
Identification of a Potent Allosteric Inhibitor of Human Protein Kinase CK2 by Bacterial Surface Display Library Screening

 and
and 
Abstract
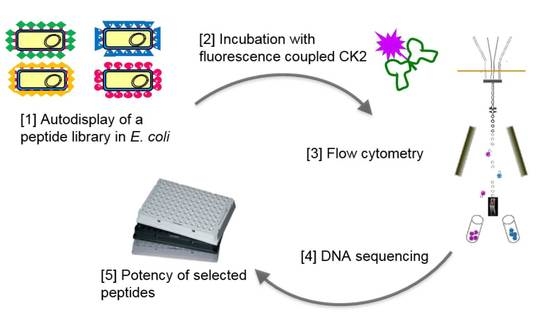
:
1. Introduction
2. Results
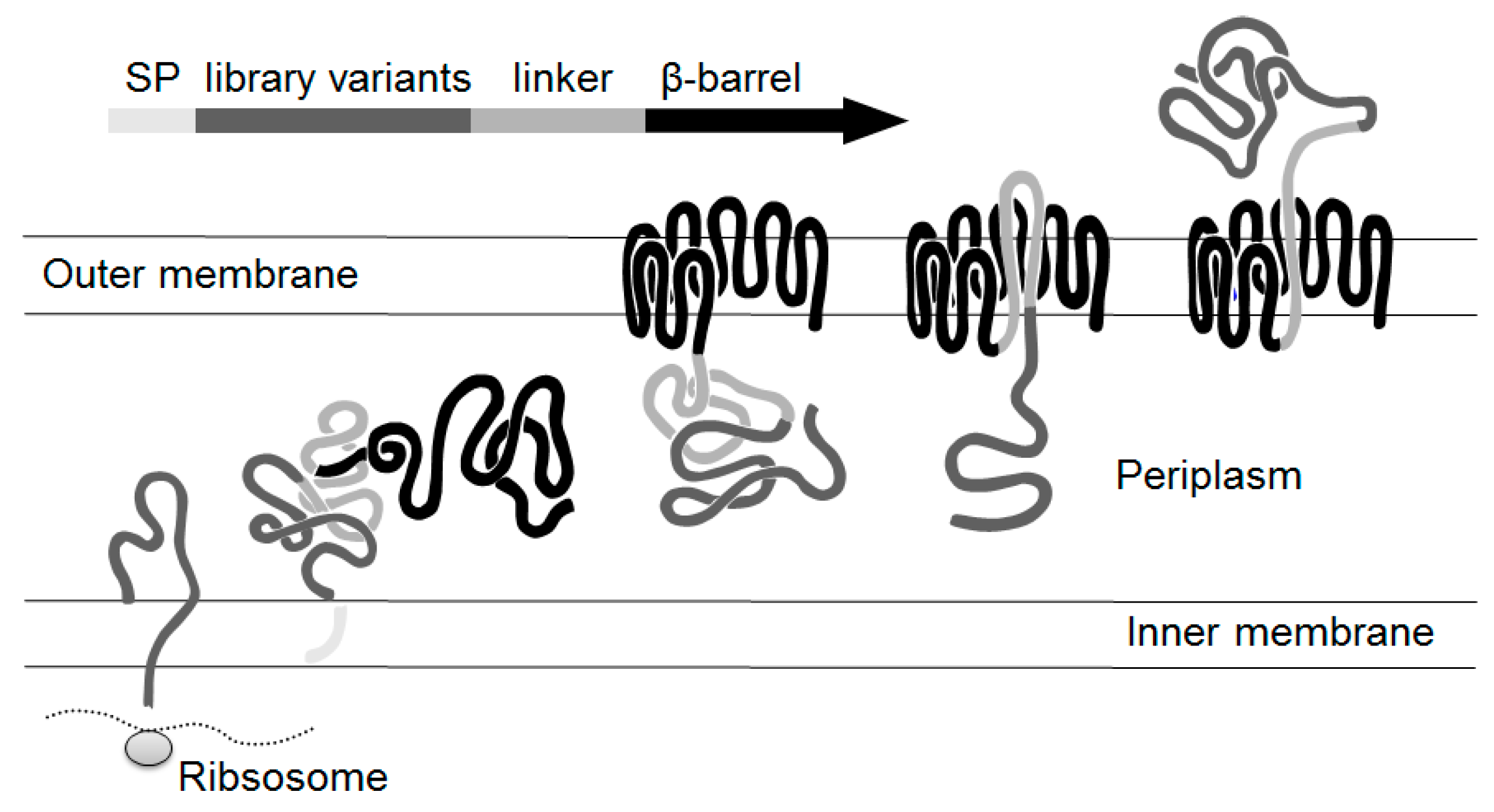
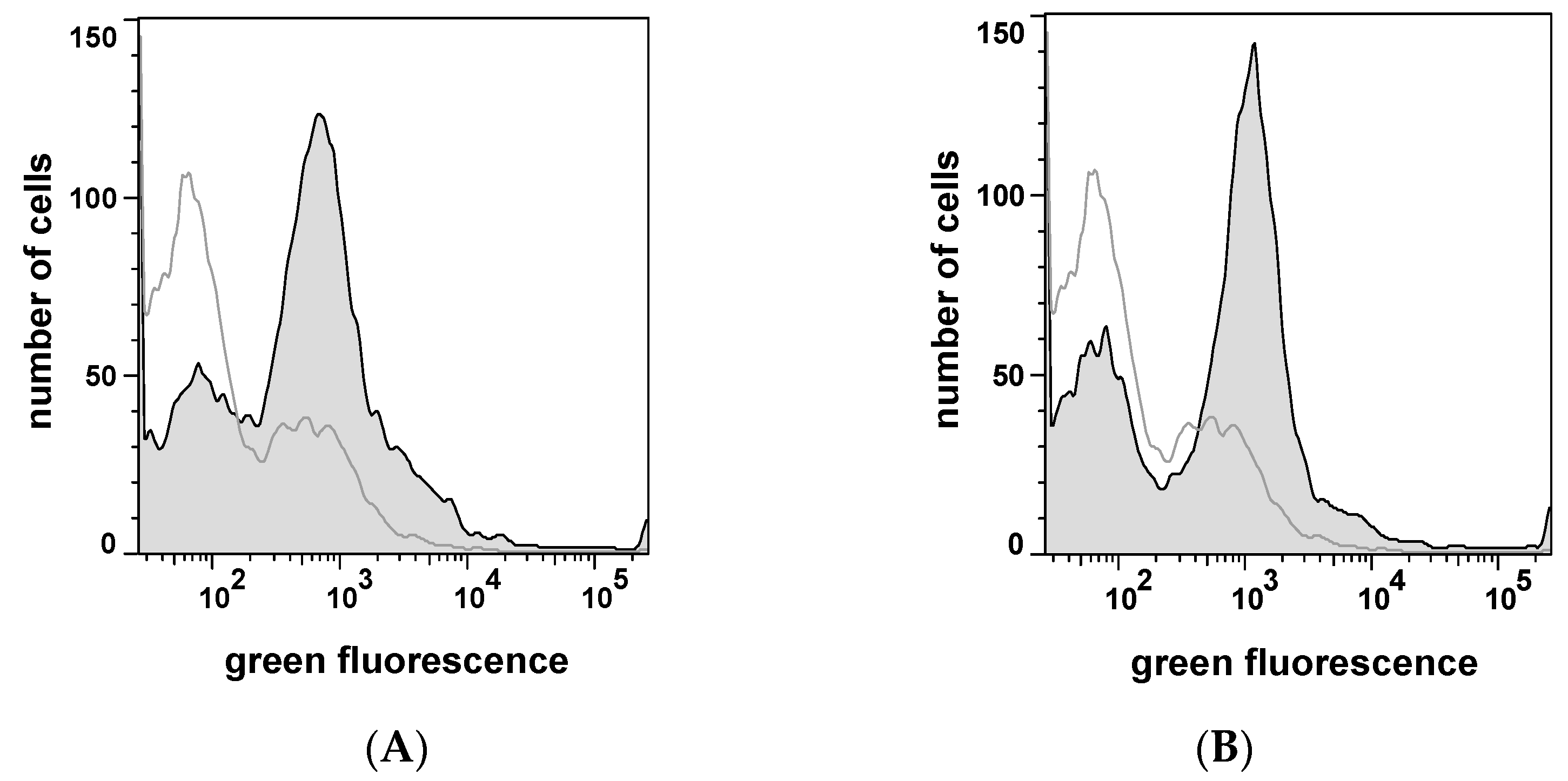
2.1. Autodisplay of Human αS1-Casein and Cellular Labeling with CK2-FITC
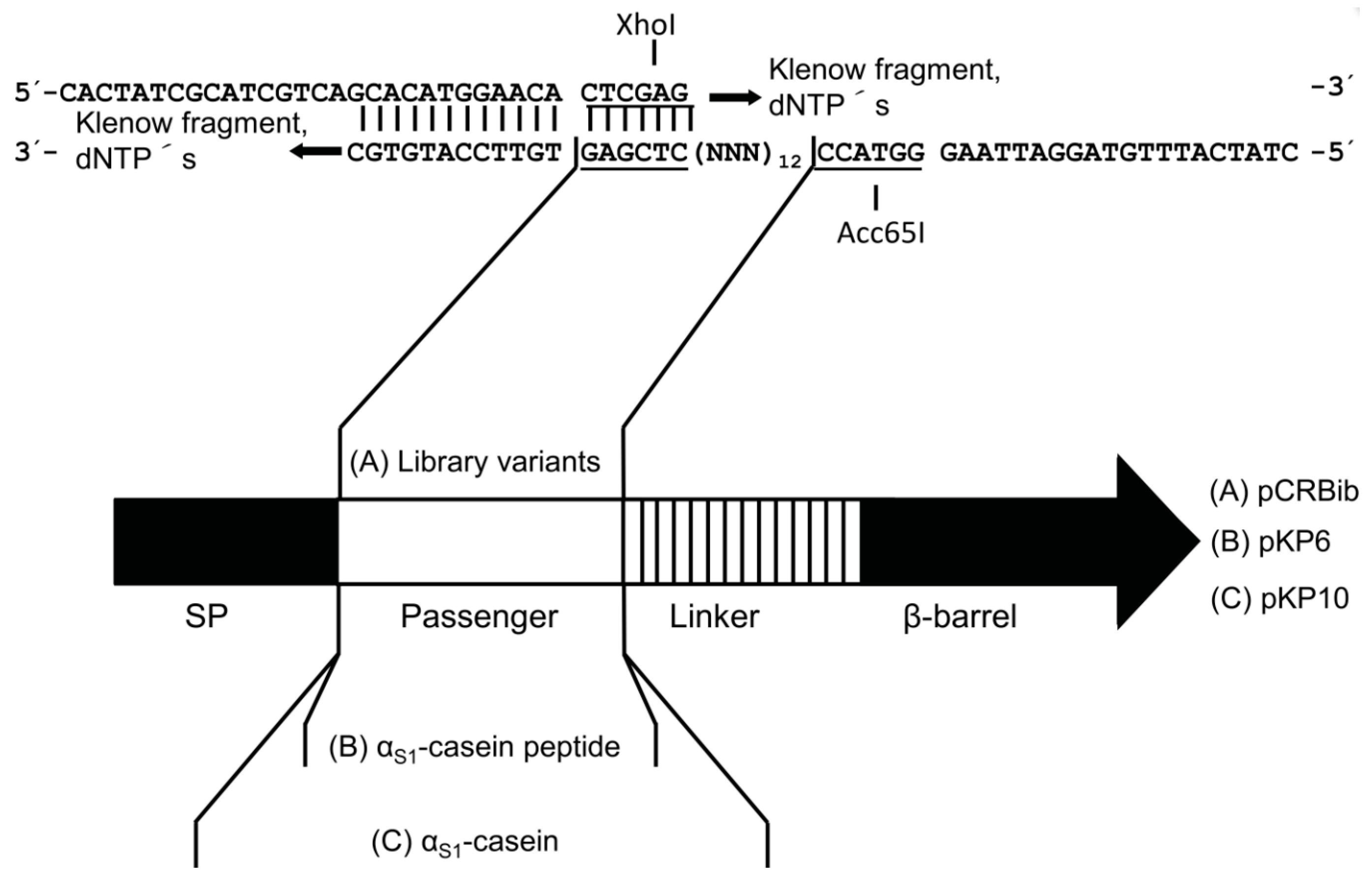
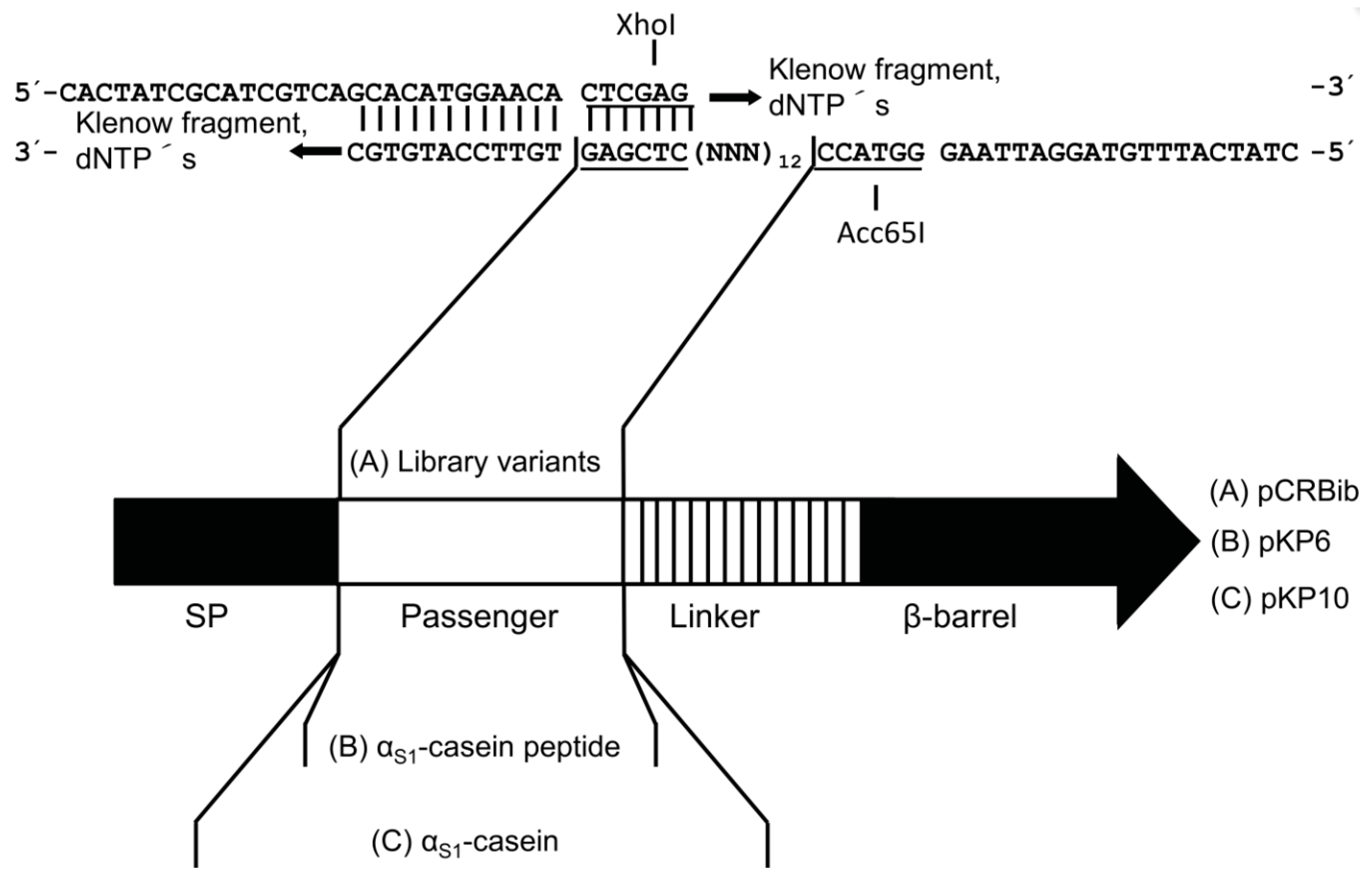
2.2. Design of a Bacterial Surface Display Library
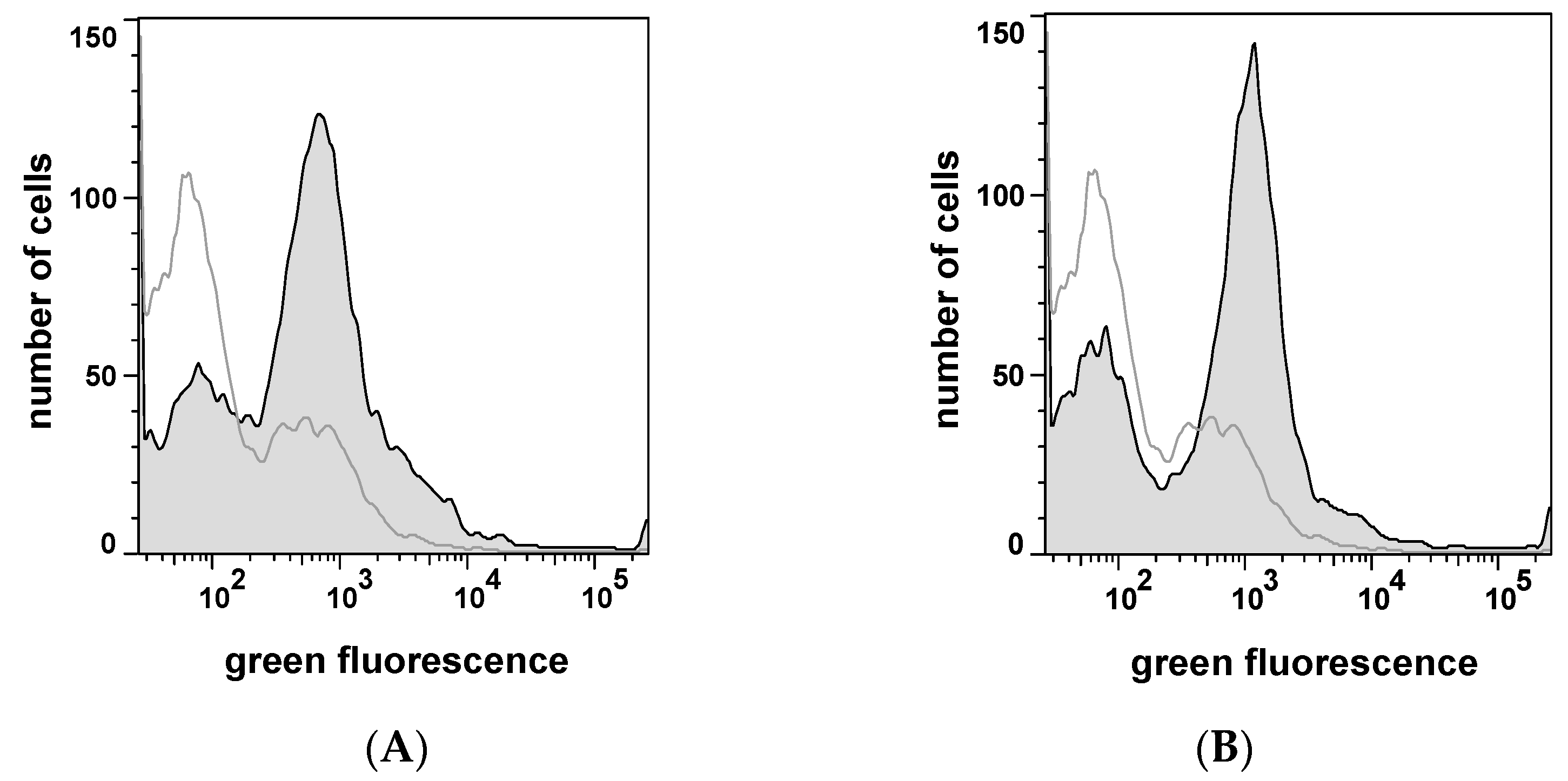
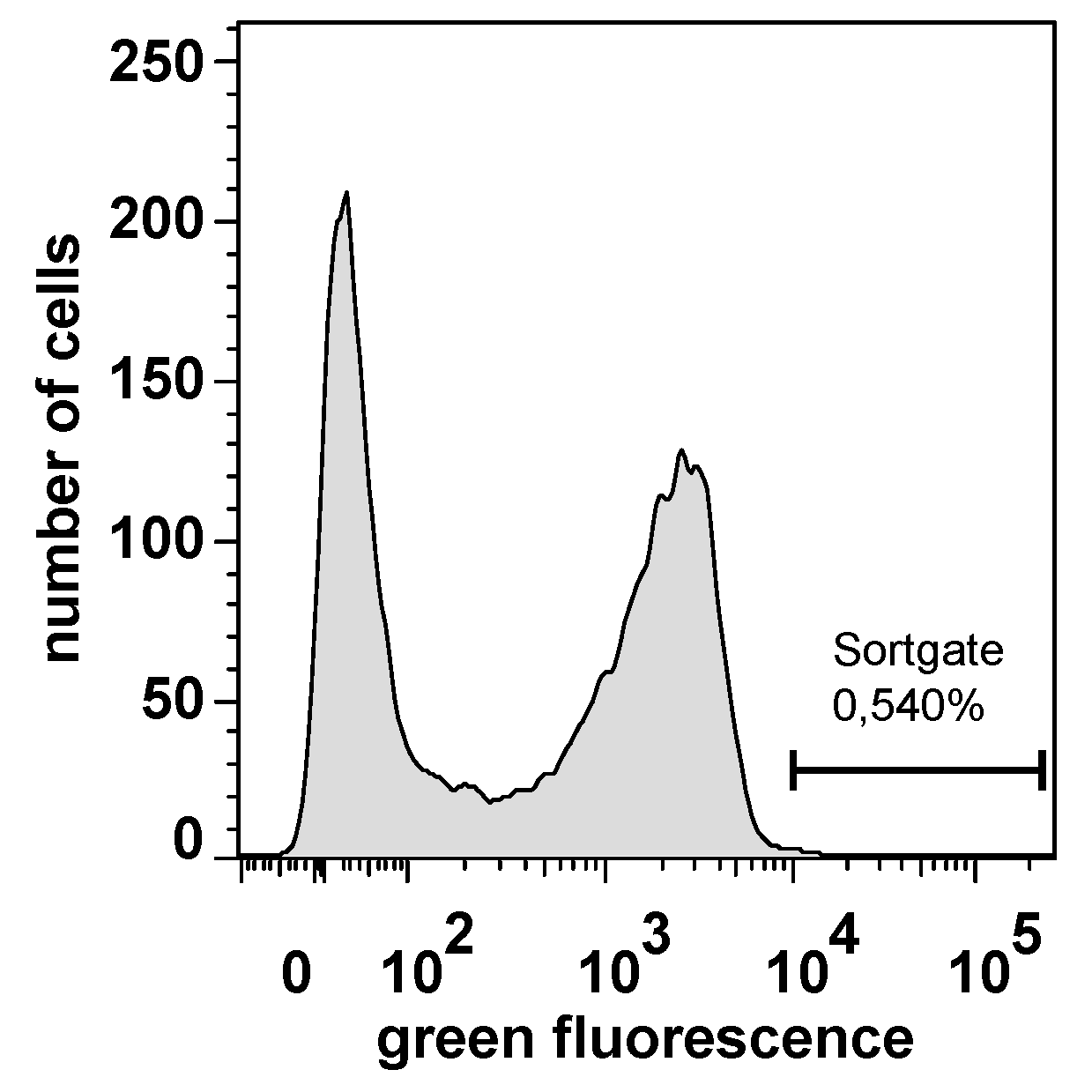
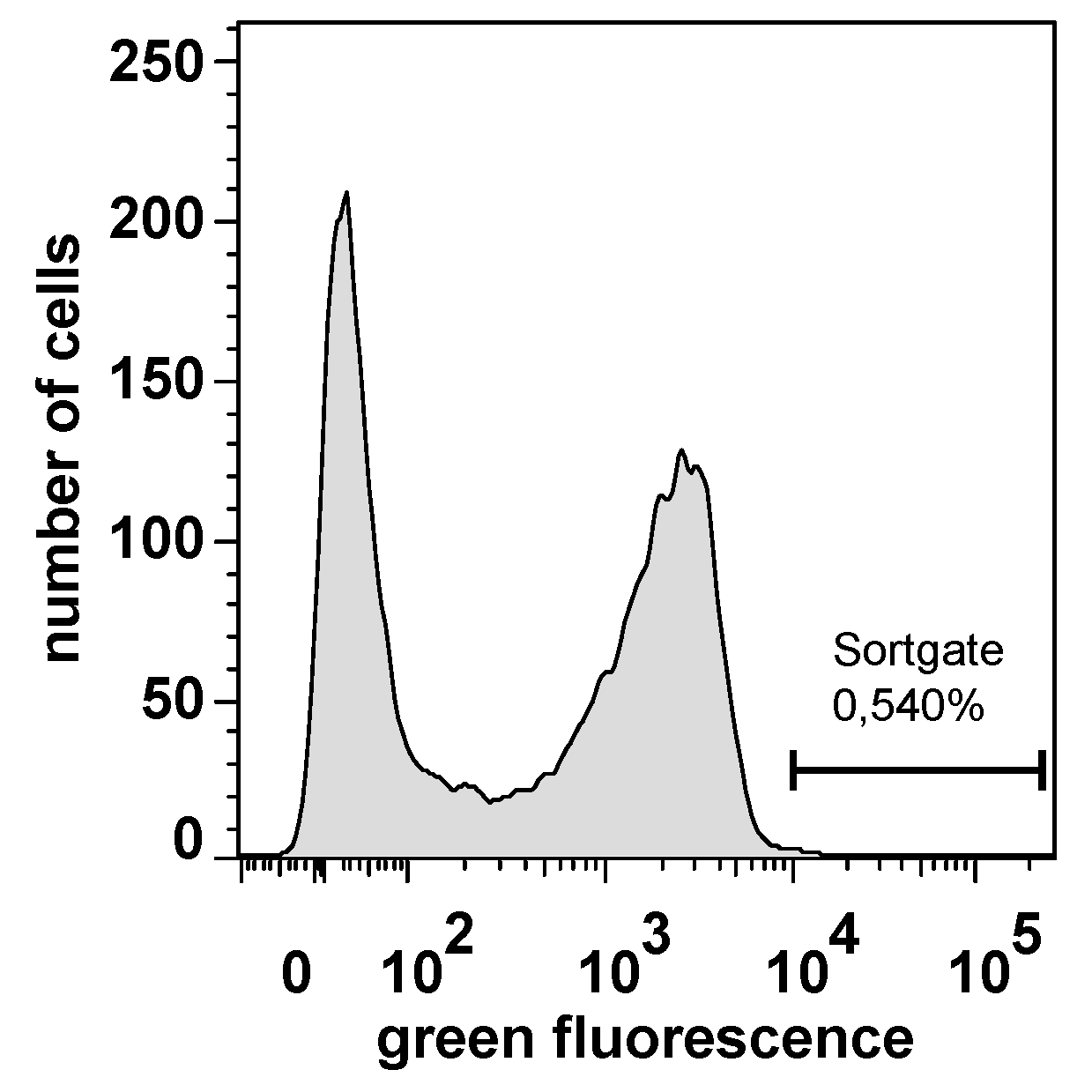
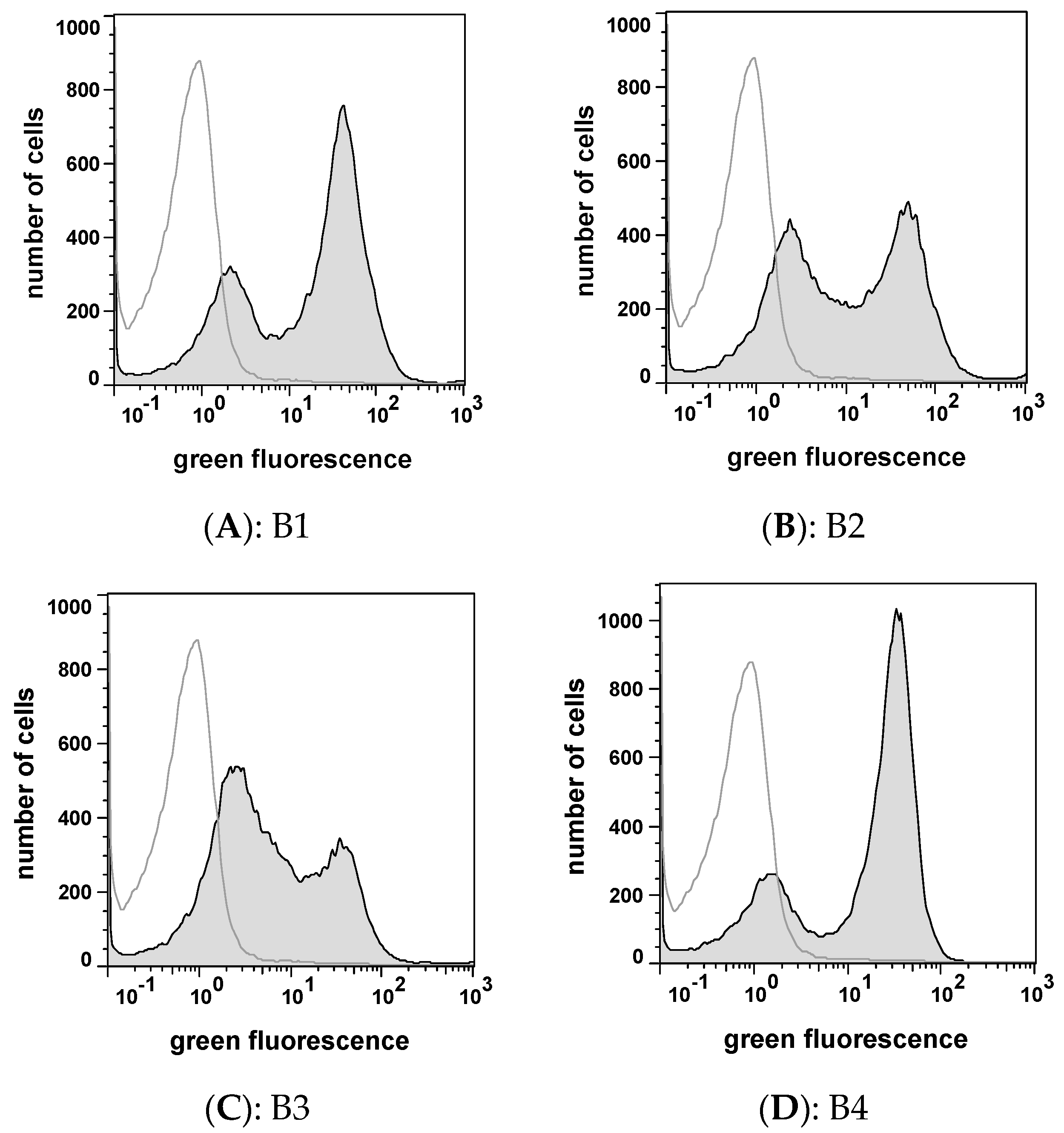
2.3. Selecting Peptides with Affinity towards CK2 by FACS
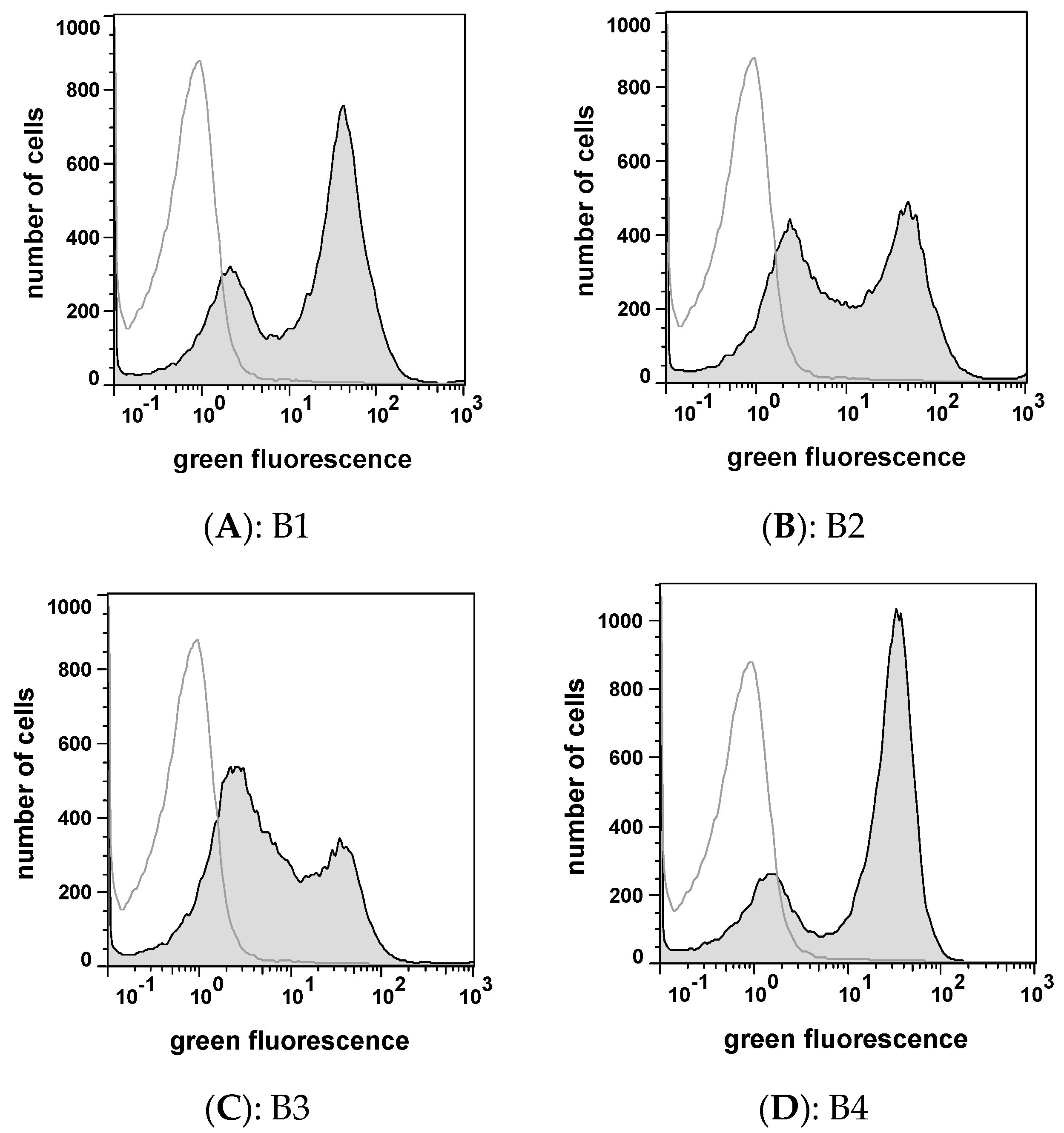
2.4. Peptide Expression and Sequence Analysis
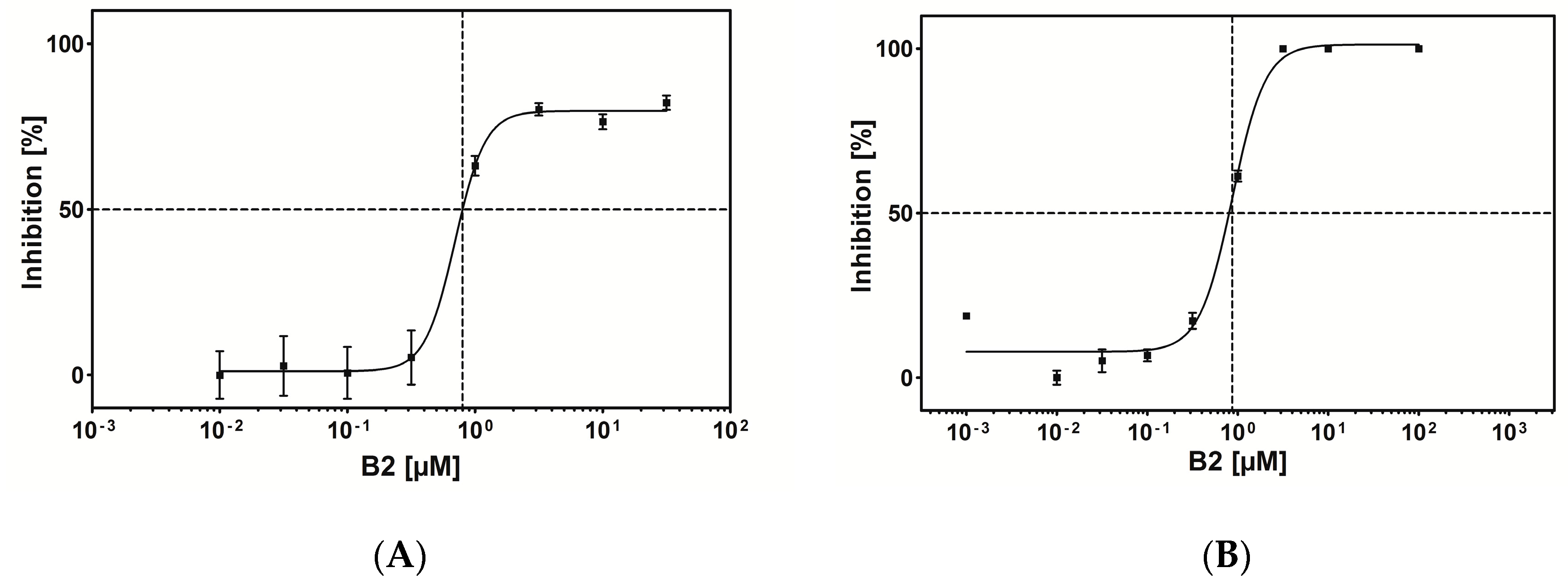
2.5. CK2 Inhibitor Testing by Capillary Electrophoresis (CE)-Assay
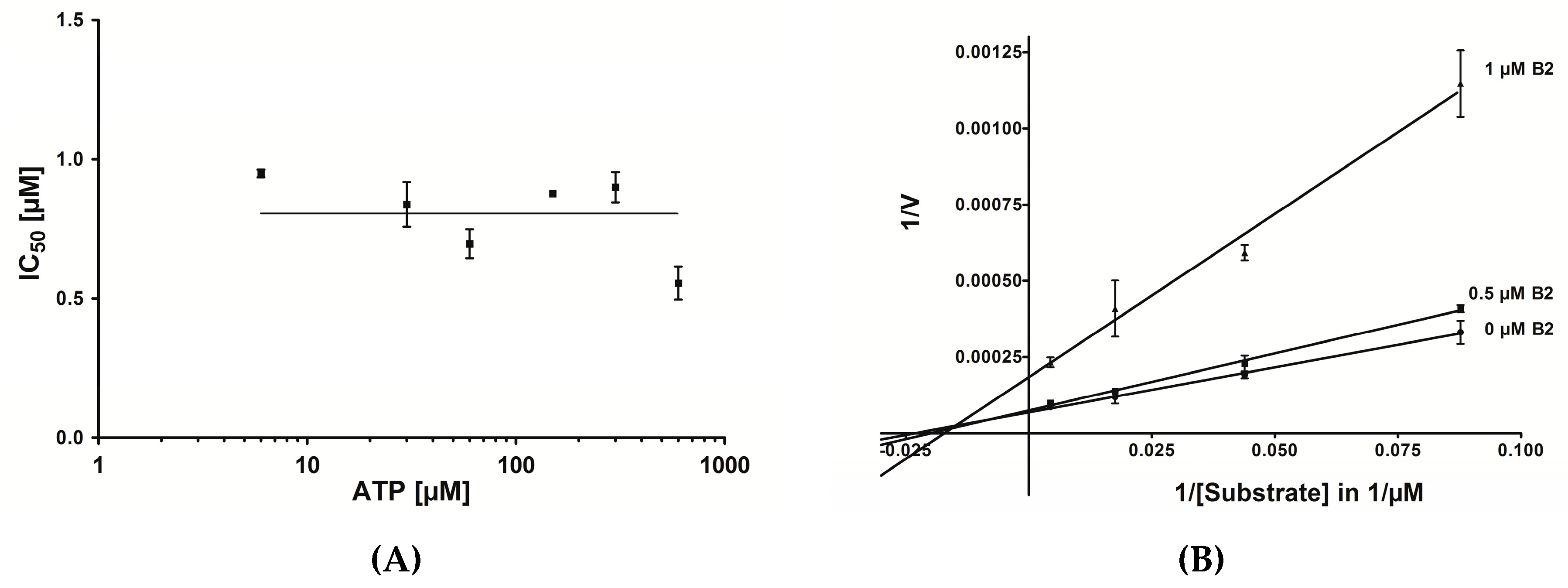
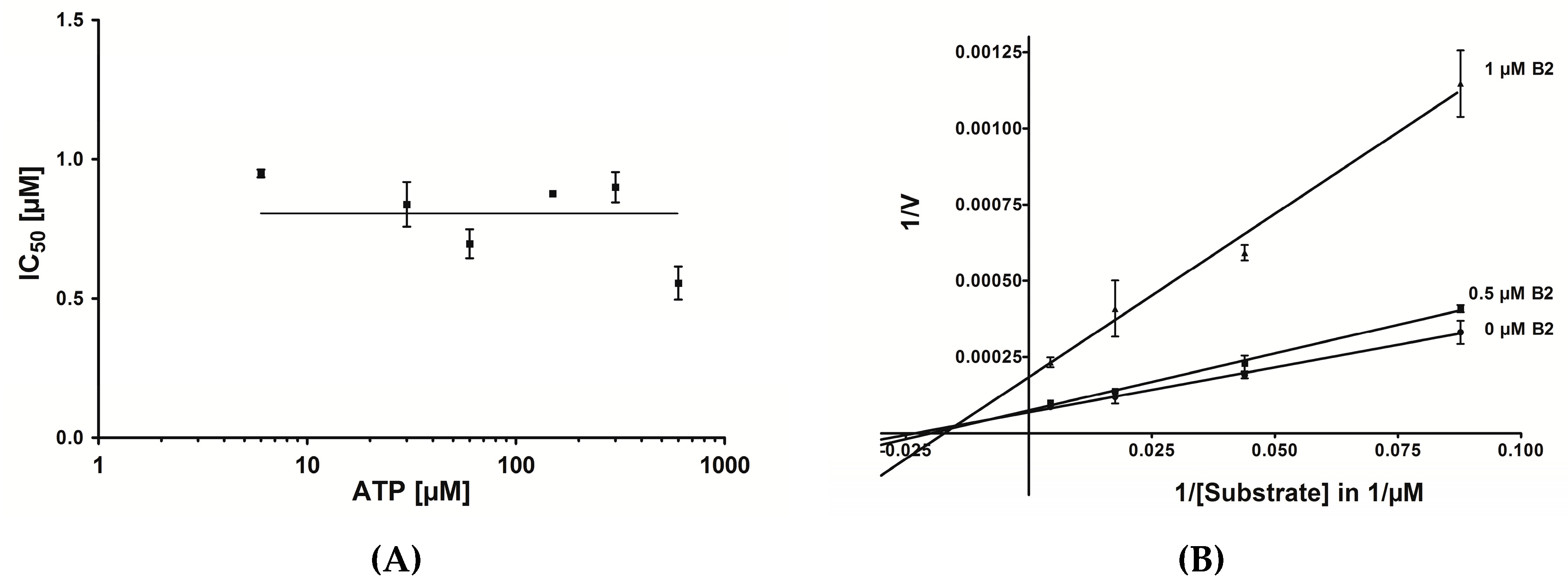
2.6. Mode of Inhibition
2.7. In Vitro Pull Down Assay
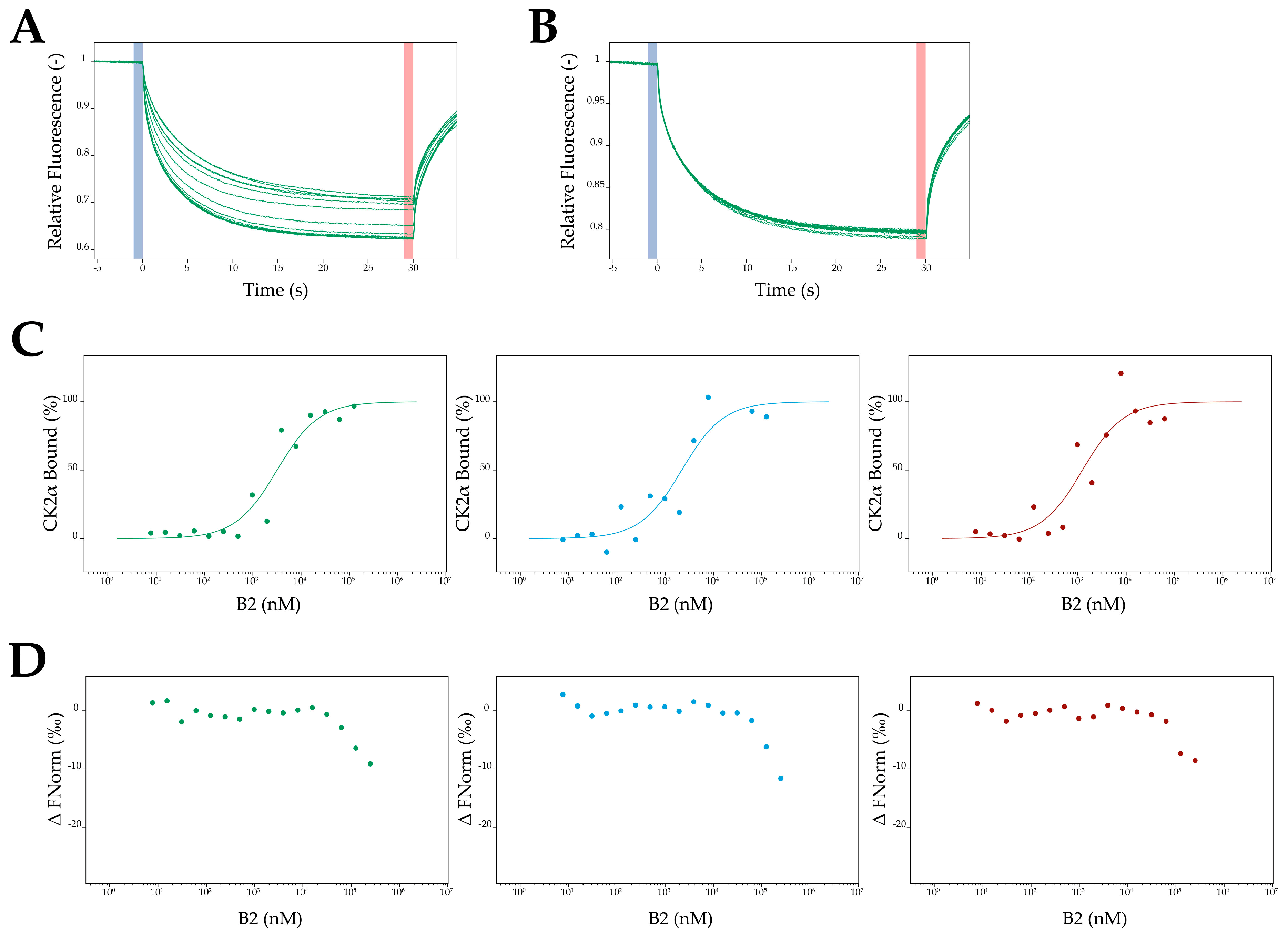
2.8. MST Measurements
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Bacteria, Plasmids, and Culture Conditions
4.3. Plasmid Construction Using Restriction Sites
4.4. Synthesis of Double Stranded DNA (dsDNA)
4.5. Construction of the Plasmid Used for Surface Display of the Library
4.6. Ligation
4.7. Transformation of E. coli by Electroporation
4.8. Outer Membrane Preparation
4.9. SDS-PAGE and Western Blot Analysis
4.10. Preparation of Recombinant Human Protein Kinase CK2
4.11. Coupling of CK2 with Fluorescein Isothiocyanate (FITC)
4.12. Flow Cytometer Analysis and Sorting
4.13. Capillary Electrophoresis (CE)-based CK2 Assay
4.14. Mode of Inhibition
4.15. Expression and Purification of GST-CK2α
4.16. In Vitro Translation of CK2β
4.17. Pull Down Assay with Recombinant Proteins
4.18. Click Reaction of CK2α-pAzF and CK2β1-193-pAzF
4.19. MST Measurements
5. Conclusions
Author Contributions
Conflicts of Interest
References
