
CK2 Molecular Targeting—Tumor Cell-Specific Delivery of RNAi in Various Models of Cancer

 ,
,  ,
,
Abstract
:1. Introduction
2. Discussion
2.1. Features of CK2 Pertinent to Cancer
2.2. CK2 Elevation in Benign Prostate Proliferation versus in Prostate Cancer
2.3. CK2 as a Target of Cancer Therapy
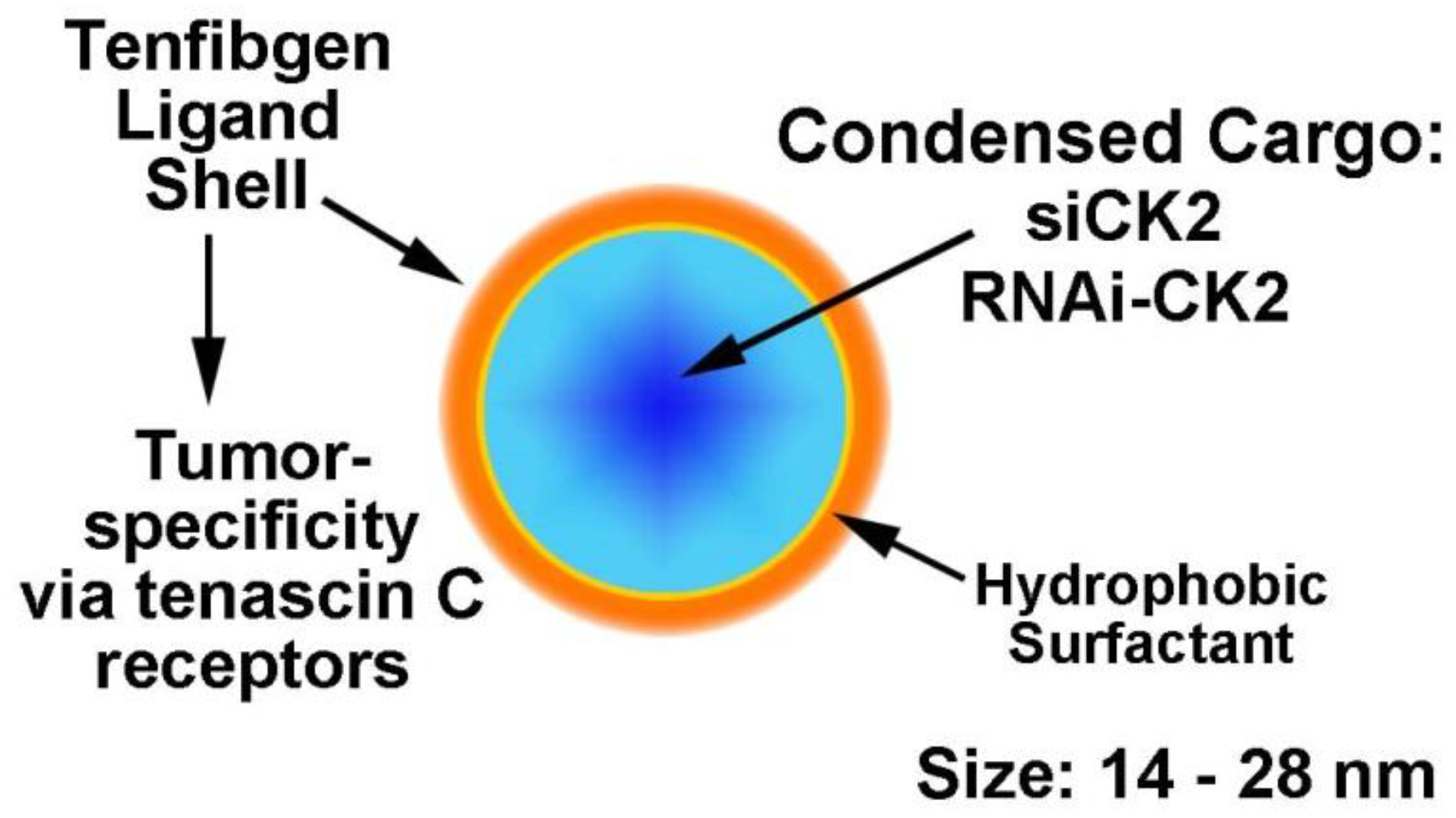
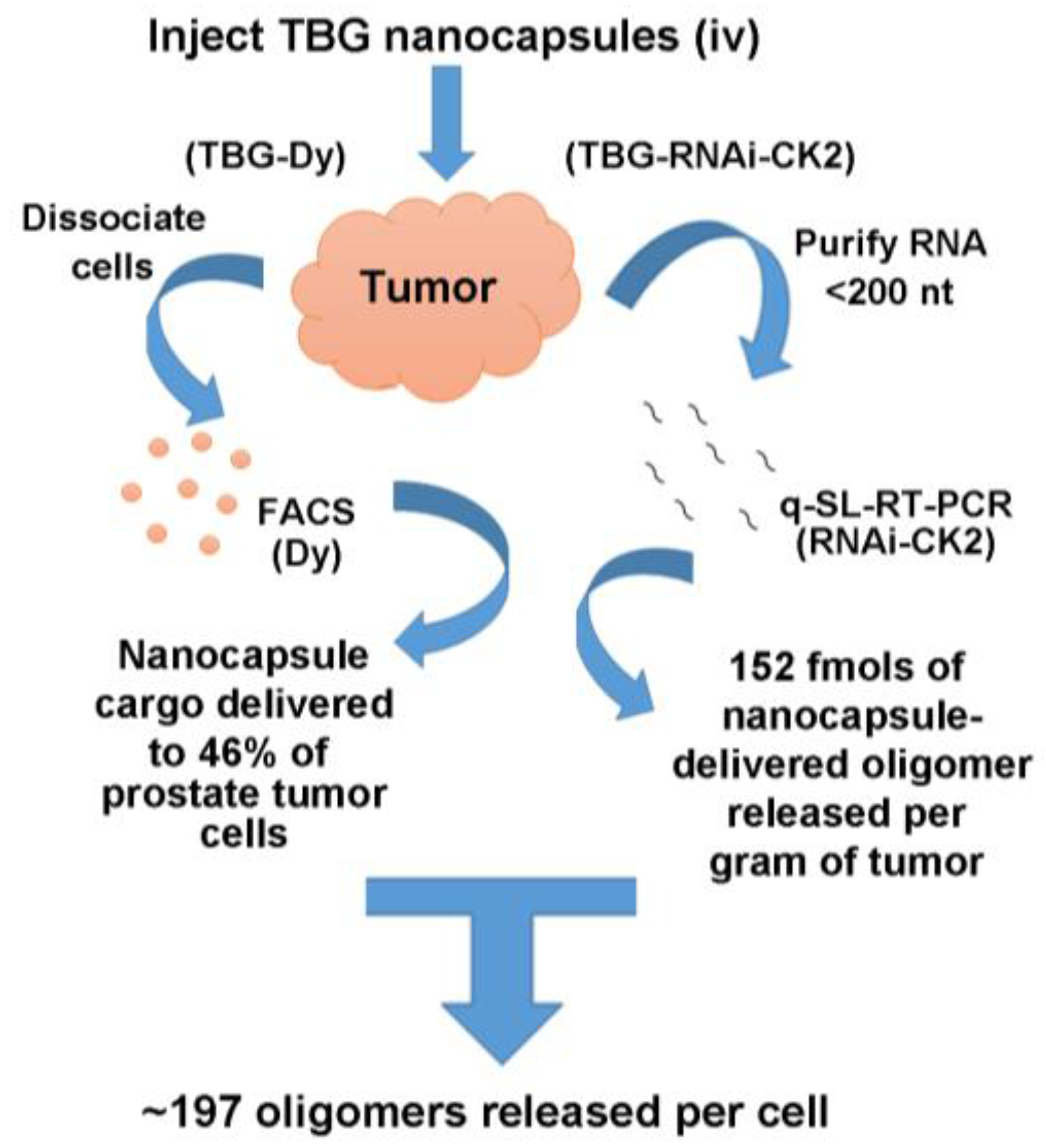
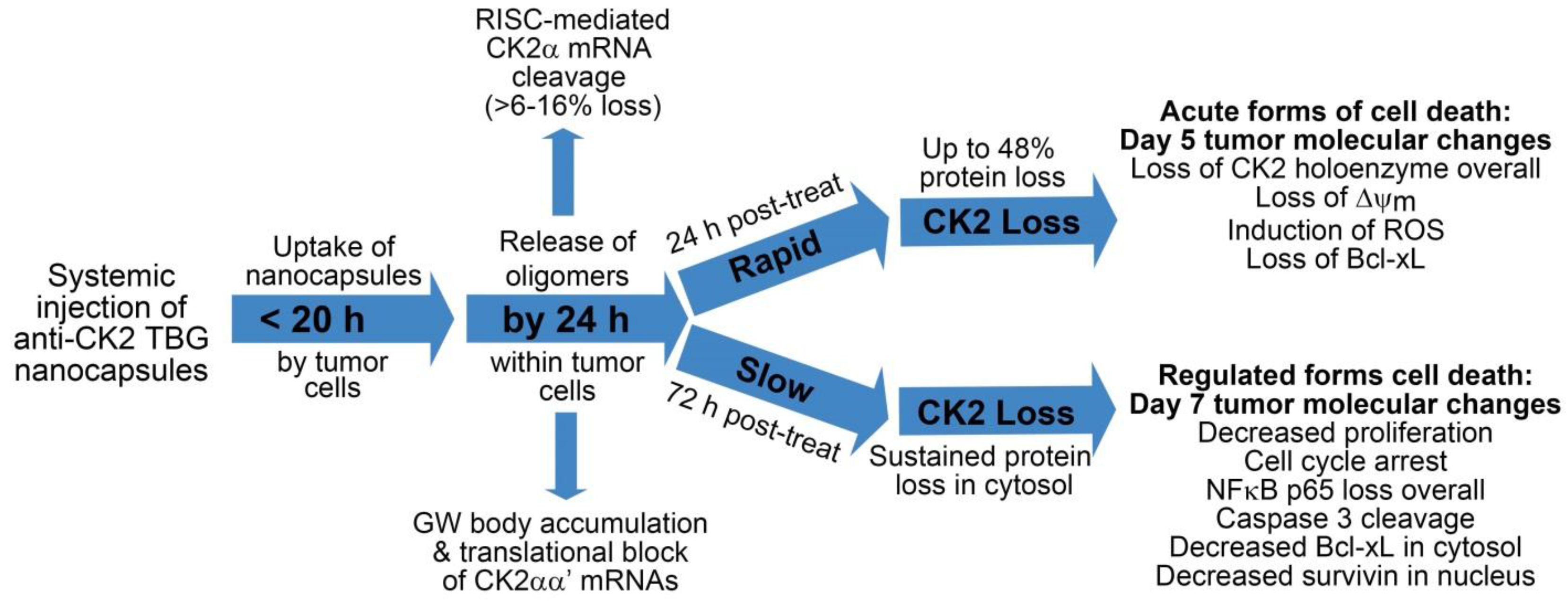
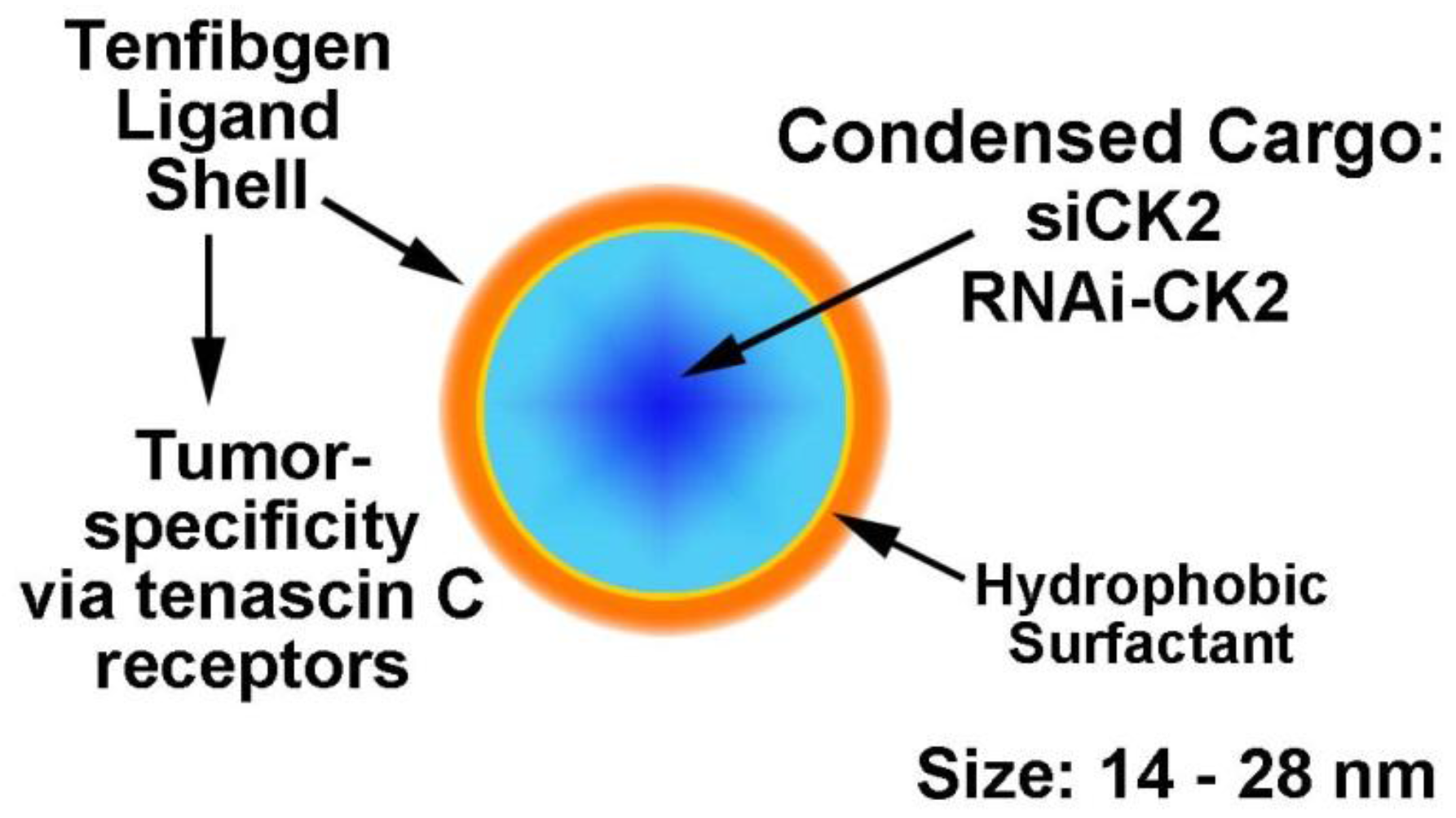
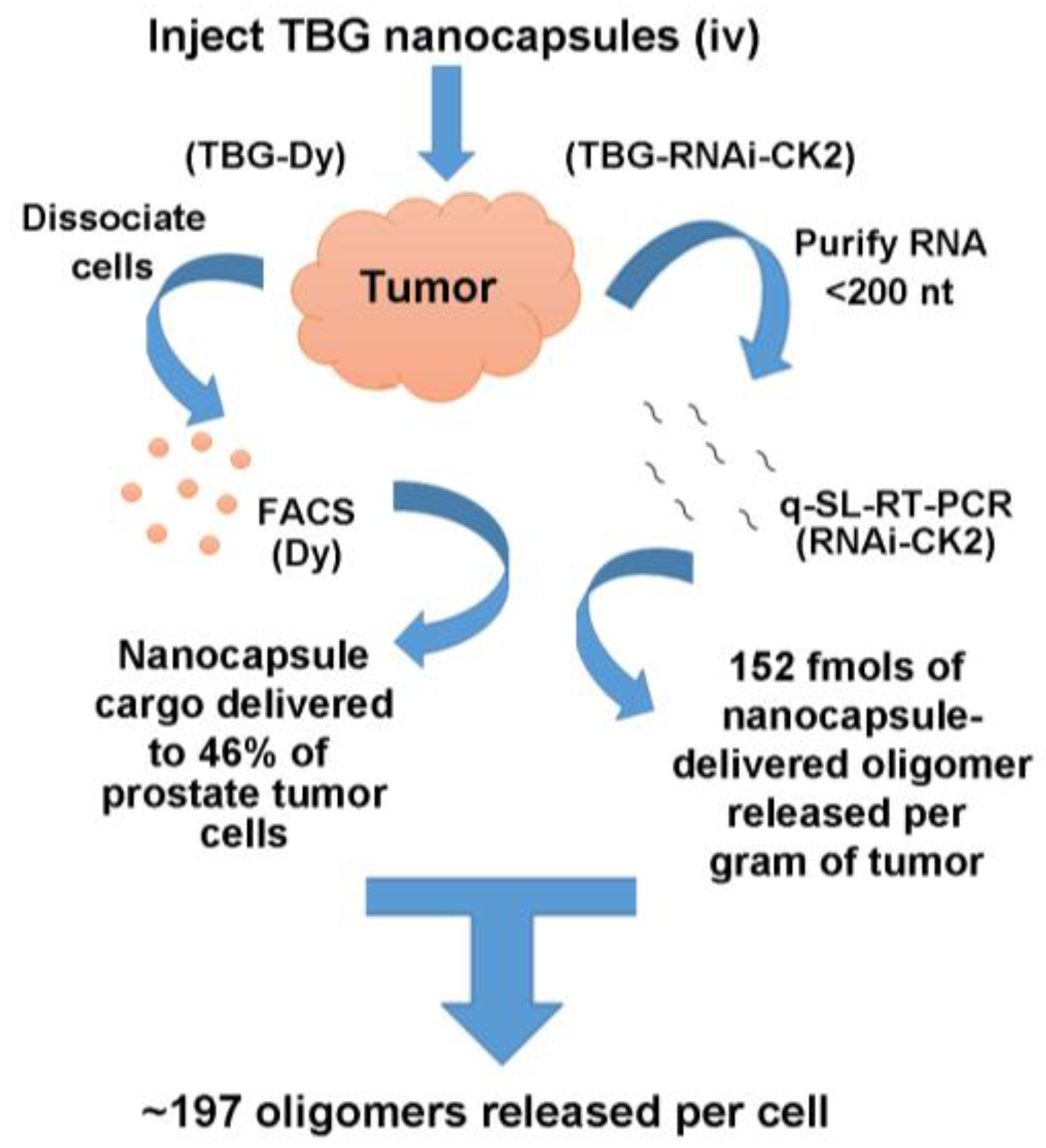
2.4. Desirability of Targeting CK2 Specifically in Cancer Cells and Utility of the TBG Nanocapsule to Accomplish This Goal
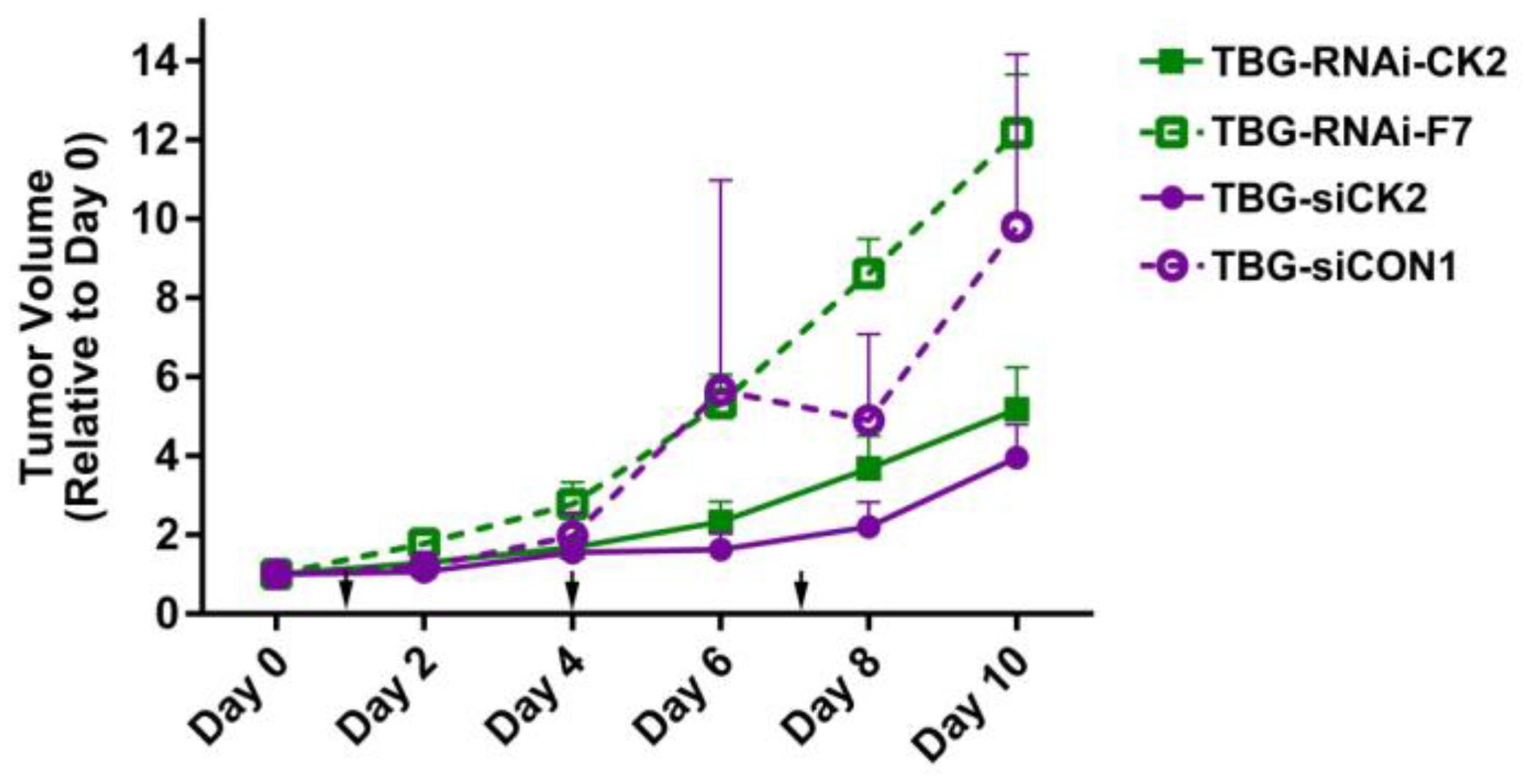
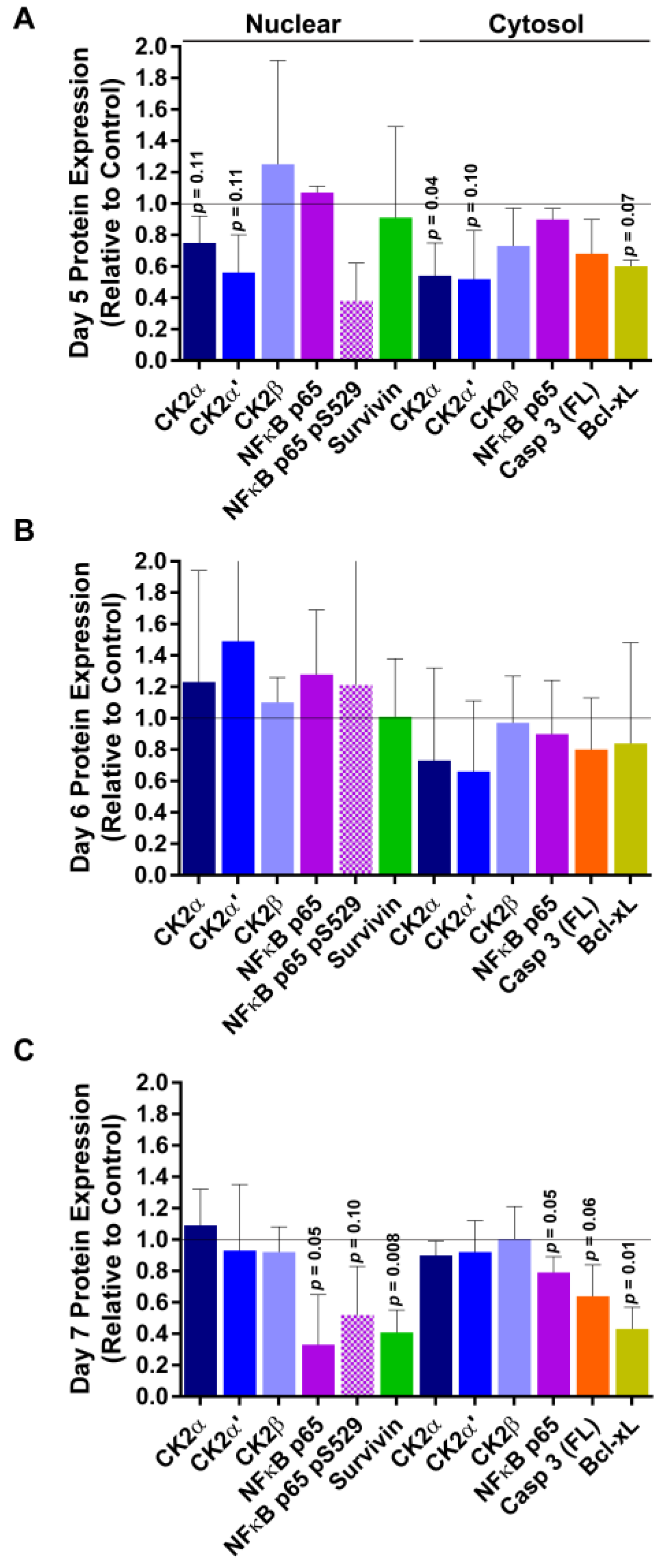
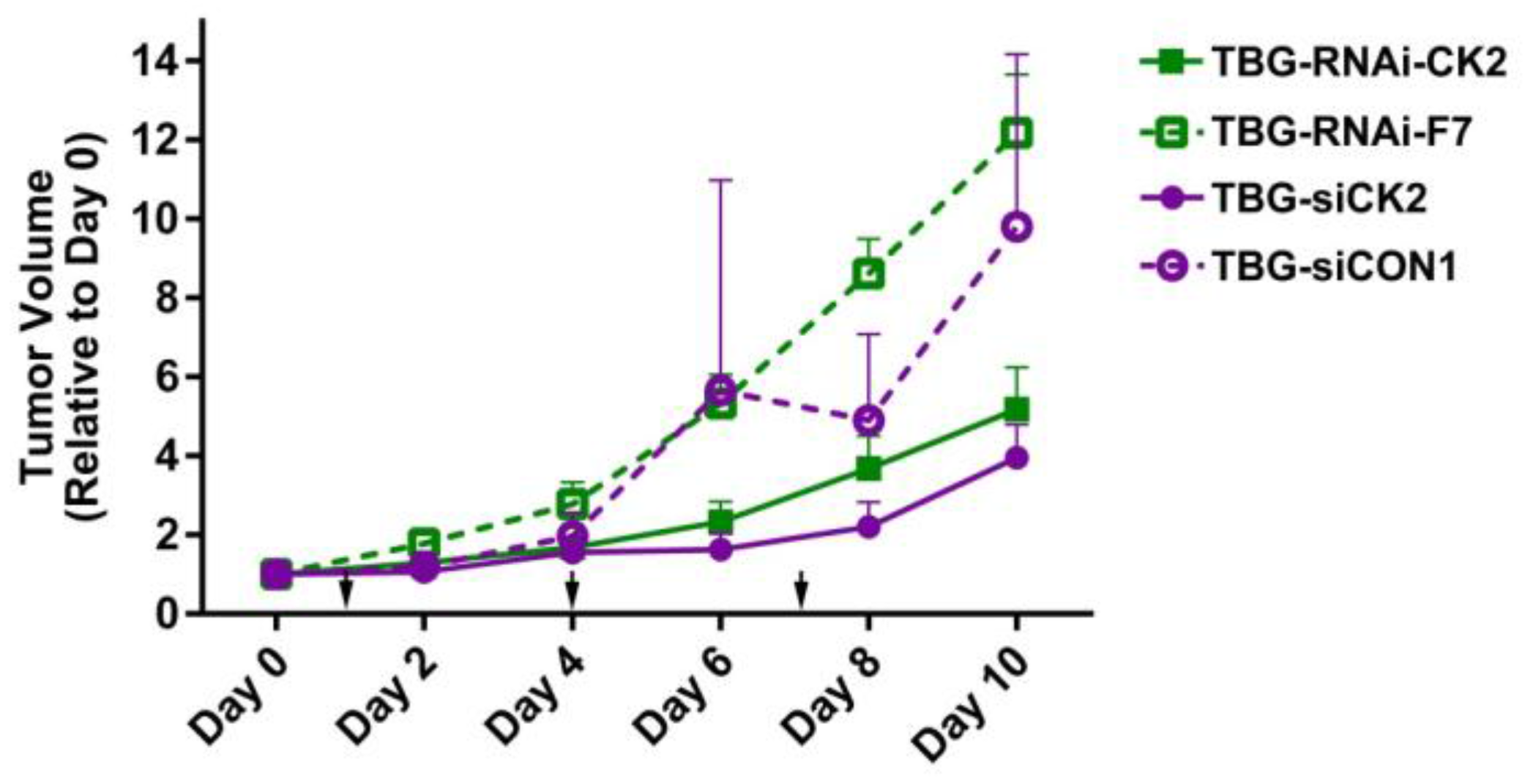
2.5. Response of Xenograft Tumors to TBG-RNAi-CK2
2.6. Phase I Trial of TBG-RNAi-CK2 in Large Animal Patients
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Guerra, B.; Issinger, O.-G. Protein kinase CK2 and its role in cellular proliferation, development and pathology. Electrophoresis 1999, 20, 391–408. [Google Scholar] [CrossRef]
- Guerra, B.; Issinger, O.-G. Protein kinase CK2 in human diseases. Curr. Med. Chem. 2008, 15, 1870–1886. [Google Scholar] [CrossRef] [PubMed]
- Pinna, L.A. Protein kinase CK2: A challenge to canons. J. Cell Sci. 2002, 115, 3873–3878. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K. Significance of the casein kinase system in cell growth and proliferation with emphasis on studies of the androgenic regulation of the prostate. Cell. Mol. Biol. Res. 1994, 40, 1–11. [Google Scholar] [PubMed]
- Ruzzene, M.; Pinna, L.A. Addiction to protein kinase CK2: A common denominator of diverse cancer cells? Biochim. Biophys. Acta 2010, 1804, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Meggio, F.; Pinna, L.A. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 2003, 17, 349–368. [Google Scholar] [CrossRef] [PubMed]
- Tawfic, S.; Yu, S.; Wang, H.; Faust, R.; Davis, A.; Ahmed, K. Protein kinase CK2 signal in neoplasia. Histol. Histopathol. 2001, 16, 573–582. [Google Scholar] [PubMed]
- Guo, C.; Yu, S.; Davis, A.T.; Wang, H.; Green, J.E.; Ahmed, K. A potential role of nuclear matrix-associated protein kinase CK2 in protection against drug-induced apoptosis in cancer cells. J. Biol. Chem. 2001, 276, 5992–5999. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.; Gerber, D.A.; Cochet, C. Joining the cell survival squad: An emerging role for protein kinase CK2. Trends Cell Biol. 2002, 12, 226–230. [Google Scholar] [CrossRef]
