
The Development of CK2 Inhibitors: From Traditional Pharmacology to in Silico Rational Drug Design


Abstract
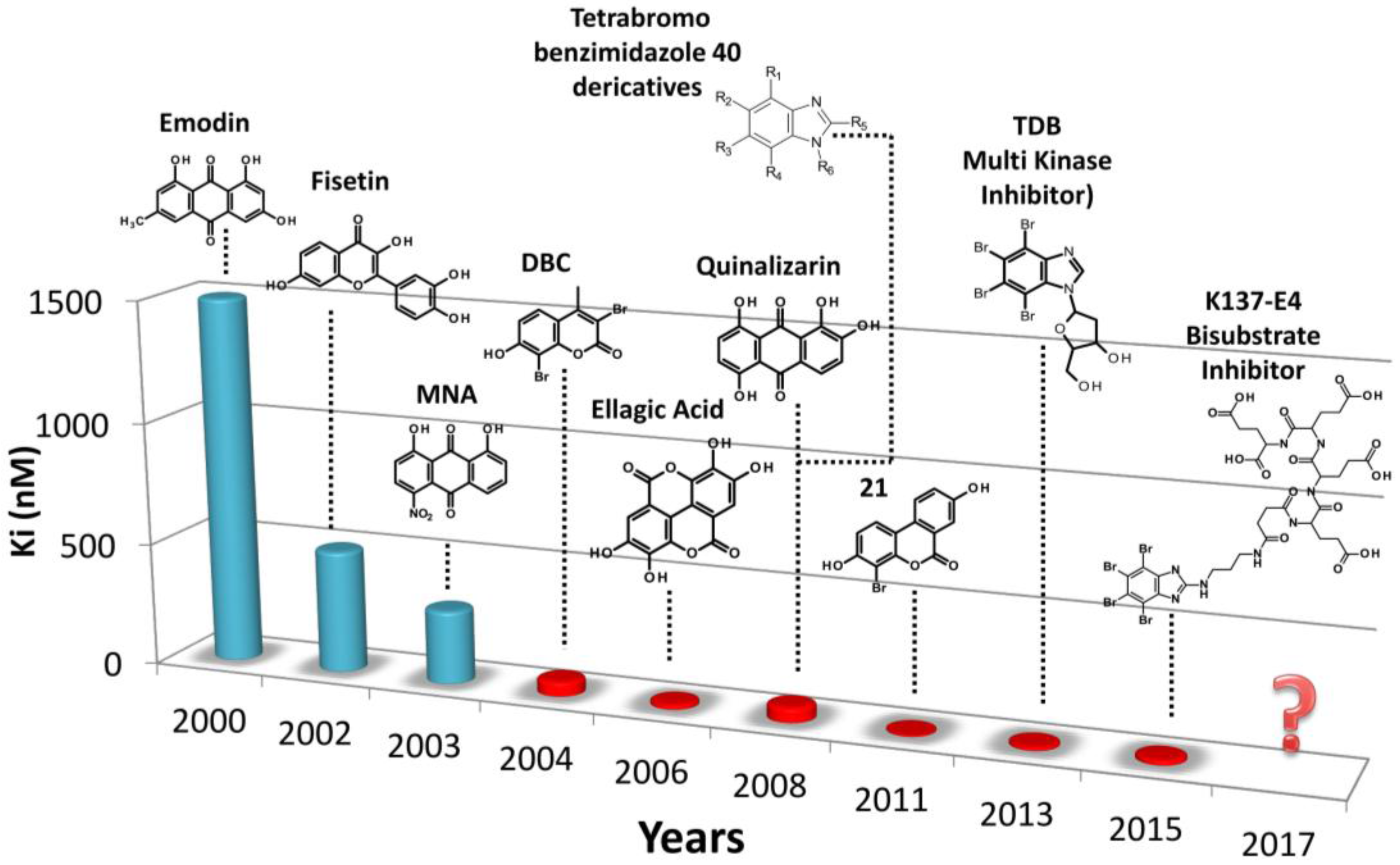