- Filhol, O.; Martiel, J.L.; Cochet, C. Protein kinase CK2: A new view of an old molecular complex. EMBO Rep. 2004, 5, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, D.W. Protein kinase CK2: Structure, regulation and role in cellular decisions of life and death. Biochem. J. 2003, 369, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Landesman-Bollag, E.; Romieu-Mourez, R.; Song, D.H.; Sonenshein, G.E.; Cardiff, R.D.; Seldin, D.C. Protein kinase CK2 in mammary gland tumorigenesis. Oncogene 2001, 20, 3247–3257. [Google Scholar] [CrossRef] [PubMed]
- Laramas, M.; Pasquier, D.; Filhol, O.; Ringeisen, F.; Descotes, J.L.; Cochet, C. Nuclear localization of protein kinase CK2 catalytic subunit (CK2α) is associated with poor prognostic factors in human prostate cancer. Eur. J. Cancer 2007, 43, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Luo, H.; Zeng, Q.; Dong, Z.; Wu, D.; Liu, L. Protein kinase CK2α is overexpressed in colorectal cancer and modulates cell proliferation and invasion via regulating emt-related genes. J. Transl. Med. 2011, 9, 97. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.S.; Litchfield, D.W. Too much of a good thing: The role of protein kinase CK2 in tumorigenesis and prospects for therapeutic inhibition of CK2. Biochim. Biophys. Acta 2008, 1784, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Eom, J.I.; Cheong, J.W.; Choi, A.J.; Lee, J.K.; Yang, W.I.; Min, Y.H. Protein kinase CK2α as an unfavorable prognostic marker and novel therapeutic target in acute myeloid leukemia. Clin. Cancer Res. 2007, 13, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Ryu, M.Y.; Kim, D.W.; Arima, K.; Mouradian, M.M.; Kim, S.U.; Lee, G. Localization of CKII β subunits in lewy bodies of parkinson's disease. J. Neurol. Sci. 2008, 266, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Trembley, J.H.; Chen, Z.; Unger, G.; Slaton, J.; Kren, B.T.; Van Waes, C.; Ahmed, K. Emergence of protein kinase CK2 as a key target in cancer therapy. Biofactors 2010, 36, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Sarno, S.; Salvi, M.; Battistutta, R.; Zanotti, G.; Pinna, L.A. Features and potentials of ATP-site directed CK2 inhibitors. Biochim. Biophys. Acta Proteins Proteom. 2005, 1754, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Pagano, M.A.; Andrzejewska, M.; Ruzzene, M.; Sarno, S.; Cesaro, L.; Bain, J.; Elliott, M.; Meggio, F.; Kazimierczuk, Z.; Pinna, L.A. Optimization of protein kinase CK2 inhibitors derived from 4,5,6,7-tetrabromobenzimidazole. J. Med. Chem. 2004, 47, 6239–6247. [Google Scholar] [CrossRef] [PubMed]
