
D11-Mediated Inhibition of Protein Kinase CK2 Impairs HIF-1α-Mediated Signaling in Human Glioblastoma Cells



Abstract
:1. Introduction
2. Results
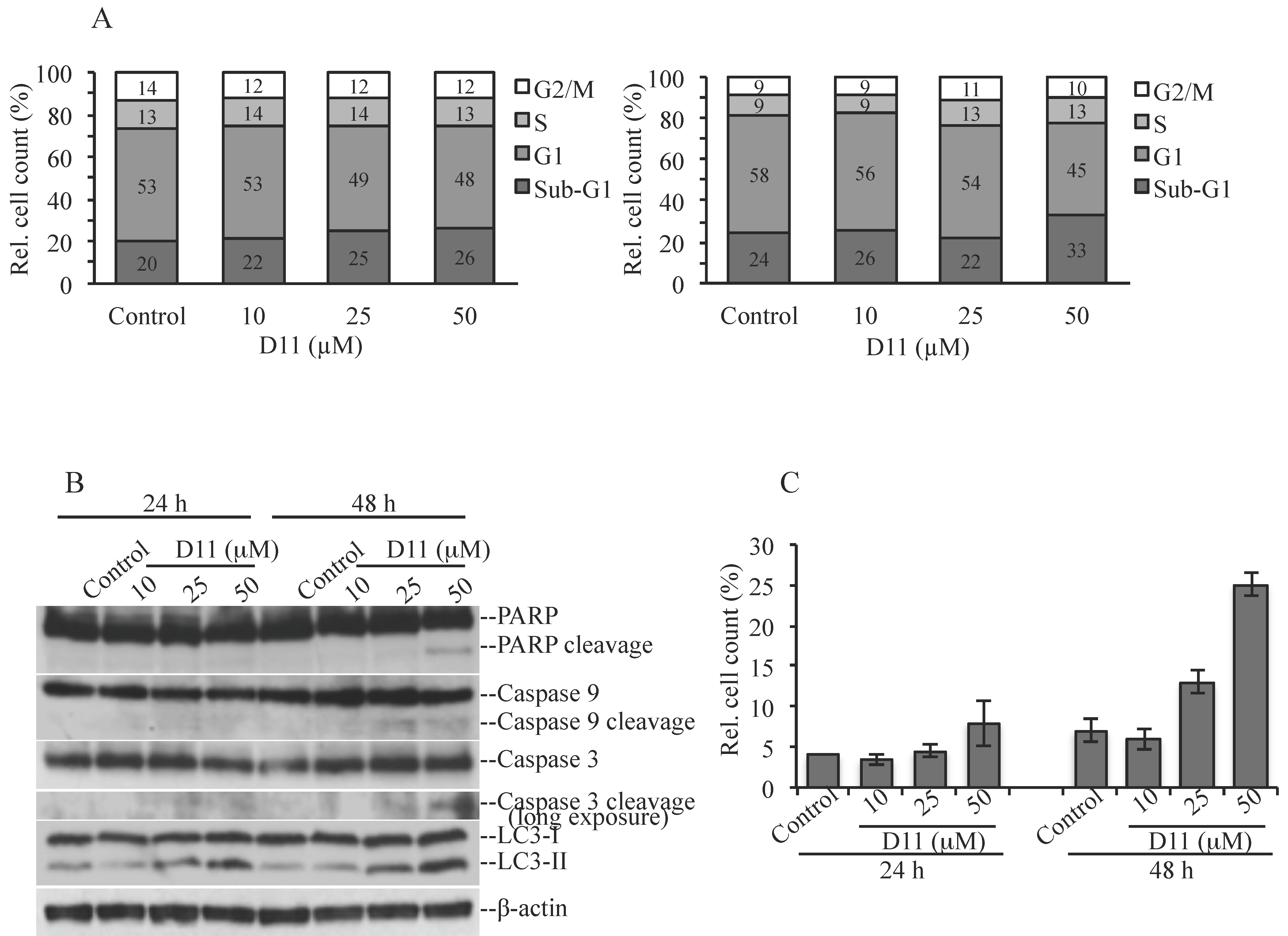
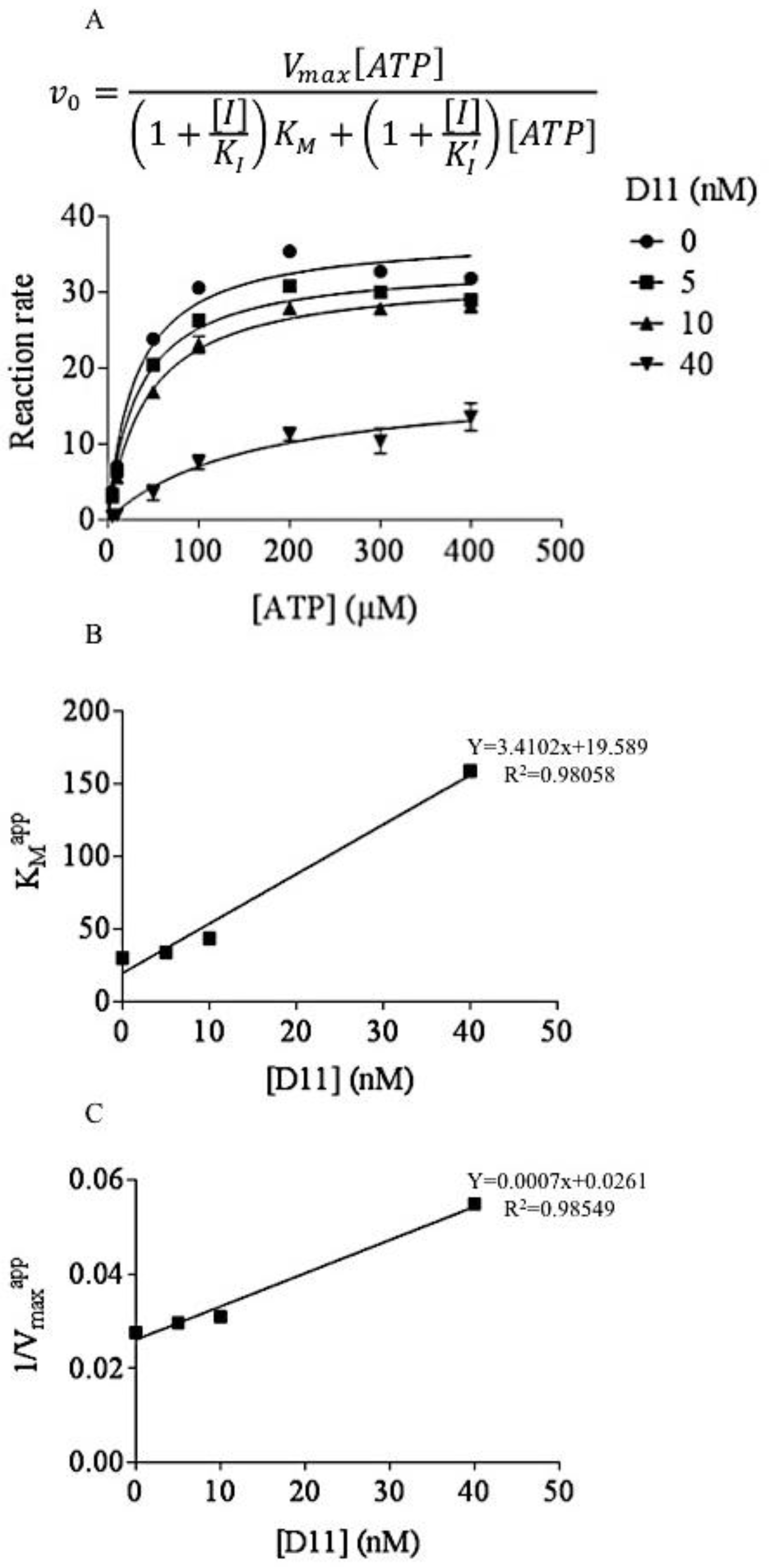
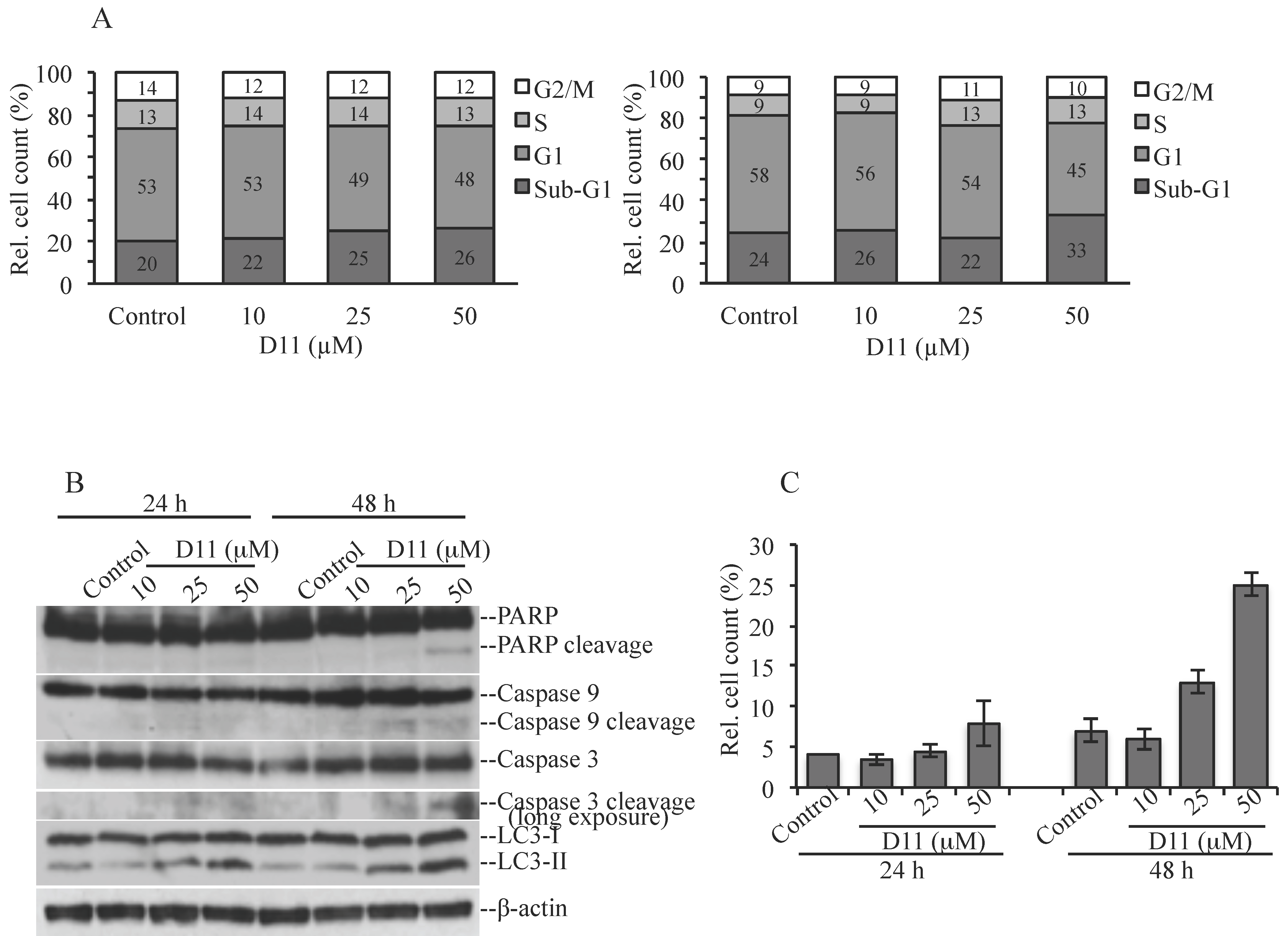
2.1. D11 is A CK2 Inhibitor That Leads to Cell Death in Glioblastoma Cells
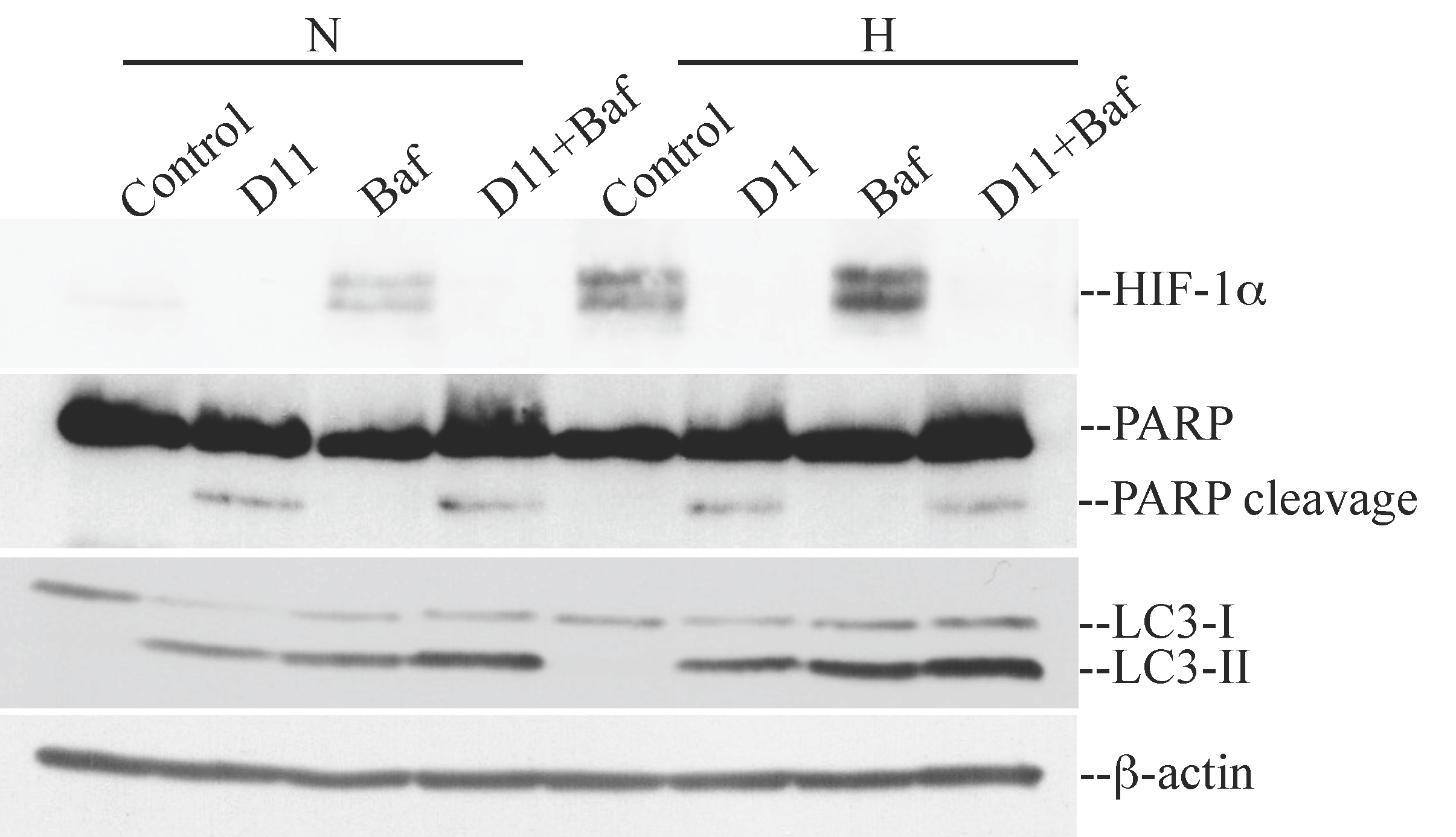
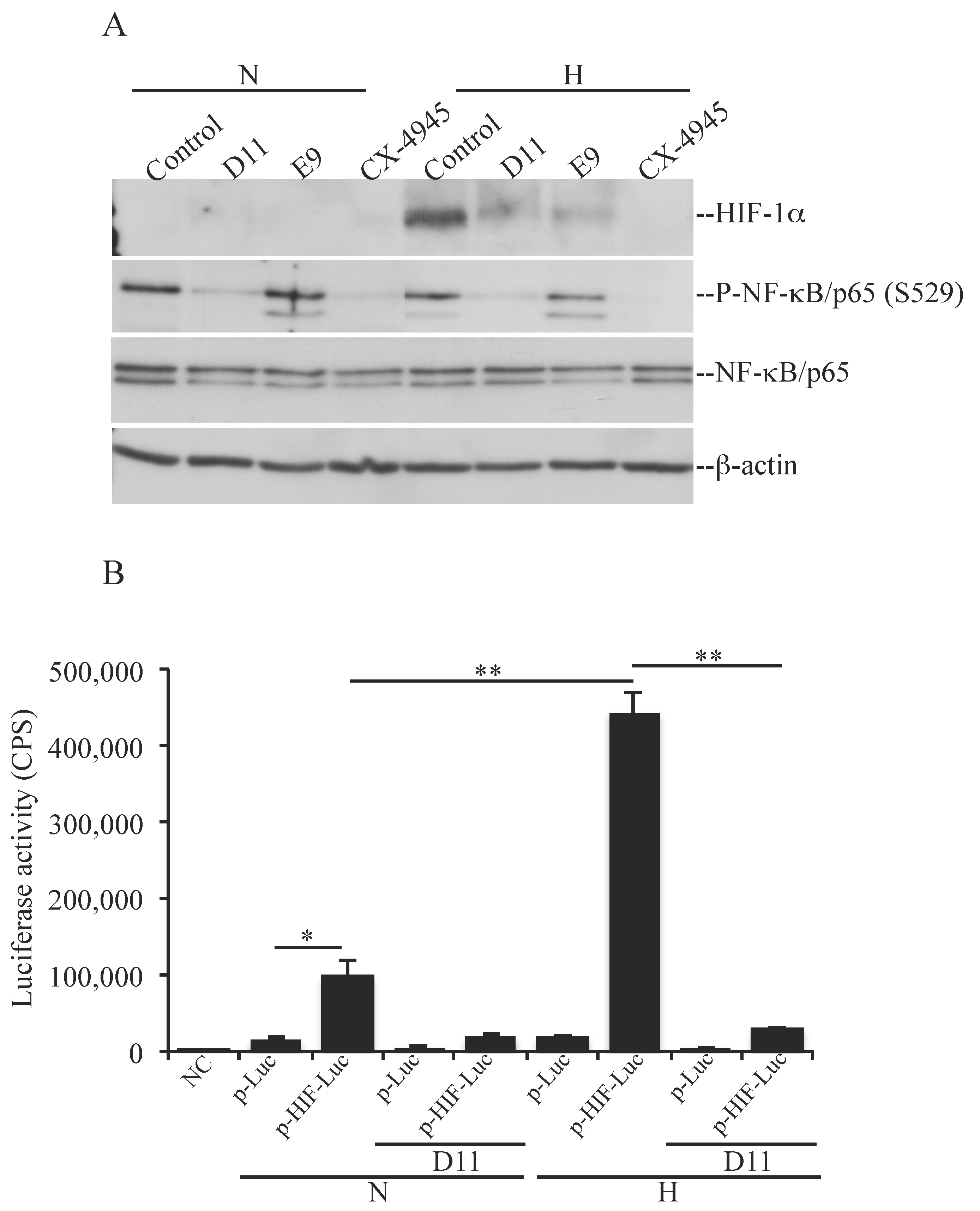
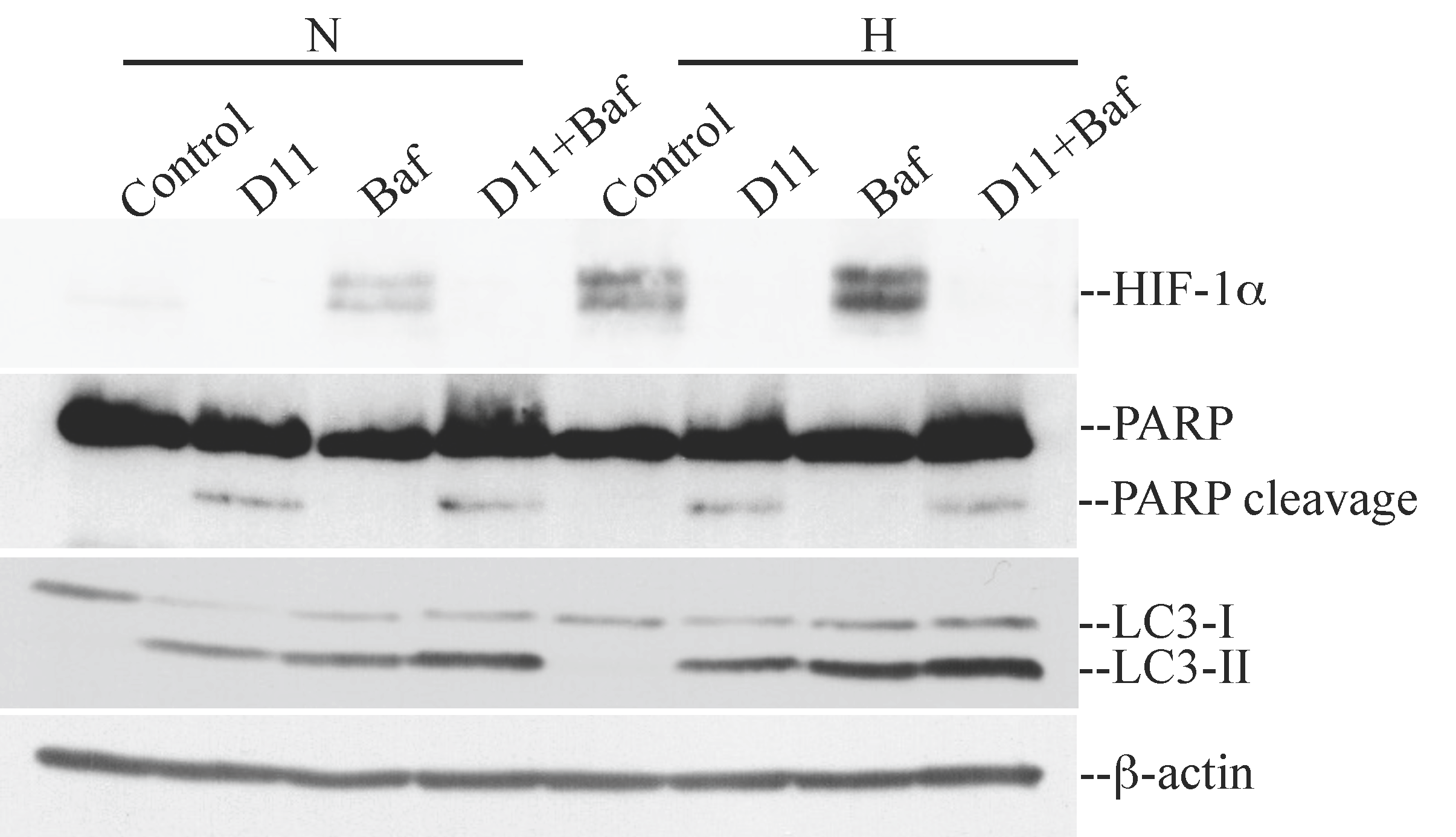
2.2. D11 Treatment Abolishes HIF-1α Transcriptional Activity and Results in Increased Activation of Autophagy under Hypoxia in Glioblastoma Cells
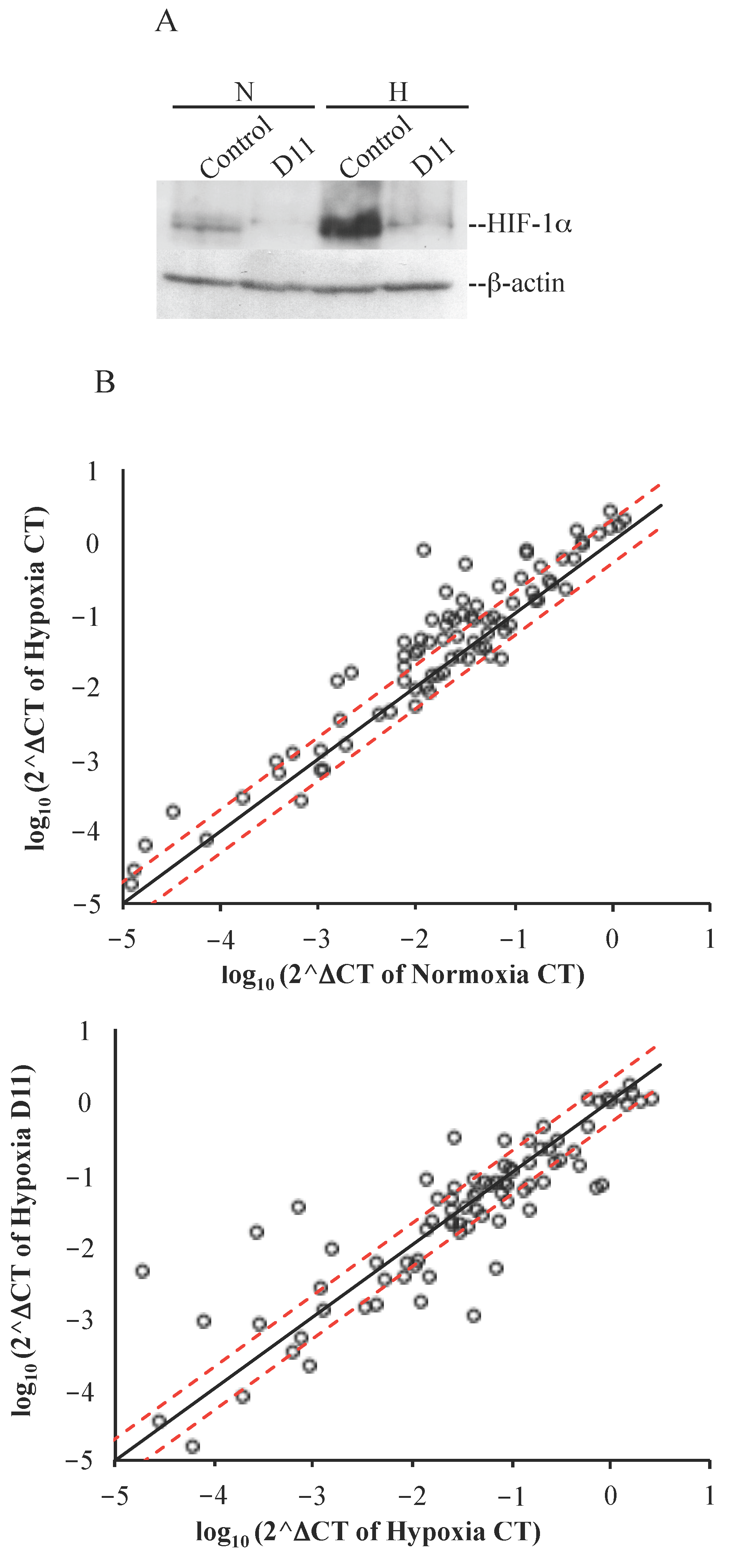
2.3. Cell Incubation with D11 Results in Altered Gene Expression Profile Induced by Hypoxia
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Hypoxia
4.2. Radioactive Kinase Assay
4.3. Cell Cycle Analysis by Flow Cytometry
4.4. Preparation of Whole Cell Lysate, Western Blot Analysis and Antibodies
4.5. Luciferase Reporter Assay
4.6. Gene Expression Analysis by Quantitative RT-PCR Array
4.7. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Zundel, W.; Schindler, C.; Haas-Kogan, D.; Koong, A.; Kaper, F.; Chen, E.; Gottschalk, A.R.; Ryan, H.E.; Johnson, R.S.; Jefferson, A.B.; et al. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev. 2000, 14, 391–396. [Google Scholar] [PubMed]
- Haas-Kogan, D.; Shalev, N.; Wong, M.; Mills, G.; Yount, G.; Stokoe, D. Protein kinase B (PKB/Akt) activity is elevated in glioblastoma cells due to mutation of the tumor suppressor PTEN/MMAC. Curr. Biol. 1998, 8, 1195–1198. [Google Scholar] [CrossRef]