- Ahmad, K.A.; Wang, G.; Unger, G.; Slaton, J.; Ahmed, K. Protein kinase CK2—A key suppressor of apoptosis. Adv. Enzyme Regul. 2008, 48, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Trembley, J.H.; Qaiser, F.; Kren, B.T.; Ahmed, K. CK2—A global regulator of cell death. In Protein Kinase CK2 Cellular Function in Normal and Disease States; Ahmed, K., Issinger, O.-G., Szyszka, R., Eds.; Springer: Cham, Switzerland, 2015; Volume 12, pp. 159–181. [Google Scholar]
- Wang, H.; Davis, A.; Yu, S.; Ahmed, K. Response of cancer cells to molecular interruption of the CK2 signal. Mol. Cell. Biochem. 2001, 227, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Perea, S.E.; Baladron, I.; Garcia, Y.; Perera, Y.; Lopez, A.; Soriano, J.L.; Batista, N.; Palau, A.; Hernández, I.; Farina, H.; et al. CIGB-300, a synthetic peptide-based drug that targets the CK2 phosphoaceptor domain. Translational and clinical research. Mol. Cell. Biochem. 2011, 356, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Pinna, L.A.; Allende, J.E. Protein kinase CK2 in health and disease: Protein kinase CK2: An ugly duckling in the kinome pond. Cell. Mol. Life Sci. 2009, 66, 1795–1799. [Google Scholar] [CrossRef] [PubMed]
- Pierre, F.; Chua, P.C.; O’Brien, S.E.; Siddiqui-Jain, A.; Bourbon, P.; Haddach, M.; Michaux, J.; Nagasawa, J.; Schwaebe, M.K.; Stefan, E.; et al. Discovery and SAR of 5-(3-chlorophenylamino)benzo[c][2,6]naphthyridine-8-carboxylic acid (CX-4945), the first clinical stage inhibitor of protein kinase CK2 for the treatment of cancer. J. Med. Chem. 2011, 54, 635–654. [Google Scholar] [CrossRef] [PubMed]
- Ortega, C.E.; Seidner, Y.; Dominguez, I. Mining CK2 in cancer. PLoS ONE 2014, 9, e115609. [Google Scholar] [CrossRef] [PubMed]
- Faust, R.A.; Niehans, G.; Gapany, M.; Hoistad, D.; Knapp, D.; Cherwitz, D.; Davis, A.; Adams, G.L.; Ahmed, K. Subcellular immunolocalization of protein kinase CK2 in normal and carcinoma cells. Int. J. Biochem. Cell Biol. 1999, 31, 941–949. [Google Scholar] [CrossRef]
- Dominguez, I.; Sonenshein, G.E.; Seldin, D.C. Protein kinase CK2 in health and disease: CK2 and its role in Wnt and NF-κB signaling: Linking development and cancer. Cell. Mol. Life Sci. 2009, 66, 1850–1857. [Google Scholar] [CrossRef] [PubMed]