- Nie, Z.; Perretta, C.; Erickson, P.; Margosiak, S.; Almassy, R.; Lu, J.; Averill, A.; Yager, K.M.; Chu, S. Structure-based design, synthesis, and study of pyrazolo[1,5-a][1,3,5]triazine derivatives as potent inhibitors of protein kinase CK2. Bioorg. Med. Chem. Lett. 2007, 17, 4191–4195. [Google Scholar] [CrossRef] [PubMed]
- Sarno, S.; de Moliner, E.; Ruzzene, M.; Pagano, M.A.; Battistutta, R.; Bain, J.; Fabbro, D.; Schoepfer, J.; Elliott, M.; Furet, P.; et al. Biochemical and three-dimensional-structural study of the specific inhibition of protein kinase CK2 by [5-oxo-5,6-dihydroindolo-(1,2-a)quinazolin-7-yl]acetic acid (IQA). Biochem. J. 2003, 374, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui-Jain, A.; Drygin, D.; Streiner, N.; Chua, P.; Pierre, F.; O’Brien, S.E.; Bliesath, J.; Omori, M.; Huser, N.; Ho, C.; et al. Cx-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010, 70, 10288–10298. [Google Scholar] [CrossRef] [PubMed]
- Prudent, R.; Moucadel, V.; Laudet, B.; Barette, C.; Lafanechère, L.; Hasenknopf, B.; Li, J.; Bareyt, S.; Lacôte, E.; Thorimbert, S.; et al. Identification of polyoxometalates as nanomolar noncompetitive inhibitors of protein kinase CK2. Chem. Biol. 2008, 15, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Allende, C.C.; Allende, J.E. Promiscuous subunit interactions: A possible mechanism for the regulation of protein kinase CK2. J. Cell. Biochem. Suppl. 1998, 30–31, 129–136. [Google Scholar] [CrossRef]
- Laudet, B.; Moucadel, V.; Prudent, R.; Filhol, O.; Wong, Y.S.; Royer, D.; Cochet, C. Identification of chemical inhibitors of protein-kinase CK2 subunit interaction. Mol. Cell. Biochem. 2008, 316, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Moucadel, V.; Prudent, R.; Sautel, C.F.; Teillet, F.; Barette, C.; Lafanechere, L.; Receveur-Brechot, V.; Cochet, C. Antitumoral activity of allosteric inhibitors of protein kinase CK2. Oncotarget 2011, 2, 997–1010. [Google Scholar] [CrossRef] [PubMed]
- Knight, Z.A.; Shokat, K.M. Features of selective kinase inhibitors. Chem. Biol. 2005, 12, 621–637. [Google Scholar] [CrossRef] [PubMed]