- Wesseling, P.; Ruiter, D.J.; Burger, P.C. Angiogenesis in brain tumors; pathobiological and clinical aspects. J. Neurooncol. 1997, 32, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Durden, D.L.; Van Meir, E.G.; Brat, D.J. “Pseudopalisading” necrosis in glioblastoma: A familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J. Neuropathol. Exp. Neurol. 2006, 65, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L. Hypoxia-a key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Guerra, B.; Hochscherf, J.; Jensen, N.B.; Issinger, O.-G. Identification of a novel potent, selective and cell permeable inhibitor of protein kinase CK2 from the NIH/NCI Diversity Set Library. Mol. Cell. Biochem. 2015, 406, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Guerra, B.; Fischer, M.; Schaefer, S.; Issinger, O.-G. The kinase inhibitor D11 induces caspase-mediated cell death in cancer cells resistant to chemotherapeutic treatment. J. Exp. Clin. Cancer Res. 2015, 34, 125. [Google Scholar] [CrossRef] [PubMed]
- Guerra, B.; Issinger, O.-G. Protein kinase CK2 and its role in cellular proliferation, development and pathology. Electrophoresis 1999, 20, 391–408. [Google Scholar] [CrossRef]
- Guerra, B.; Issinger, O.-G. Protein kinase CK2 in human diseases. Curr. Med. Chem. 2008, 15, 1870–1886. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.S.; Litchfield, D.W. Too much of a good thing: The role of protein kinase CK2 in tumorigenesis and prospects for therapeutic inhibition of CK2. Biochim. Biophys. Acta 2008, 1784, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Trembley, J.H.; Wang, G.; Unger, G.; Slaton, J.; Ahmed, K. Protein kinase CK2 in health and disease: CK2: A key player in cancer biology. Cell. Mol. Life Sci. 2009, 66, 1858–1867. [Google Scholar] [CrossRef] [PubMed]