:1. Introduction
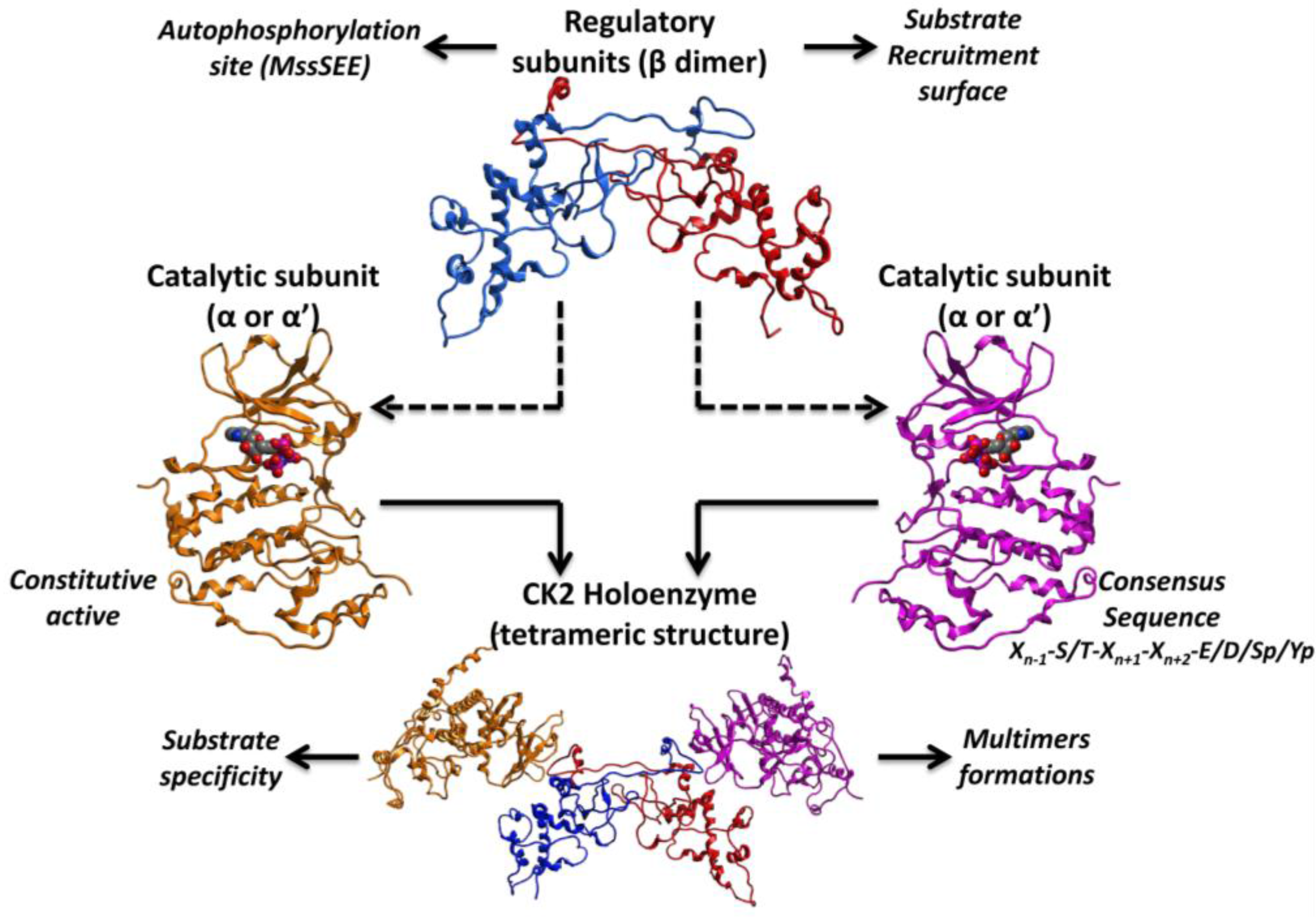
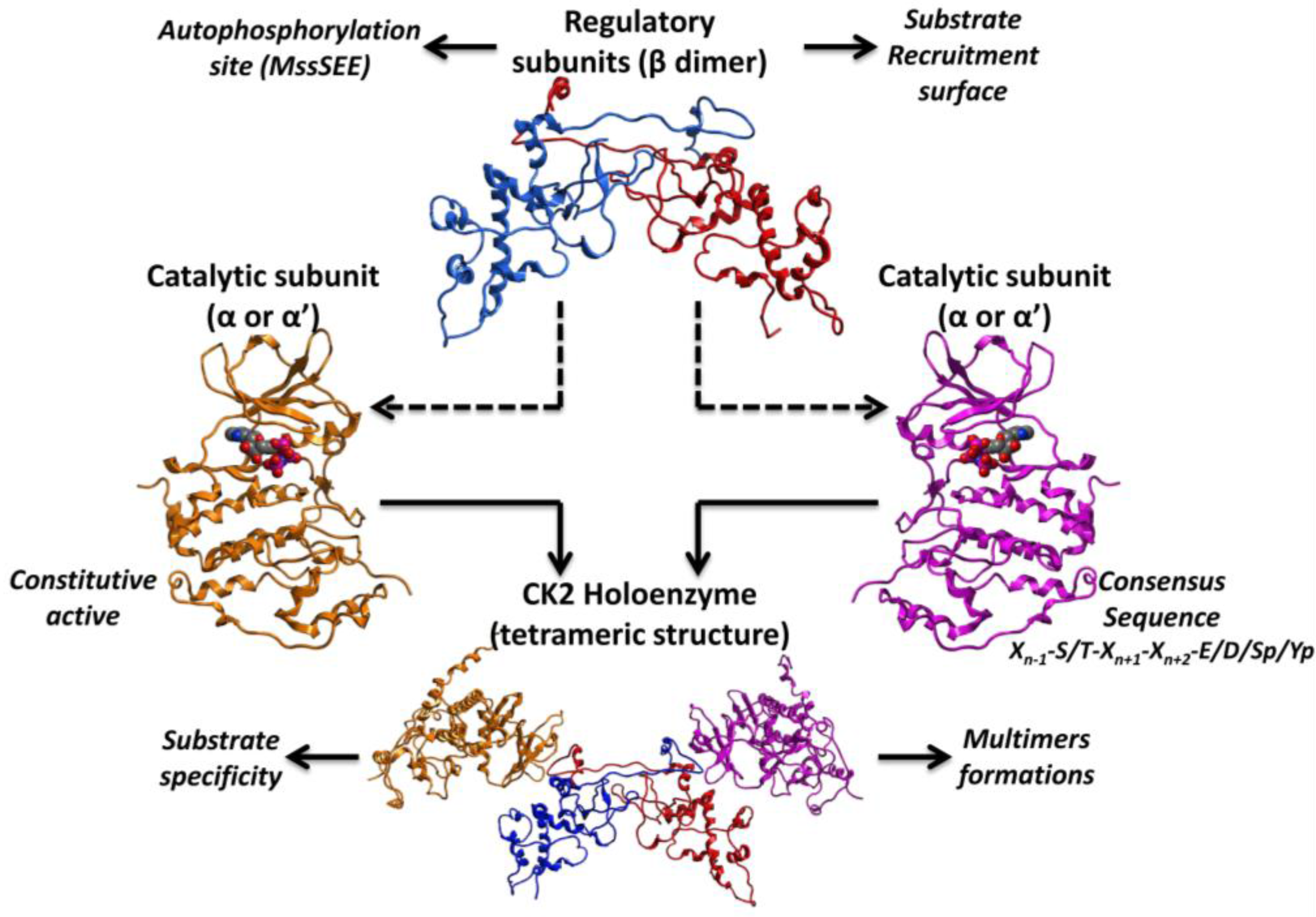
2. Structure and Biological Roles of CK2