- Laudet, B.; Barette, C.; Dulery, V.; Renaudet, O.; Dumy, P.; Metz, A.; Prudent, R.; Deshiere, A.; Dideberg, O.; Filhol, O.; et al. Structure-based design of small peptide inhibitors of protein kinase CK2 subunit interaction. Biochem. J. 2007, 408, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Perea, S.E.; Reyes, O.; Baladron, I.; Perera, Y.; Farina, H.; Gil, J.; Rodriguez, A.; Bacardi, D.; Marcelo, J.L.; Cosme, K.; et al. CIGB-300, a novel proapoptotic peptide that impairs the CK2 phosphorylation and exhibits anticancer properties both in vitro and in vivo. Mol. Cell. Biochem. 2008, 316, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Perea, S.E.; Baladron, I.; Garcia, Y.; Perera, Y.; Lopez, A.; Soriano, J.L.; Batista, N.; Palau, A.; Hernández, I.; Farina, H.; et al. CIGB-300, a synthetic peptide-based drug that targets the CK2 phosphoaceptor domain. Translational and clinical research. Mol. Cell. Biochem. 2011, 356, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Martel, V.; Filhol, O.; Colas, P.; Cochet, C. P53-dependent inhibition of mammalian cell survival by a genetically selected peptide aptamer that targets the regulatory subunit of protein kinase CK2. Oncogene 2006, 25, 7343–7353. [Google Scholar] [CrossRef] [PubMed]
- Bibby, A.C.; Litchfield, D.W. The multiple personalities of the regulatory subunit of protein kinase CK2: CK2 dependent and CK2 independent roles reveal a secret identity for CK2β. Int. J. Biol. Sci. 2005, 1, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef] [PubMed]
- Wernerus, H.; Stahl, S. Biotechnological applications for surface-engineered bacteria. Biotechnol. Appl. Biochem. 2004, 40, 209–228. [Google Scholar] [PubMed]
- Szabo, A.; Heja, D.; Szakacs, D.; Zboray, K.; Kekesi, K.A.; Radisky, E.S.; Sahin-Toth, M.; Pal, G. High affinity small protein inhibitors of human chymotrypsin C (CTRC) selected by phage display reveal unusual preference for p4′ acidic residues. J. Biol. Chem. 2011, 286, 22535–22545. [Google Scholar] [CrossRef] [PubMed]
- de Marco, R.; Azzolini, S.S.; Lovato, D.V.; Torquato, R.J.; Amino, R.; de Miranda, A.; Tanaka, A.S. Validation of phage display method for protease inhibitor selection using synthetic hybrid peptides. Comb. Chem. High. Throughput Screen. 2010, 13, 829–835. [Google Scholar] [CrossRef] [PubMed]
- Rajik, M.; Jahanshiri, F.; Omar, A.R.; Ideris, A.; Hassan, S.S.; Yusoff, K. Identification and characterisation of a novel anti-viral peptide against avian influenza virus H9N2. Virol J. 2009, 6, 74. [Google Scholar] [CrossRef] [PubMed]