- Ruzzene, M.; Pinna, L.A. Addiction to protein kinase CK2: A common denominator of diverse cancer cells? Biochim. Biophys. Acta 2010, 1804, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Hubert, A.; Paris, S.; Piret, J.-P.; Ninane, N.; Raes, M.; Michiels, C. Casein kinase 2 inhibition decreases hypoxia-inducible factor-1 activity under hypoxia through elevated p53 protein level. J. Cell. Sci. 2006, 119, 3351–3362. [Google Scholar] [CrossRef] [PubMed]
- Mazure, N.M.; Chen, E.Y.; Laderoute, K.R.; Giaccia, A.J. Induction of vascular endothelial growth factor by hypoxia is modulated by a phosphatidylinositol 3-kinase/Akt signaling pathway in Ha-ras-transformed cells through a hypoxia inducible factor-1 transcriptional element. Blood 1997, 90, 3322–3331. [Google Scholar] [PubMed]
- Jiang, B.H.; Jiang, G.; Zheng, J.Z.; Lu, Z.; Hunter, T.; Vogt, P.K. Phosphatidylinositol 3-kinase signaling controls levels of hypoxia-inducible factor 1. Cell Growth Differ. 2001, 12, 363–369. [Google Scholar] [PubMed]
- Pore, N.; Liu, S.; Haas-Kogan, D.A.; O’Rourke, D.M.; Maity, A. PTEN mutation and epidermal growth factor receptor activation regulate vascular endothelial growth factor (VEGF) mRNA expression in human glioblastoma cells by transactivating the proximal VEGF promoter. Cancer Res. 2003, 63, 236–241. [Google Scholar] [PubMed]
- Wang, D.; Westerheide, S.D.; Hanson, J.L.; Baldwin, A.S. Tumor necrosis factor α-induced phosphorylation of RelA/p65 on Ser529 is controlled by casein kinase II. J. Biol. Chem. 2000, 275, 32592–32597. [Google Scholar] [CrossRef] [PubMed]