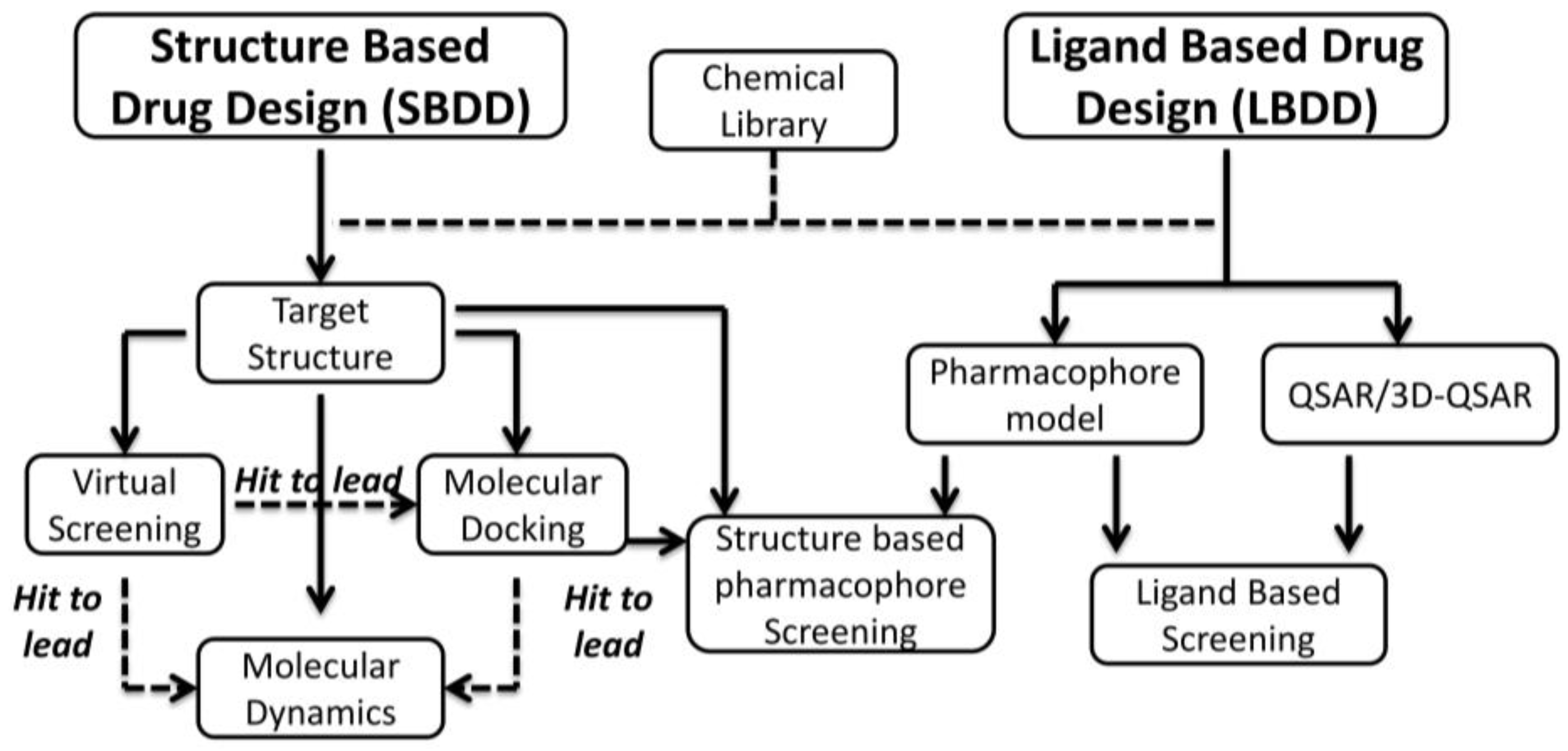
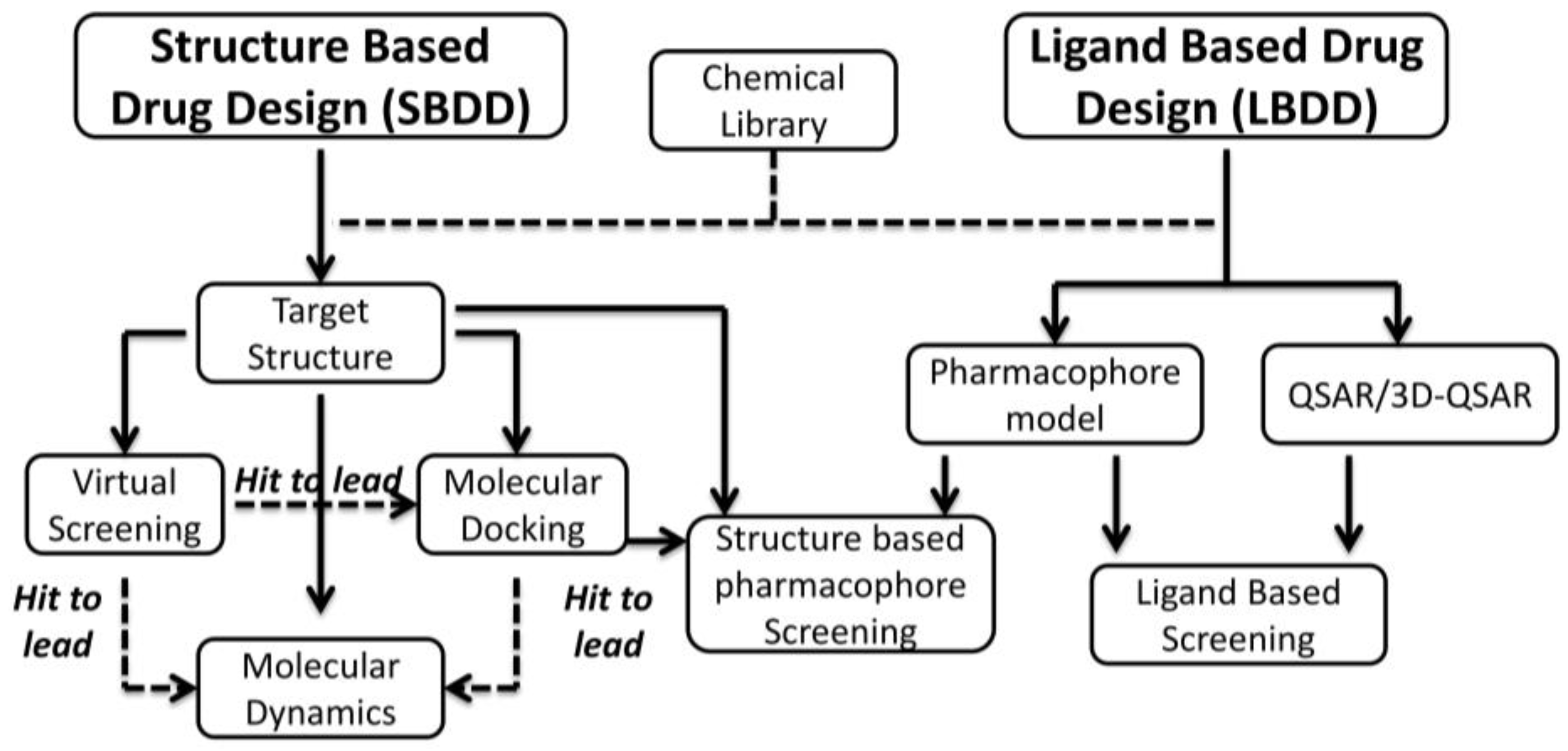
3. Rational Drug Design of CK2 Inhibitors: Structure Based Drug Design
3.1. Protein and Ligand Preparation
3.2. Virtual Screening Approach
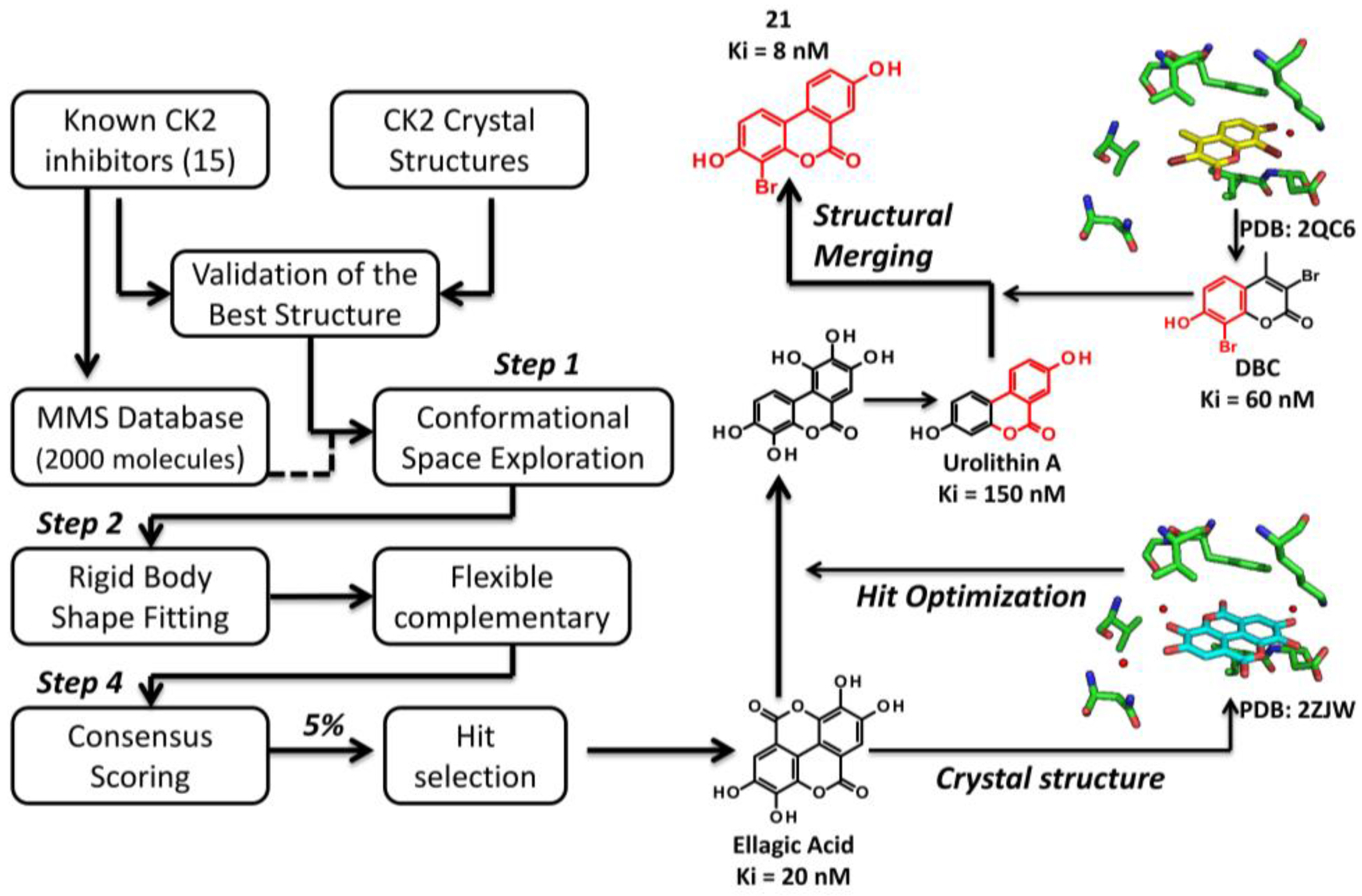