- Welch, B.D.; VanDemark, A.P.; Heroux, A.; Hill, C.P.; Kay, M.S. Potent d-peptide inhibitors of HIV-1 entry. Proc. Natl. Acad. Sci. USA 2007, 104, 16828–16833. [Google Scholar] [CrossRef] [PubMed]
- Jose, J.; Betscheider, D.; Zangen, D. Bacterial surface display library screening by target enzyme labeling: Identification of new human cathepsin g inhibitors. Anal. Biochem. 2005, 346, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Gratz, A.; Jose, J. Protein domain library generation by overlap extension (PDLGO): A tool for enzyme engineering. Anal. Biochem. 2008, 378, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Gratz, A.; Jose, J. Focussing mutations to defined domains: Protein domain library generation by overlap extension (PDLGO). In Cdna Libraries: New Techniques and Applications; Lu, C., Browse, J., Wallis, J.G., Eds.; Humana Press: London, UK, 2011; Volume 279, pp. 153–166. [Google Scholar]
- Jose, J.; Jähnig, F.; Meyer, T.F. Common structural features of IGA1 protease-like outer membrane protein autotransporters. Mol. Microbiol. 1995, 18, 378–380. [Google Scholar] [CrossRef] [PubMed]
- Burnett, G.; Kennedy, E.P. The enzymatic phosphorylation of proteins. J. Biol. Chem. 1954, 211, 969–980. [Google Scholar] [PubMed]
- Petermann, K.; Vordenbaumen, S.; Maas, R.; Braukmann, A.; Bleck, E.; Saenger, T.; Schneider, M.; Jose, J. Autoantibodies to αs1-casein are induced by breast-feeding. PLoS ONE 2012, 7, e32716. [Google Scholar] [CrossRef] [PubMed]
- Jose, J.; Bernhardt, R.; Hannemann, F. Cellular surface display of dimeric adx and whole cell p450-mediated steroid synthesis on E. coli. J. Biotechnol. 2002, 95, 257–268. [Google Scholar] [CrossRef]
- Gratz, A.; Götz, C.; Jose, J. A ce-based assay for human protein kinase CK2 activity measurement and inhibitor screening. Electrophoresis 2010, 31, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Copeland, R.A. Evaluation of enzyme inhibitors in drug discovery: A guide for medicinal chemists and pharmakologists. John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2005. [Google Scholar]
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [PubMed]