- Seldin, D.C.; Landesman-Bollag, E.; Farago, M.; Currier, N.; Lou, D.; Dominguez, I. CK2 as a positive regulator of Wnt signalling and tumourigenesis. Mol. Cell. Biochem. 2005, 274, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-X.; Jiang, S.-S.; Zhang, X.-F.; Zhou, Z.-Q.; Pan, Q.-Z.; Chen, C.-L.; Zhao, J.-J.; Tang, Y.; Xia, J.-C.; Weng, D.-S. Protein kinase CK2α catalytic subunit is overexpressed and serves as an unfavorable prognostic marker in primary hepatocellular carcinoma. Oncotarget 2015, 6, 34800. [Google Scholar] [PubMed]
- Ahmed, K.; Davis, A.T.; Wang, H.; Faust, R.A.; Yu, S.; Tawfic, S. Significance of protein kinase CK2 nuclear signaling in neoplasia. J. Cell. Biochem. Suppl. 2000, 35, 130–135. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Ahmad, K.A.; Ahmed, K. Modulation of death receptor-mediated apoptosis by CK2. Mol. Cell. Biochem. 2005, 274, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Trembley, J.H.; Wang, G.; Unger, G.; Slaton, J.; Ahmed, K. Protein kinase CK2 in health and disease: CK2: A key player in cancer biology. Cell. Mol. Life Sci. 2009, 66, 1858–1867. [Google Scholar] [CrossRef] [PubMed]
- Trembley, J.H.; Wu, J.; Unger, G.M.; Kren, B.T.; Ahmed, K. CK2 suppression of apoptosis and its implications in cancer biology and therapy. In Protein Kinase CK2; the Wiley-IUBMB Series on Biochemistry and Molecular Biology; Pinna, L.A., Ed.; Wiley: Hoboken, NJ, USA, 2013; pp. 319–333. [Google Scholar]
- Gapany, M.; Faust, R.A.; Tawfic, S.; Davis, A.; Adams, G.L.; Ahmed, K. Association of elevated protein kinase CK2 activity with aggressive behavior of squamous cell carcinoma of the head and neck. Mol. Med. 1995, 1, 659–666. [Google Scholar] [PubMed]
- Yenice, S.; Davis, A.T.; Goueli, S.A.; Akdas, A.; Limas, C.; Ahmed, K. Nuclear casein kinase 2 (CK-2) activity in human normal, benign hyperplastic, and cancerous prostate. Prostate 1994, 24, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Faust, R.A.; Gapany, M.; Tristani, P.; Davis, A.; Adams, G.L.; Ahmed, K. Elevated protein kinase CK2 activity in chromatin of head and neck tumors: Association with malignant transformation. Cancer Lett. 1996, 101, 31–35. [Google Scholar] [CrossRef]
- Giusiano, S.; Cochet, C.; Filhol, O.; Duchemin-Pelletier, E.; Secq, V.; Bonnier, P.; Carcopino, X.; Boubli, L.; Birnbaum, D.; Garcia, S.; et al. Protein kinase CK2α subunit over-expression correlates with metastatic risk in breast carcinomas: Quantitative immunohistochemistry in tissue microarrays. Eur. J. Cancer 2011, 47, 792–801. [Google Scholar] [CrossRef] [PubMed]