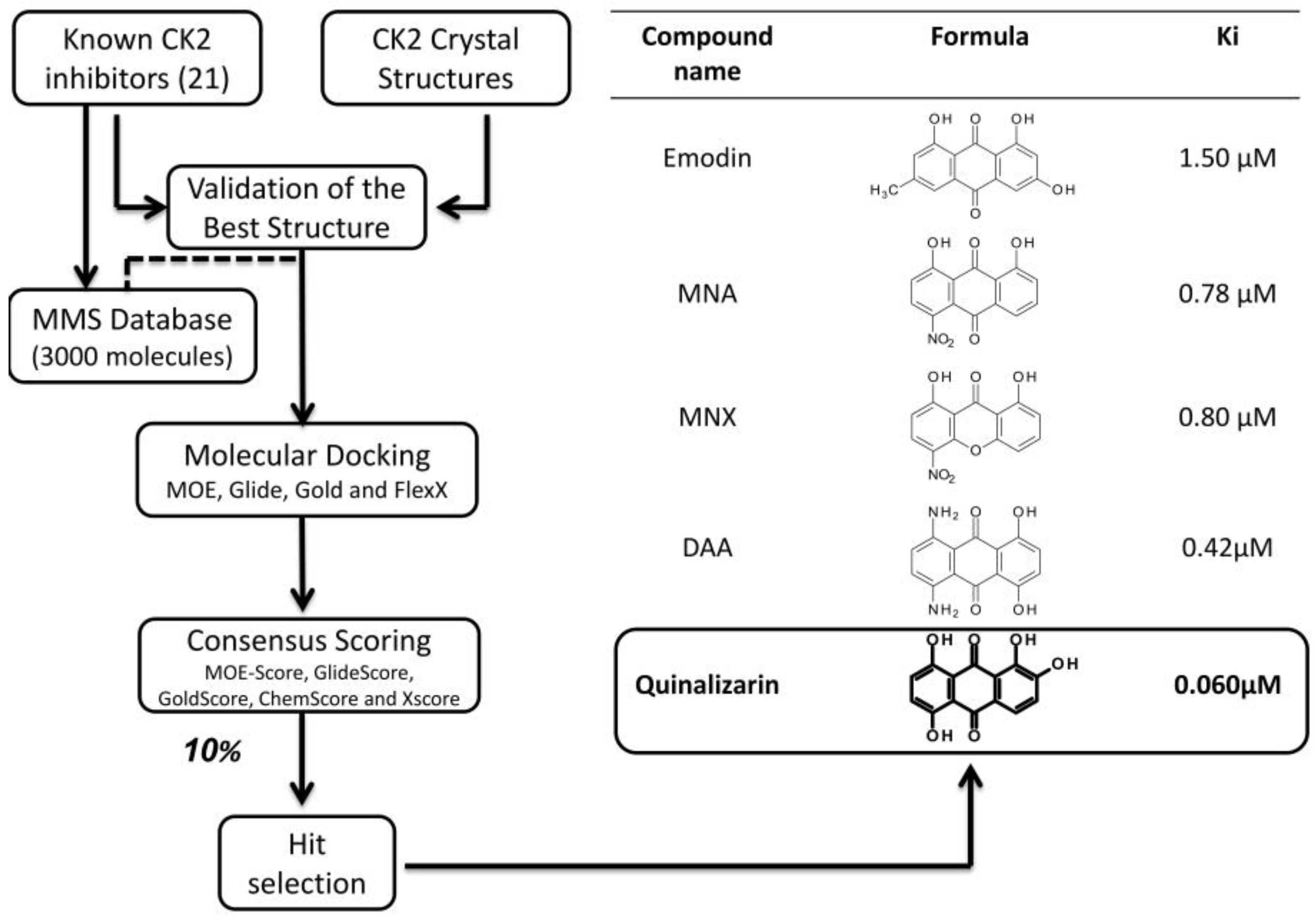
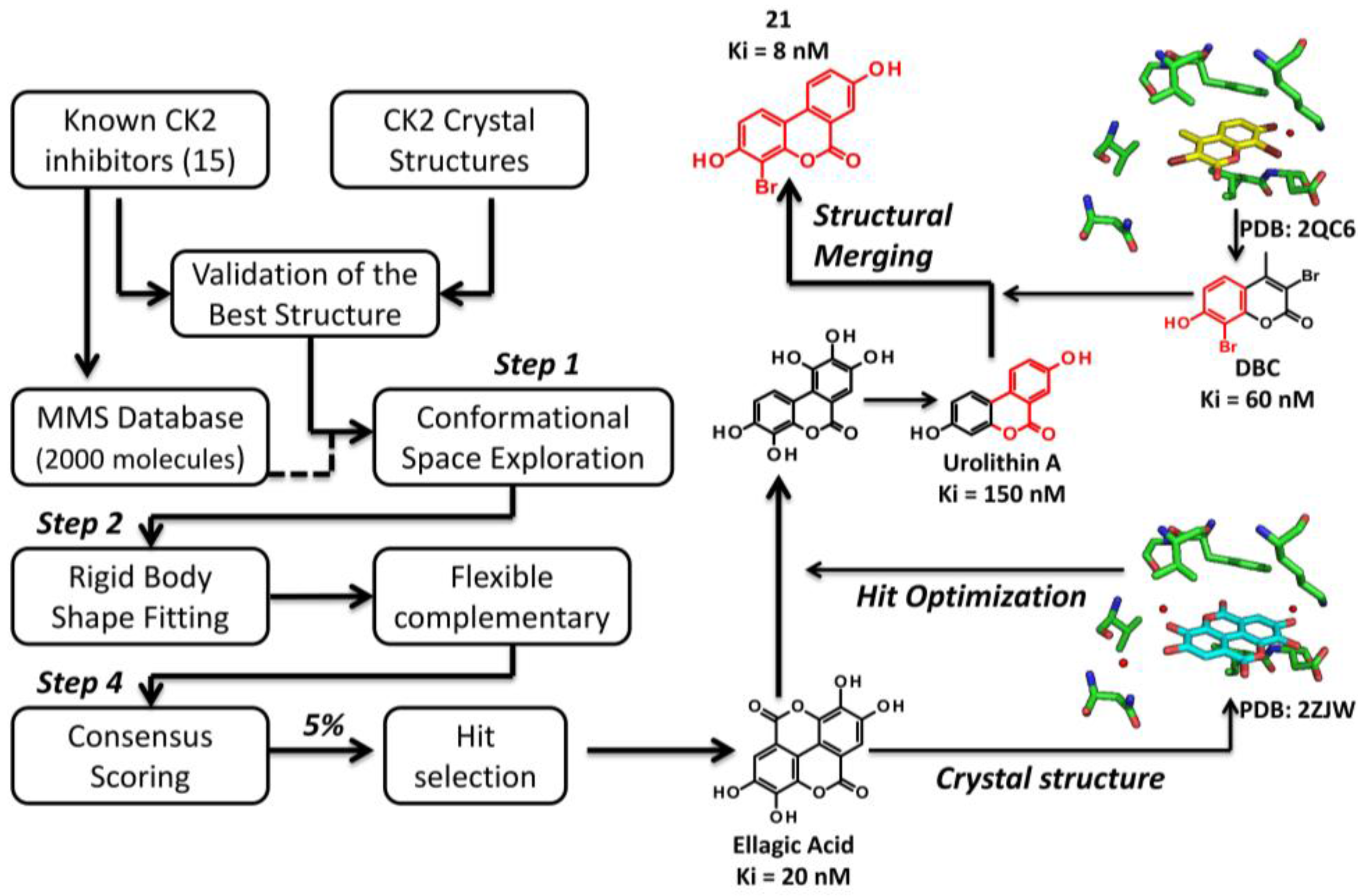
3.2.1. Virtual Screening Example 1: The Discovery of Ellagic Acid