- Jerabek-Willemsen, M.; Wienken, C.J.; Braun, D.; Baaske, P.; Duhr, S. Molecular interaction studies using microscale thermophoresis. Assay Drug Dev. Technol. 2011, 9, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Nienberg, C.; Retterath, A.; Becher, K.S.; Saenger, T.; Mootz, H.D.; Jose, J. Site-specific labeling of protein kinase CK2: Combining surface display and click chemistry for drug discovery applications. Pharmaceuticals 2016, 9, 36. [Google Scholar] [CrossRef] [PubMed]
- Raaf, J.; Brunstein, E.; Issinger, O.G.; Niefind, K. The interaction of CK2α and CK2β, the subunits of protein kinase CK2, requires CK2β in a preformed conformation and is enthalpically driven. Protein Sci. 2008, 17, 2180–2186. [Google Scholar] [CrossRef] [PubMed]
- Chin, J.W.; Santoro, S.W.; Martin, A.B.; King, D.S.; Wang, L.; Schultz, P.G. Addition of p-azido-l-phenylalanine to the genetic code of escherichia coli. J. Am. Chem. Soc. 2002, 124, 9026–9027. [Google Scholar] [CrossRef] [PubMed]
- Georgiou, G.; Stathopoulos, C.; Daugherty, P.S.; Nayak, A.R.; Iverson, B.L.; Curtiss, R., 3rd. Display of heterologous proteins on the surface of microorganisms: From the screening of combinatorial libraries to live recombinant vaccines. Nat. Biotechnol. 1997, 15, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Boder, E.T.; Midelfort, K.S.; Wittrup, K.D. Directed evolution of antibody fragments with monovalent femtomolar antigen-binding affinity. Proc. Natl. Acad. Sci. USA 2000, 97, 10701–10705. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, P.S.; Chen, G.; Olsen, M.J.; Iverson, B.L.; Georgiou, G. Antibody affinity maturation using bacterial surface display. Protein Eng. 1998, 11, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Christmann, A.; Wentzel, A.; Meyer, C.; Meyers, G.; Kolmar, H. Epitope mapping and affinity purification of monospecific antibodies by Escherichia coli cell surface display of gene-derived random peptide libraries. J. Immunol. Methods 2001, 257, 163–173. [Google Scholar] [CrossRef]
