chondrocyte hypertrophy. | [36,37,38,39,41,75,128,165] | TGFβ1 but not TGFβ3 has been shown to bind to HS; effects of the interaction remain conflicting. | [166,167,168,169] |
| TGFβ3 better supports chondrogenic differentiation than TGFβ1. | TGFβ binds to the small leucine rich PGs, decorin, biglycan and fibromodulin, but via their protein core; CS/DS chains interfere with this binding. | [170] | |||
| BMP | GDF5 | Important role in joint formation and organisation of articular cartilage; GDF5 expressed in healthy pre-hypertrophic cartilage, but not as OA develops; GDF5 dominant negative mutation results in articular cartilage degeneration. | [23,171,172,173,174,175] | Heparin binding sequence predicted. | [182] |
| Increases cartilaginous ECM production in vitro. | [176,177,178,179] | ||||
| Supplementation with TGFβ3 shown to promote hypertrophy. However, a combinatorial study with TGFβ1, BMP2 and GDF5 suggest that it is the TGFβ actually promoting hypertrophy. | [180,181] | ||||
| BMP2/4/6/7 | Promotes chondrogenic differentiation of MSCs, especially when used in combination with TGFβ. BMP2/7 indicated as particularly useful for inducing chondrogenesis. However, most studies indicate that BMP supplementation also leads to hypertrophy. | [153,181,183,184,185,186] | Use of heparin/HS to potentiate the activity of BMPs has been widely studied; especially in the case of BMP2 for bone TE. | [187,188,189,190,191,192,193,194,195] | |
| Wnt | Wnt3a, Wnt5a | Promotes chondrogenic differentiation. | [196,197,198] | Glypican3 is strongly linked to the Wnt pathway; HS chains bind to Wnts with different affinities to fine-tune access to Wnt receptors; 6-O-desulfation of HS reduces ability of Glypican1 HS chains to bind Wnt, and therefore facilitates Wnt-receptor interaction. | [200,201,202] |
| Inhibits hypertrophy. | [199] | ||||
| However, Wnt5a also reported to promote hypertrophy during early stages of differentiation. | |||||
| Wnt11 | Promotes chondrogenic differentiation and hypertrophy. | [203] | |||
| Wnt4, Wnt8 | Inhibits chondrogenic differentiation. Promotes hypertrophy. | [196,204] | |||
| Wnt9a | Inhibits chondrogenic differentiation. Inhibits hypertrophy. | [205] | |||
| IGF | IGF1 | When used in combination with TGFβ3 collagen I production is reduced. | [183] | Heparin/HS/DS stimulate the release of free and bioactive IGF1 from IGF binding proteins | [207] |
| Promotes hypertrophic differentiation. | [206] | ||||
| PTHrP | PTHrP (1–34) isoform | Inhibits TGFβ induced hypertrophic differentiation. | [39,153,208,209] | PTHrP is activated by Ihh signalling (feedback loop). HS binds Ihh and negatively regulates signalling. | [210,211] |
| Molecular Category | TGFβ Sub-Family Pathway 1 | BMP Sub-Family Pathway |
|---|---|---|
| Ligands | TGFβs, ActivinsGDF8/9/10/11, BMP3, Nodal | BMP2/4/5/6/7/8/9/10, GDF1/3/5/6/7, MIS |
| Type II receptors | TβRII, ActRIIA, ActRIIB | BMPRII, ActRIIA, ActRIIB |
| Type I receptors | ALK4, TβRI (ALK5), ALK7 | ALK1/2, BMPR1A (ALK3), BMPR1B (ALK6) |
| R-Smad | Smad2/3 | Smad1/5/8 |
| Co-Smad | Smad4 | Smad4 |
| I-Smad | Smad7 | Smad6/7 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ayerst, B.I.; Merry, C.L.R.; Day, A.J. The Good the Bad and the Ugly of Glycosaminoglycans in Tissue Engineering Applications. Pharmaceuticals 2017, 10, 54. https://doi.org/10.3390/ph10020054
Ayerst BI, Merry CLR, Day AJ. The Good the Bad and the Ugly of Glycosaminoglycans in Tissue Engineering Applications. Pharmaceuticals. 2017; 10(2):54. https://doi.org/10.3390/ph10020054
Chicago/Turabian StyleAyerst, Bethanie I., Catherine L.R. Merry, and Anthony J. Day. 2017. "The Good the Bad and the Ugly of Glycosaminoglycans in Tissue Engineering Applications" Pharmaceuticals 10, no. 2: 54. https://doi.org/10.3390/ph10020054
APA StyleAyerst, B. I., Merry, C. L. R., & Day, A. J. (2017). The Good the Bad and the Ugly of Glycosaminoglycans in Tissue Engineering Applications. Pharmaceuticals, 10(2), 54. https://doi.org/10.3390/ph10020054