- Wenger, R.H.; Kvietikova, I.; Rolfs, A.; Gassmann, M.; Marti, H.H. Hypoxia-inducible factor-1 α is regulated at the post-mRNA level. Kidney Int. 1997, 51, 560–563. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.E.; Gu, J.; Schau, M.; Bunn, H.F. Regulation of hypoxia-inducible factor 1α is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 7987–7992. [Google Scholar] [CrossRef] [PubMed]
- Guerra, B.; Rasmussen, T.D.L.; Schnitzler, A.; Jensen, H.H.; Boldyreff, B.S.; Miyata, Y.; Marcussen, N.; Niefind, K.; Issinger, O.-G. Protein kinase CK2 inhibition is associated with the destabilization of HIF-1α in human cancer cells. Cancer Lett. 2015, 356, 751–761. [Google Scholar] [CrossRef] [PubMed]
- Yeo, E.-J.; Chun, Y.-S.; Cho, Y.-S.; Kim, J.; Lee, J.-C.; Kim, M.-S.; Park, J.-W. YC-1: A potential anticancer drug targeting hypoxia-inducible factor 1. J. Natl. Cancer Inst. 2003, 95, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, J.S.; Jung, Y.-J.; Mimnaugh, E.G.; Martinez, A.; Cuttitta, F.; Neckers, L.M. Hsp90 regulates a von Hippel Lindau-independent hypoxia-inducible factor 1α-degradative pathway. J. Biol. Chem. 2002, 277, 29936–29944. [Google Scholar] [CrossRef] [PubMed]
- Mabjeesh, N.J.; Post, D.E.; Willard, M.T.; Kaur, B.; Van Meir, E.G.; Simons, J.W.; Zhong, H. Geldanamycin induces degradation of hypoxia-inducible factor 1α protein via the proteosome pathway in prostate cancer cells. Cancer Res. 2002, 62, 2478–2482. [Google Scholar] [PubMed]