- Bessette, P.H.; Rice, J.J.; Daugherty, P.S. Rapid isolation of high-affinity protein binding peptides using bacterial display. Protein Eng. Des. Sel. 2004, 17, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Kenrick, S.A.; Daugherty, P.S. Bacterial display enables efficient and quantitative peptide affinity maturation. Protein Eng. Des. Sel. 2010, 23, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Perea, S.E.; Reyes, O.; Puchades, Y.; Mendoza, O.; Vispo, N.S.; Torrens, I.; Santos, A.; Silva, R.; Acevedo, B.; Lopez, E.; et al. Antitumor effect of a novel proapoptotic peptide that impairs the phosphorylation by the protein kinase 2 (casein kinase 2). Cancer Res. 2004, 64, 7127–7129. [Google Scholar] [CrossRef] [PubMed]
- Prudent, R.; Cochet, C. New protein kinase CK2 inhibitors: Jumping out of the catalytic box. Chem. Biol. 2009, 16, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Prudent, R.; Sautel, C.F.; Cochet, C. Structure-based discovery of small molecules targeting different surfaces of protein-kinase CK2. Biochim. Biophys. Acta Proteins Proteom. 2010, 1804, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Grankowski, N.; Boldyreff, B.; Issinger, O.G. Isolation and characterization of recombinant human casein kinase II subunits α and β from bacteria. Eur. J. Biochem. 1991, 198, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Petermann, K.; Vordenbäumen, S.; Pyun, J.C.; Braukmann, A.; Bleck, E.; Schneider, M.; Jose, J. Autodisplay of 60-kDa Ro/SS-A antigen and development of a surface display enzyme-linked immunosorbent assay for systemic lupus erythematosus patient sera screening. Anal. Biochem. 2010, 407, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Zuo, R.; Ornek, D.; Wood, T.K. Aluminum- and mild steel-binding peptides from phage display. Appl. Microbiol. Biotechnol. 2005, 68, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Chung, B.H.; Yoon, S.; Lee, K.K.; Lönnerdal, B.; Yu, D.Y. High-level expression of human αs1-casein in Escherichia coli. Biotechnol. Tech. 1997, 11, 675–678. [Google Scholar] [CrossRef]
- Hantke, K. Regulation of ferric iron transport in Escherichia coli K12: Isolation of a constitutive mutant. Mol. Gen. Genet. 1981, 182, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Schultheiss, E.; Paar, C.; Schwab, H.; Jose, J. Functional esterase surface display by the autotransporter pathway in Escherichia coli. J. Mol. Catal. 2002, 18, 89–97. [Google Scholar] [CrossRef]