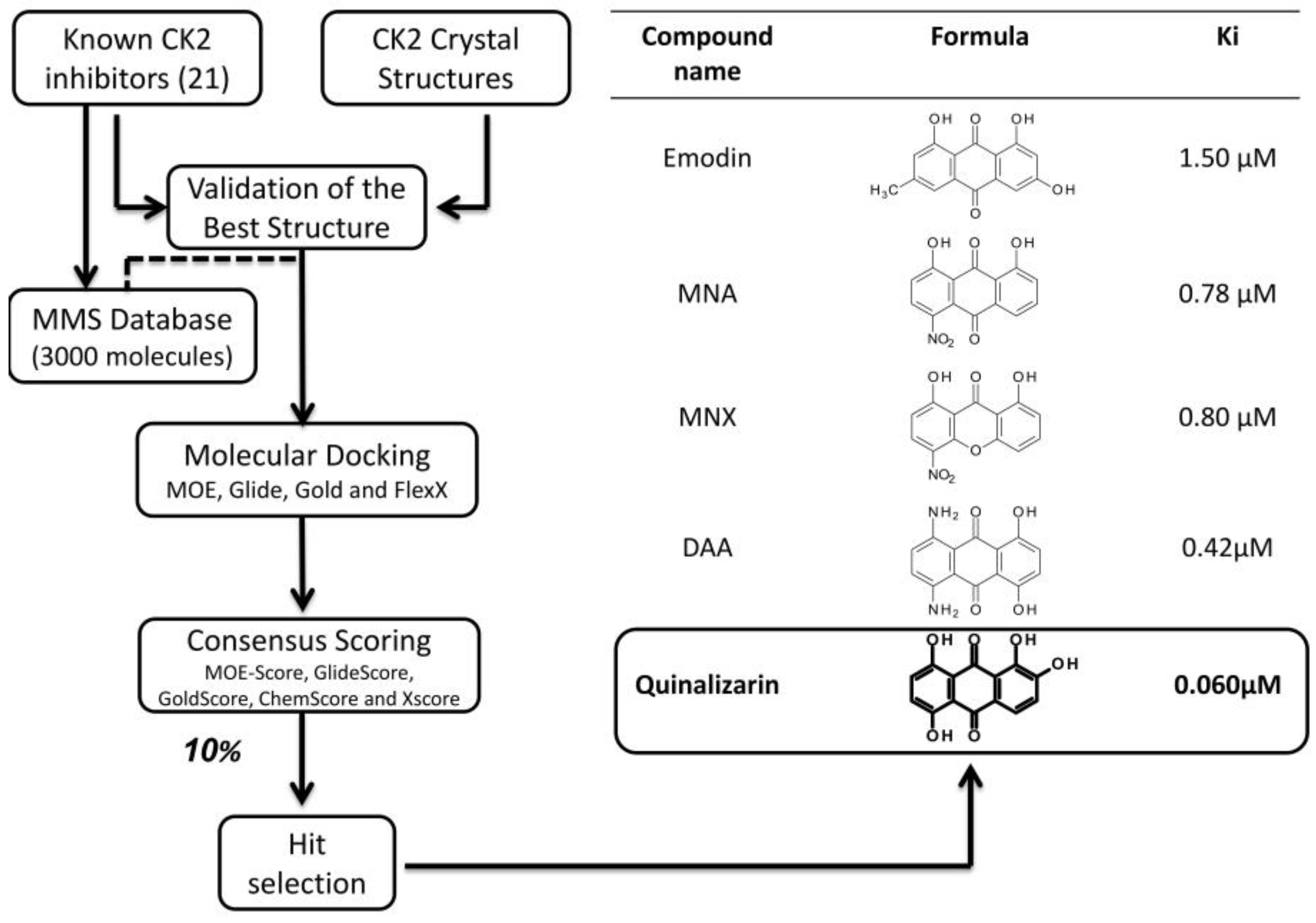
3.2.2. Virtual Screening Example 2: The Discovery of Quinalizarin
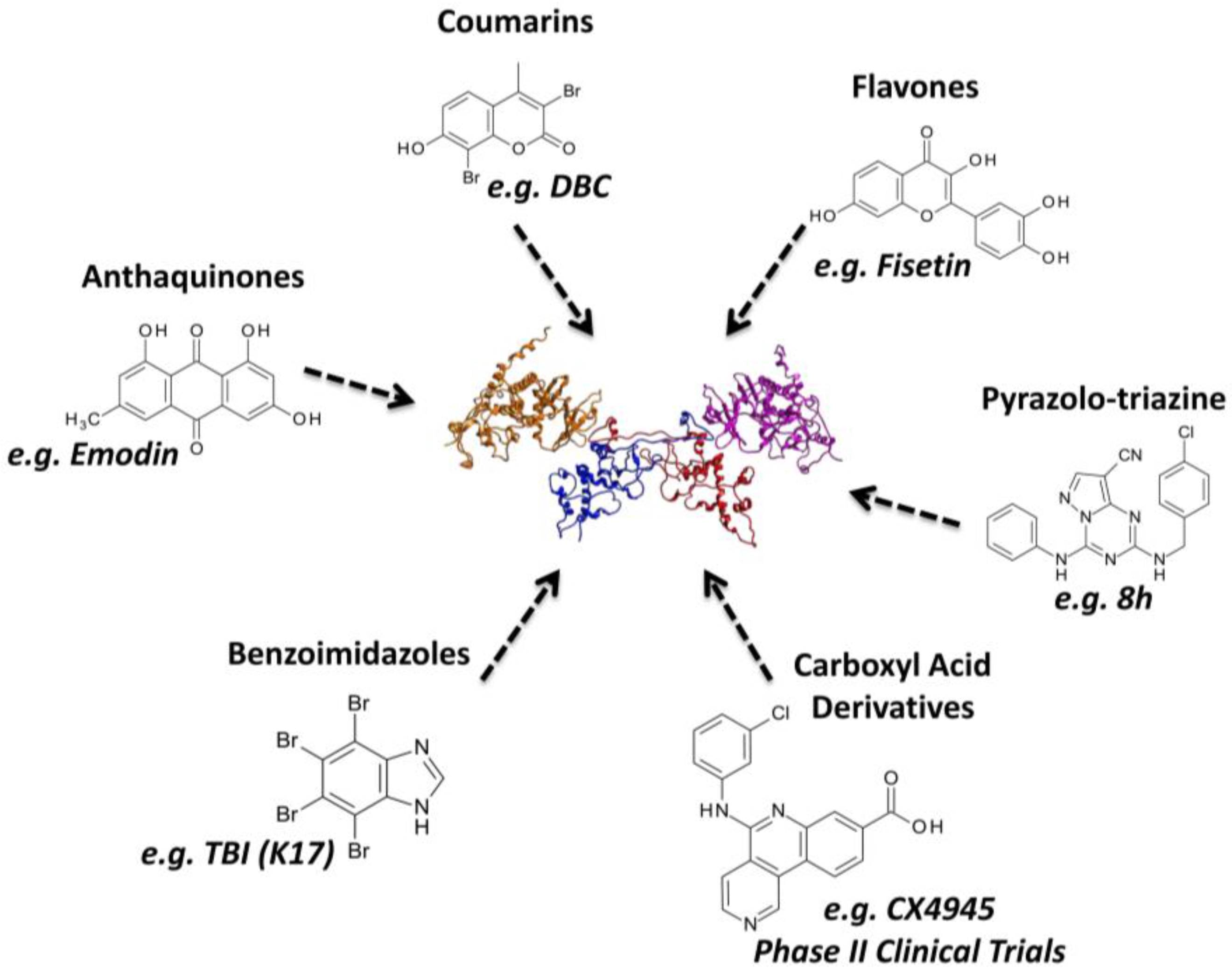