- Laramas, M.; Pasquier, D.; Filhol, O.; Ringeisen, F.; Descotes, J.L.; Cochet, C. Nuclear localization of protein kinase CK2 catalytic subunit (CK2α) is associated with poor prognostic factors in human prostate cancer. Eur. J. Cancer 2007, 43, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.; Yenice, S.; Davis, A.; Goueli, S.A. Association of casein kinase 2 with nuclear chromatin in relation to androgenic regulation of rat prostate. Proc. Natl. Acad. Sci. USA 1993, 90, 4426–4430. [Google Scholar] [CrossRef] [PubMed]
- Rayan, A.; Goueli, S.A.; Lange, P.; Ahmed, K. Chromatin-associated protein kinases in human normal and benign hyperplastic prostate. Cancer Res. 1985, 45, 2277–2282. [Google Scholar] [PubMed]
- Qaiser, F.; Trembley, J.H.; Sadiq, S.; Muhammad, I.; Younis, R.; Hashmi, S.N.; Murtaza, B.; Rector, T.S.; Naveed, A.K.; Ahmed, K. Examination of CK2α and NF-κB p65 expression in human benign prostatic hyperplasia and prostate cancer tissues. Mol. Cell. Biochem. 2016, 420, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Westerheide, S.D.; Hanson, J.L.; Baldwin, A.S. Tumor necrosis factor α-induced phosphorylation of RelA/p65 on Ser529 is controlled by casein kinase II. J. Biol. Chem. 2000, 275, 32592–32597. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell 2015, 163, 1011–1025. [Google Scholar]
- Slaton, J.W.; Unger, G.M.; Sloper, D.T.; Davis, A.T.; Ahmed, K. Induction of apoptosis by antisense CK2 in human prostate cancer xenograft model. Mol. Cancer Res. 2004, 2, 712–721. [Google Scholar] [PubMed]
- Wang, G.; Unger, G.; Ahmad, K.A.; Slaton, J.W.; Ahmed, K. Downregulation of CK2 induces apoptosis in cancer cells—A potential approach to cancer therapy. Mol. Cell. Biochem. 2005, 274, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Trembley, J.H.; Chen, Z.; Unger, G.; Slaton, J.; Kren, B.T.; Van Waes, C.; Ahmed, K. Emergence of protein kinase CK2 as a key target in cancer therapy. Biofactors 2010, 36, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Sarno, S.; Pinna, L.A. Protein kinase CK2 as a druggable target. Mol. Biosyst. 2008, 4, 889–894. [Google Scholar] [CrossRef] [PubMed]
- Sarno, S.; Ruzzene, M.; Frascella, P.; Pagano, M.A.; Meggio, F.; Zambon, A.; Mazzorana, M.; Di Maira, G.; Lucchini, V.; Pinna, L.A. Development and exploitation of CK2 inhibitors. Mol. Cell. Biochem. 2005, 274, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Zanin, S.; Sarno, S.; Costa, E.; Girardi, C.; Ribaudo, G.; Salvi, M.; Zagotto, G.; Ruzzene, M.; Pinna, L.A. Design, validation and efficacy of bisubstrate inhibitors specifically affecting ecto-CK2 kinase activity. Biochem. J. 2015, 471, 415–430. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui-Jain, A.; Drygin, D.; Streiner, N.; Chua, P.; Pierre, F.; O’Brien, S.E.; Bliesath, J.; Omori, M.; Huser, N.; Ho, C.; et al. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010, 70, 10288–10298. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui-Jain, A.; Bliesath, J.; Macalino, D.; Omori, M.; Huser, N.; Streiner, N.; Ho, C.B.; Anderes, K.; Proffitt, C.; O’Brien, S.E.; et al. CK2 inhibitor CX-4945 suppresses DNA repair response triggered by DNA-targeted anticancer drugs and augments efficacy: Mechanistic rationale for drug combination therapy. Mol. Cancer Ther. 2012, 11, 994–1005. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.; Kren, B.T.; Abedin, M.J.; Vogel, R.I.; Shaughnessy, D.P.; Nacusi, L.; Korman, V.L.; Li, Y.; Dehm, S.M.; Zimmerman, C.L.; et al. CK2 targeted RNAi therapeutic delivered via malignant cell-directed tenfibgen nanocapsule: Dose and molecular mechanisms of response in xenograft prostate tumors. Oncotarget 2016, 7, 61789–61805. [Google Scholar] [CrossRef] [PubMed]
- Unger, G.M.; Kren, B.T.; Korman, V.L.; Kimbrough, T.G.; Vogel, R.I.; Ondrey, F.G.; Trembley, J.H.; Ahmed, K. Mechanism and efficacy of sub-50-nm tenfibgen nanocapsules for cancer cell-directed delivery of anti-CK2 RNAi to primary and metastatic squamous cell carcinoma. Mol. Cancer Ther. 2014, 13, 2018–2029. [Google Scholar] [CrossRef] [PubMed]
- Kren, B.; Unger, G.; Abedin, M.; Vogel, R.; Henzler, C.; Ahmed, K.; Trembley, J. Preclinical evaluation of cyclin dependent kinase 11 and casein kinase 2 survival kinases as RNA interference targets for triple negative breast cancer therapy. Breast Cancer Res. 2015, 17, 19. [Google Scholar] [CrossRef] [PubMed]
- Qaiser, F.; Trembley, J.H.; Kren, B.T.; Wu, J.J.; Naveed, A.K.; Ahmed, K. Protein kinase CK2 inhibition induces cell death via early impact on mitochondrial function. J. Cell. Biochem. 2014, 115, 2103–2115. [Google Scholar] [CrossRef] [PubMed]
- Girardi, C.; Ottaviani, D.; Pinna, L.A.; Ruzzene, M. Different persistence of the cellular effects promoted by protein kinase CK2 inhibitors CX-4945 and TDB. Biomed. Res. Int. 2015, 2015, 185736. [Google Scholar] [CrossRef] [PubMed]
- Cree, I.A.; Charlton, P. Molecular chess? Hallmarks of anti-cancer drug resistance. BMC Cancer 2017, 17, 10. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.; Unger, G.M.; Kren, B.T.; Trembley, J.H. Targeting CK2 for Cancer Therapy Using A Nanomedicine Approach. In Protein Kinase CK2 in Cellular Function in Normal and Disease States; Ahmed, K., Issinger, O.-G., Szyszka, R., Eds.; Springer International Publishing Switzerland: Gewerbestrasse, Switzerland, 2015; Volume 12, pp. 299–315. [Google Scholar]