- Hastie, C.J.; McLauchlan, H.J.; Cohen, P. Assay of protein kinases using radiolabeled ATP: A protocol. Nat. Protoc. 2006, 1, 968–971. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.C.; Hessenauer, A.; Gotz, C.; Montenarh, M. Dmat, an inhibitor of protein kinase CK2 induces reactive oxygen species and DNA double strand breaks. Oncol. Rep. 2009, 21, 1593–1597. [Google Scholar] [PubMed]
- Aberle, H.; Butz, S.; Stappert, J.; Weissig, H.; Kemler, R.; Hoschuetzky, H. Assembly of the cadherin-catenin complex in vitro with recombinant proteins. J. Cell. Sci. 1994, 107, 3655–3663. [Google Scholar] [PubMed]
- Spohrer, S.; Dimova, E.Y.; Kietzmann, T.; Montenarh, M.; Gotz, C. The nuclear fraction of protein kinase CK2 binds to the upstream stimulatory factors (USFS) in the absence of DNA. Cell. Signal. 2016, 28, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Yu, X.P.; Degraff, D.J.; Matusik, R.J. Upstream stimulatory factor 2, a novel foxa1-interacting protein, is involved in prostate-specific gene expression. Mol. Endocrinol. 2009, 23, 2038–2047. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequence | Inhibition* (%) | IC50 Value (µM) |
|---|---|---|---|
| B1 | KHTKGPTAYCPL | < 5 | n.d. |
| B2 | DCRGLIVMIKLH | 79 | 0.8 |
| B3 | YRKPHWFIHTRI | < 5 | n.d. |
| B4 | PCPAPRAPKLSI | 29 | n.d. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nienberg, C.; Garmann, C.; Gratz, A.; Bollacke, A.; Götz, C.; Jose, J. Identification of a Potent Allosteric Inhibitor of Human Protein Kinase CK2 by Bacterial Surface Display Library Screening. Pharmaceuticals 2017, 10, 6. https://doi.org/10.3390/ph10010006
Nienberg C, Garmann C, Gratz A, Bollacke A, Götz C, Jose J. Identification of a Potent Allosteric Inhibitor of Human Protein Kinase CK2 by Bacterial Surface Display Library Screening. Pharmaceuticals. 2017; 10(1):6. https://doi.org/10.3390/ph10010006
Chicago/Turabian StyleNienberg, Christian, Claudia Garmann, Andreas Gratz, Andre Bollacke, Claudia Götz, and Joachim Jose. 2017. "Identification of a Potent Allosteric Inhibitor of Human Protein Kinase CK2 by Bacterial Surface Display Library Screening" Pharmaceuticals 10, no. 1: 6. https://doi.org/10.3390/ph10010006
APA StyleNienberg, C., Garmann, C., Gratz, A., Bollacke, A., Götz, C., & Jose, J. (2017). Identification of a Potent Allosteric Inhibitor of Human Protein Kinase CK2 by Bacterial Surface Display Library Screening. Pharmaceuticals, 10(1), 6. https://doi.org/10.3390/ph10010006