- Zagzag, D.; Nomura, M.; Friedlander, D.R.; Blanco, C.Y.; Gagner, J.-P.; Nomura, N.; Newcomb, E.W. Geldanamycin inhibits migration of glioma cells in vitro: A potential role for hypoxia-inducible factor (HIF-1α) in glioma cell invasion. J. Cell. Physiol. 2003, 196, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Zuehlke, A.; Johnson, J.L. Hsp90 and co-chaperones twist the functions of diverse client proteins. Biopolymers 2010, 93, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Miyata, Y. Protein kinase CK2 in health and disease: CK2: The kinase controlling the Hsp90 chaperone machinery. Cell. Mol. Life Sci. 2009, 66, 1840–1849. [Google Scholar] [CrossRef] [PubMed]
- Espert, L.; Denizot, M.; Grimaldi, M.; Robert-Hebmann, V.; Gay, B.; Varbanov, M.; Codogno, P.; Biard-Piechaczyk, M. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J. Clin. Investig. 2006, 116, 2161–2172. [Google Scholar] [CrossRef] [PubMed]
- Wykoff, C.C.; Beasley, N.; Watson, P.H.; Campo, L.; Chia, S.K.; English, R.; Pastorek, J.; Sly, W.S.; Ratcliffe, P.; Harris, A.L. Expression of the hypoxia-inducible and tumor-associated carbonic anhydrases in ductal carcinoma in situ of the breast. Am. J. Pathol. 2001, 158, 1011–1019. [Google Scholar] [CrossRef]
- Verma, S.; Dixit, R.; Pandey, K.C. Cysteine Proteases: Modes of Activation and Future Prospects as Pharmacological Targets. Front. Pharmacol. 2016, 7, 107. [Google Scholar] [CrossRef] [PubMed]
- Ellen, T.P.; Ke, Q.; Zhang, P.; Costa, M. NDRG1, a growth and cancer related gene: Regulation of gene expression and function in normal and disease states. Carcinogenesis 2008, 29, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Guan, R.J.; Ford, H.L.; Fu, Y.; Li, Y.; Shaw, L.M.; Pardee, A.B. Drg-1 as a differentiation-related, putative metastatic suppressor gene in human colon cancer. Cancer Res. 2000, 60, 749–755. [Google Scholar] [PubMed]