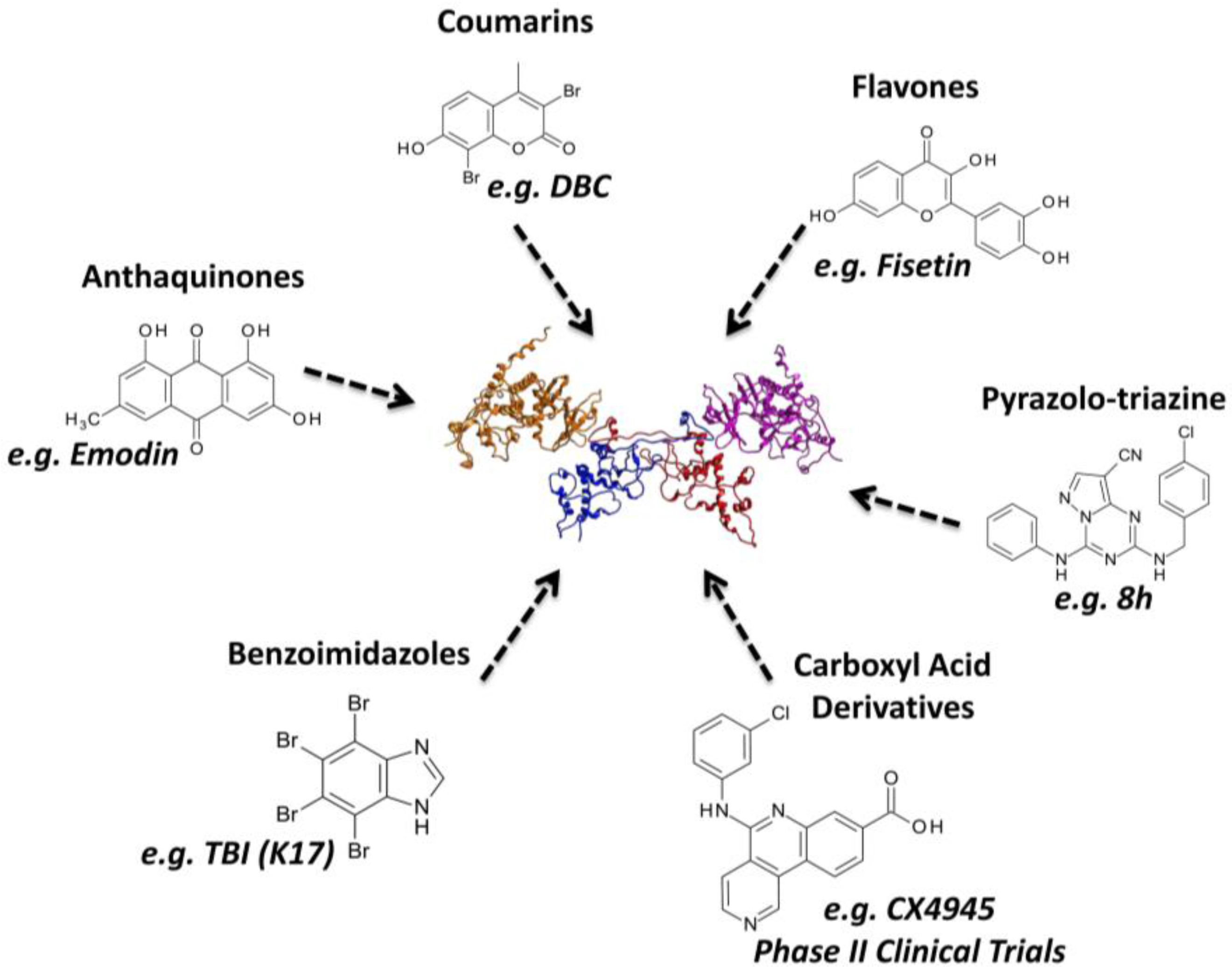
3.2.3. Virtual Screening: Other Examples
3.3. Molecular Docking, Molecular Dynamics and Hit