- Unger, G.; Trembley, J.; Kren, B.; Ahmed, K. Nanoparticles in cancer therapy. In Encyclopedia of Cancer: SpringerReference; Schwab, M., Ed.; Springer: Heidelberg, Germany, 2012; pp. 1–4. [Google Scholar]
- Trembley, J.H.; Unger, G.M.; Korman, V.L.; Tobolt, D.K.; Kazimierczuk, Z.; Pinna, L.A.; Kren, B.T.; Ahmed, K. Nanoencapsulated anti-CK2 small molecule drug or sirna specifically targets malignant cancer but not benign cells. Cancer Lett. 2012, 315, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Tuxhorn, J.A.; Ayala, G.E.; Rowley, D.R. Reactive stroma in prostate cancer progression. J. Urol. 2001, 166, 2472–2483. [Google Scholar] [CrossRef]
- Erickson, H.P.; Bourdon, M.A. Tenascin: An extracellular matrix protein prominent in specialized embryonic tissues and tumors. Annu. Rev. Cell Biol. 1989, 5, 71–92. [Google Scholar] [CrossRef] [PubMed]
- Chiquet-Ehrismann, R.; Chiquet, M. Tenascins: Regulation and putative functions during pathological stress. J. Pathol. 2003, 200, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, K.; Erickson, H.P.; Ikeda, Y.; Takada, Y. Identification of amino acid sequences in fibrinogen gamma -chain and tenascin C C-terminal domains critical for binding to integrin αvβ3. J. Biol. Chem. 2000, 275, 16891–16898. [Google Scholar] [CrossRef] [PubMed]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Guttery, D.; Shaw, J.; Lloyd, K.; Pringle, J.; Walker, R. Expression of tenascin-C and its isoforms in the breast. Cancer Metastasis Rev. 2010, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Oskarsson, T.; Acharyya, S.; Zhang, X.H.; Vanharanta, S.; Tavazoie, S.F.; Morris, P.G.; Downey, R.J.; Manova-Todorova, K.; Brogi, E.; Massague, J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 2011, 17, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Diallo, O.T.; Hu, M.; Ehsanian, R.; Yang, X.; Arun, P.; Lu, H.; Korman, V.; Unger, G.; Ahmed, K.; et al. CK2 modulation of NF-κB, TP53, and the malignant phenotype in head and neck cancer by anti-CK2 oligonucleotides in vitro or in vivo via sub-50-nm nanocapsules. Clin. Cancer Res. 2010, 16, 2295–2307. [Google Scholar] [CrossRef] [PubMed]