- Bandyopadhyay, S.; Pai, S.K.; Gross, S.C.; Hirota, S.; Hosobe, S.; Miura, K.; Saito, K.; Commes, T.; Hayashi, S.; Watabe, M.; et al. The Drg-1 gene suppresses tumor metastasis in prostate cancer. Cancer Res. 2003, 63, 1731–1736. [Google Scholar] [PubMed]
- Salnikow, K.; Costa, M.; Figg, W.D.; Blagosklonny, M.V. Hyperinducibility of hypoxia-responsive genes without p53/p21-dependent checkpoint in aggressive prostate cancer. Cancer Res. 2000, 60, 5630–5634. [Google Scholar] [PubMed]
- Guerra, B.; Iwabuchi, K.; Issinger, O.-G. Protein kinase CK2 is required for the recruitment of 53BP1 to sites of DNA double-strand break induced by radiomimetic drugs. Cancer Lett. 2014, 345, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Sandholt, I.S.; Olsen, B.B.; Guerra, B.; Issinger, O.-G. Resorufin: A lead for a new protein kinase CK2 inhibitor. Anticancer Drugs 2009, 20, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Yde, C.W.; Olsen, B.B.; Meek, D.; Watanabe, N.; Guerra, B. The regulatory beta-subunit of protein kinase CK2 regulates cell-cycle progression at the onset of mitosis. Oncogene 2008, 27, 4986–4997. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Gene Symbol | Fold-Change | Fold-Change |
|---|---|---|---|
| CTH vs. CTN | D11H vs. CTH | ||
| Adrenomedullin | ADM | 4.358 | 1.226 |
| Angiopoietin-like 4 | ANGPTL4 | 7.646 | −7.888 |
| Ankyrin repeat domain 37 | ANKRD37 | 2.880 | −2.106 |
| Basic helix-loop-helix family member e40 | BHLHE40 | 3.229 | −2.275 |
| Bloom syndrome, RecQ helicase-like | BLM | −1.904 | −1.549 |
| Bcl2 interacting protein 3 | BNIP3 | 5.984 | 1.282 |
| Bcl2 interacting protein 3-like | BNIP3L | 3.364 | −1.058 |
| Carbonic anhydrase IX | CA9 | 10.768 | −2.924 |
| Cyclin G2 | CCNG2 | 3.367 | 2.445 |
| DNA-damage-inducible transcript 4 | DDIT4 | 7.233 | −4.314 |
| Egl nine homolog 1 (C. elegans) | EGLN1 | 1.953 | −2.032 |
| Early growth response 1 | EGR1 | −2.474 | 54.951 |
| Erythropoietin | EPO | 2.150 | 1.275 |
| ERO1-like (S. cerevisiae) | ERO1A | 2.033 | −1.566 |
| Coagulation factor III | F3 | 5.802 | −41.328 |
| Glucan branching enzyme 1 | GBE1 | 2.523 | −2.082 |
| Glycogen synthase | GYS1 | 2.051 | −2.645 |
| Hypoxia inducible factor 3 α subunit | HIF3A | 3.719 | −3.992 |
| Hexokinase 2 | HK2 | 3.152 | 1.100 |
| Insulin-like growth factor binding protein 3 | IGFBP3 | 67.054 | −11.170 |
| Lactate dehydrogenase A | LDHA | 2.602 | −2.361 |
| Lectin, galactoside-binding, soluble, 3 | LGALS3 | 2.363 | −2.141 |
| Lysyl oxidase | LOX | 5.199 | −4.730 |
| Macrophage migration inhibitory factor | MIF | 1.986 | −1.040 |
| Matrix Metallopeptidase 9 | MMP9 | 6.044 | −2.535 |
| MAX interactor 1 | MXI1 | 4.017 | −1.462 |
| N-myc downstream-regulated 1 | NDRG1 | 15.150 | −3.611 |
| Ornithine decarboxylase 1 | ODC1 | −2.231 | 11.551 |
| Prolyl 4-hydroxylase, α 1 | P4HA1 | 3.362 | 1.053 |
| Pyruvate dehydrogenase kinase | PDK1 | 2.946 | −1.522 |
| 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 | PFKFB3 | 2.394 | 1.146 |
| 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 4 | PFKFB4 | 3.304 | −14.057 |