- Trembley, J.H.; Unger, G.M.; Gomez, O.C.; Abedin, J.; Korman, V.L.; Vogel, R.I.; Niehans, G.; Kren, B.T.; Ahmed, K. Tenfibgen-DMAT nanocapsule delivers CK2 inhibitor dmat to prostate cancer xenograft tumors causing inhibition of cell proliferation. Mol. Cell. Pharmacol. 2014, 6, 15–25. [Google Scholar] [PubMed]
- Trembley, J.H.; Unger, G.M.; Korman, V.L.; Abedin, M.J.; Nacusi, L.P.; Vogel, R.I.; Slaton, J.W.; Kren, B.T.; Ahmed, K. Tenfibgen ligand nanoencapsulation delivers bi-functional anti-CK2 RNAi oligomer to key sites for prostate cancer targeting using human xenograft tumors in mice. PLoS ONE 2014, 9, e109970. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, D.W.; Davis, M.E. Impact of tumor-specific targeting and dosing schedule on tumor growth inhibition after intravenous administration of sirna-containing nanoparticles. Biotechnol. Bioeng. 2008, 99, 975–985. [Google Scholar] [CrossRef] [PubMed]
- Kren, B.T.; Korman, V.L.; Tobolt, D.K.; Unger, G.M. Subcutaneous delivery of hepatocyte targeted sub-50 nm nanoencapsulated sirna mediates gene silencing. Mol. Ther. 2011, 19, S319–S320. [Google Scholar]
- Cannon, C.M.; Trembley, J.H.; Kren, B.T.; Unger, G.M.; O’Sullivan, M.G.; Cornax, I.; Modiano, J.F.; Ahmed, K. Protein kinase CK2 as a promising new therapeutic target in feline squamous cell carcinoma. Am. J. Vet. Res. 2017, in press. [Google Scholar]
- Wypij, J.M. A naturally occurring feline model of head and neck squamous cell carcinoma. Pathol. Res. Int. 2013, 2013, 7. [Google Scholar] [CrossRef] [PubMed]
- Hanif, I.M.; Ahmad, K.A.; Ahmed, K.; Pervaiz, S. Involvement of reactive oxygen species in apoptosis induced by pharmacological inhibition of protein kinase CK2. Ann. N. Y. Acad. Sci. 2009, 1171, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Tapia, J.C.; Torres, V.A.; Rodriguez, D.A.; Leyton, L.; Quest, A.F. Casein kinase 2 (CK2) increases survivin expression via enhanced beta-catenin-T cell factor/lymphoid enhancer binding factor-dependent transcription. Proc. Natl. Acad. Sci. USA 2006, 103, 15079–15084. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yu, S.; Davis, A.T.; Ahmed, K. Cell cycle dependent regulation of protein kinase CK2 signaling to the nuclear matrix. J. Cell. Biochem. 2003, 88, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, K.A.; Wang, G.; Ahmed, K. Intracellular hydrogen peroxide production is an upstream event in apoptosis induced by down-regulation of casein kinase 2 in prostate cancer cells. Mol. Cancer Res. 2006, 4, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, K.A.; Harris, N.H.; Johnson, A.D.; Lindvall, H.C.; Wang, G.; Ahmed, K. Protein kinase CK2 modulates apoptosis induced by resveratrol and epigallocatechin-3-gallate in prostate cancer cells. Mol. Cancer Ther. 2007, 6, 1006–1012. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Wang, H.; Davis, A.; Ahmed, K. Consequences of CK2 signaling to the nuclear matrix. Mol. Cell. Biochem. 2001, 227, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Ahmad, K.A.; Harris, N.H.; Ahmed, K. Impact of protein kinase CK2 on inhibitor of apoptosis proteins in prostate cancer cells. Mol. Cell. Biochem. 2008, 316, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Trembley, J.H.; Unger, G.M.; Tobolt, D.K.; Korman, V.L.; Wang, G.; Ahmad, K.A.; Slaton, J.W.; Kren, B.T.; Ahmed, K. Systemic administration of antisense oligonucleotides simultaneously targeting CK2α and α’ subunits reduces orthotopic xenograft prostate tumors in mice. Mol. Cell. Biochem. 2011, 356, 21–35. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor Model | Treatment | Tumor Volume on Final Day a | p-Value |
|---|---|---|---|
| PC3-LN4 | TBG-RNAi-CK2—0.01 mg/kg | 5.2 ± 3.2 | 0.005 |
| TBG-RNAi-F7—0.01 mg/kg | 12.2 ± 4.2 | ||
| PC3-LN4 | TBG-siCK2—0.01 mg/kg | 4.0 ± 2.5 | 0.007 |
| TBG-siCON1—1.0 mg/kg | 10.6 ± 5.5 | ||
| 22Rv1 | TBG-RNAi-CK2—0.1 mg/kg | 2.5 ± 1.5 | 0.11 |
| TBG-RNAi-F7—1.0 mg/kg | 4.0 ± 1.5 | ||
| MDA-MB-231 | TBG-siCK2—0.01 mg/kg | 1.4 ± 0.32 | 0.026 |
| TBG-siCON1—0.01 mg/kg | 2.1 ± 0.55 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trembley, J.H.; Kren, B.T.; Abedin, M.J.; Vogel, R.I.; Cannon, C.M.; Unger, G.M.; Ahmed, K. CK2 Molecular Targeting—Tumor Cell-Specific Delivery of RNAi in Various Models of Cancer. Pharmaceuticals 2017, 10, 25. https://doi.org/10.3390/ph10010025
Trembley JH, Kren BT, Abedin MJ, Vogel RI, Cannon CM, Unger GM, Ahmed K. CK2 Molecular Targeting—Tumor Cell-Specific Delivery of RNAi in Various Models of Cancer. Pharmaceuticals. 2017; 10(1):25. https://doi.org/10.3390/ph10010025
Chicago/Turabian StyleTrembley, Janeen H., Betsy T. Kren, Md. Joynal Abedin, Rachel I. Vogel, Claire M. Cannon, Gretchen M. Unger, and Khalil Ahmed. 2017. "CK2 Molecular Targeting—Tumor Cell-Specific Delivery of RNAi in Various Models of Cancer" Pharmaceuticals 10, no. 1: 25. https://doi.org/10.3390/ph10010025
APA StyleTrembley, J. H., Kren, B. T., Abedin, M. J., Vogel, R. I., Cannon, C. M., Unger, G. M., & Ahmed, K. (2017). CK2 Molecular Targeting—Tumor Cell-Specific Delivery of RNAi in Various Models of Cancer. Pharmaceuticals, 10(1), 25. https://doi.org/10.3390/ph10010025