| Phosphofructokinase, liver | PFKL | 2.452 | −2.309 |
| Placental growth factor | PGF | 2.242 | 2.089 |
| Phosphoglycerate kinase 1 | PGK1 | 3.159 | −1.581 |
| Plasminogen activator, urokinase | PLAU | −2.926 | 1.674 |
| Serpin peptidase inhibitor, clade E member 1 | SERPINE1 | 1.911 | 1.744 |
| Solute carrier family 16 member 3 | SLC16A3 | 2.454 | −4.346 |
| Solute carrier family 2 member 1 (GLUT1) | SLC2A1 | 5.890 | −1.162 |
| Solute carrier family 2 member 3 (GLUT 3) | SLC2A3 | 2.462 | 2.460 |
| Thioredoxin interacting protein | TXNIP | 3.422 | 1.559 |
| Vascular endothelial growth factor A | VEGFA | 5.367 | −11.028 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schaefer, S.; Svenstrup, T.H.; Fischer, M.; Guerra, B. D11-Mediated Inhibition of Protein Kinase CK2 Impairs HIF-1α-Mediated Signaling in Human Glioblastoma Cells. Pharmaceuticals 2017, 10, 5. https://doi.org/10.3390/ph10010005
Schaefer S, Svenstrup TH, Fischer M, Guerra B. D11-Mediated Inhibition of Protein Kinase CK2 Impairs HIF-1α-Mediated Signaling in Human Glioblastoma Cells. Pharmaceuticals. 2017; 10(1):5. https://doi.org/10.3390/ph10010005
Chicago/Turabian StyleSchaefer, Susanne, Tina H. Svenstrup, Mette Fischer, and Barbara Guerra. 2017. "D11-Mediated Inhibition of Protein Kinase CK2 Impairs HIF-1α-Mediated Signaling in Human Glioblastoma Cells" Pharmaceuticals 10, no. 1: 5. https://doi.org/10.3390/ph10010005
APA StyleSchaefer, S., Svenstrup, T. H., Fischer, M., & Guerra, B. (2017). D11-Mediated Inhibition of Protein Kinase CK2 Impairs HIF-1α-Mediated Signaling in Human Glioblastoma Cells. Pharmaceuticals, 10(1), 5. https://doi.org/10.3390/ph10010005