Optimization (Hit to Lead)
3.3.1. Hit to Lead: Ellagic Acid
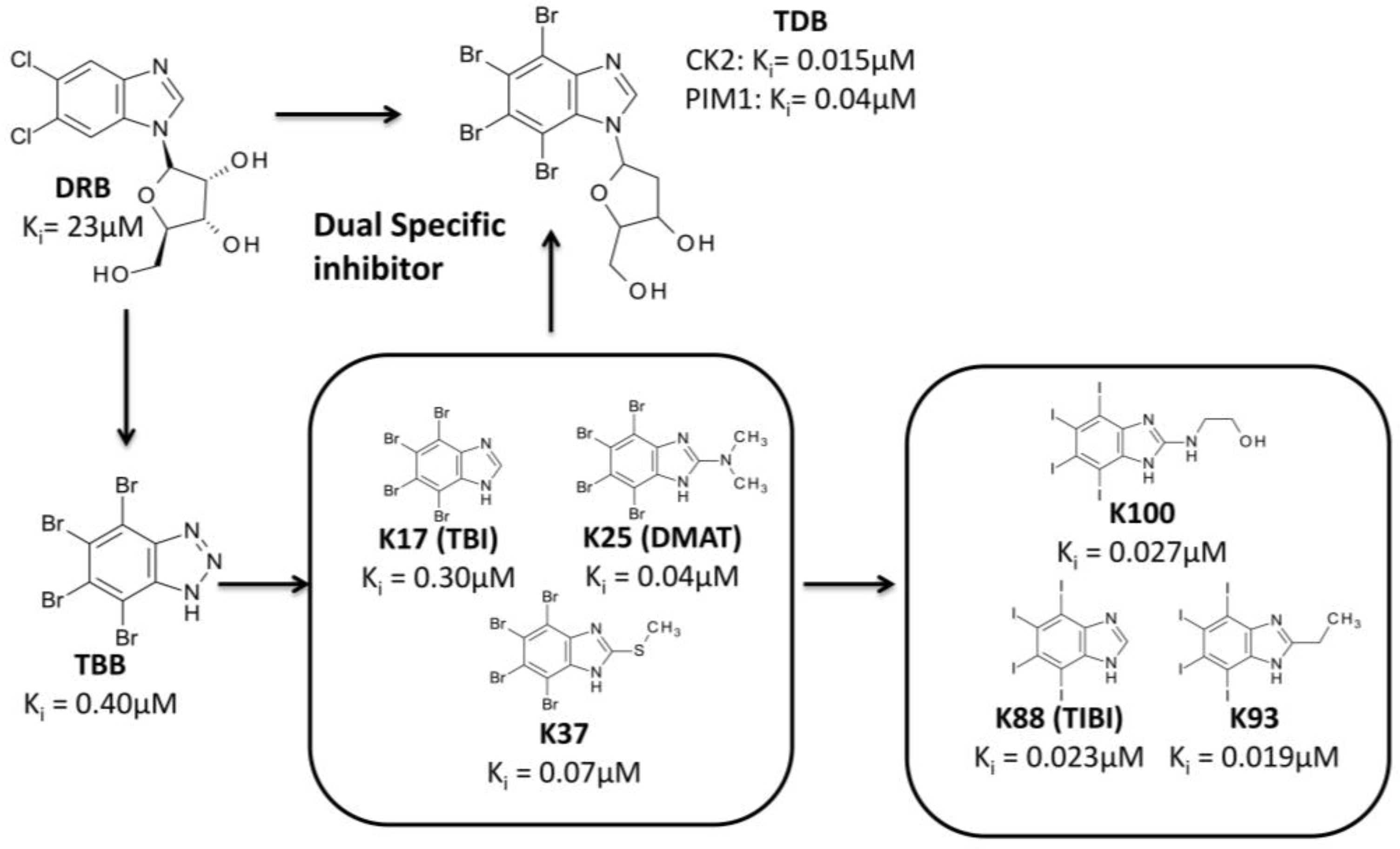
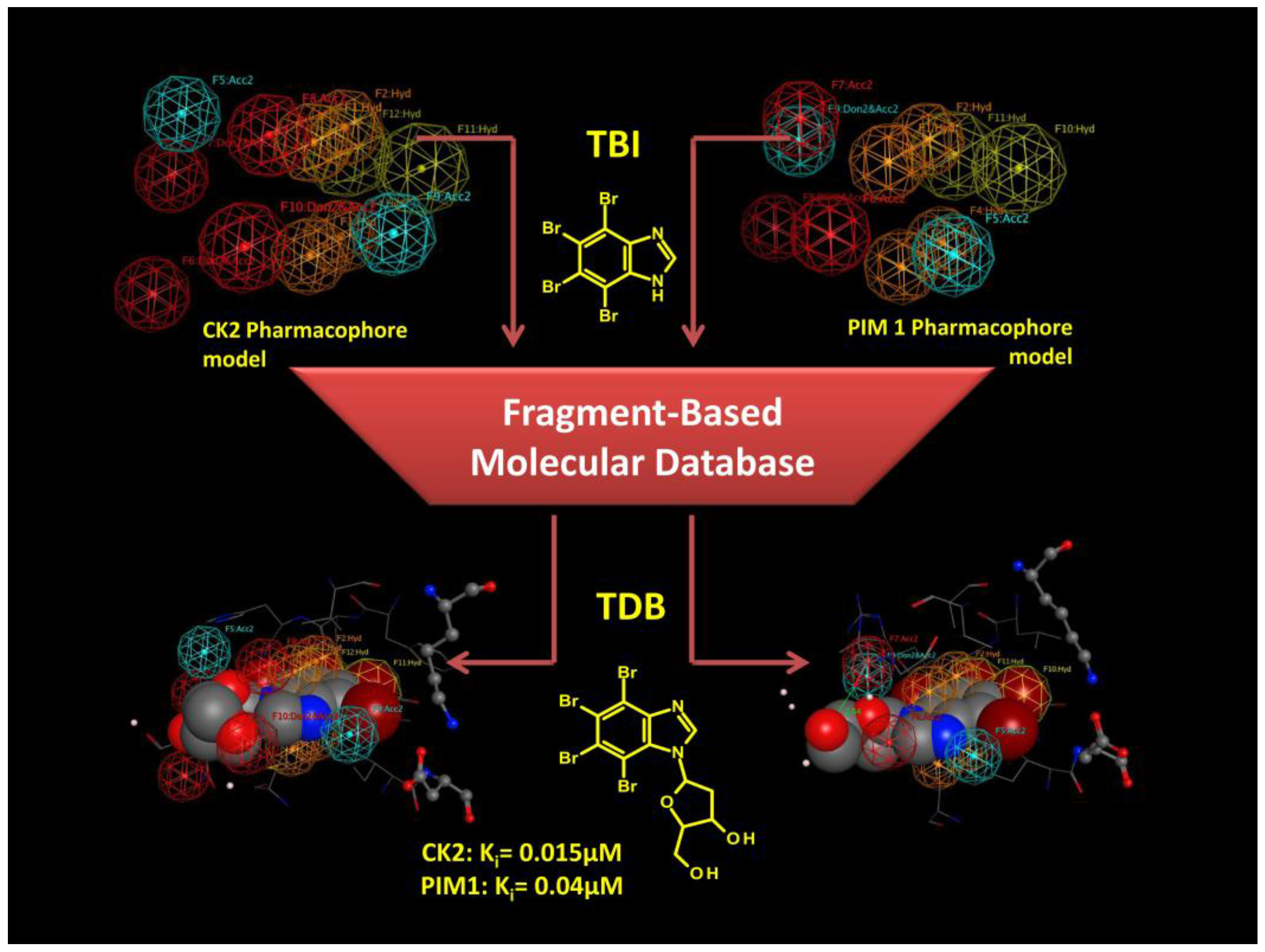
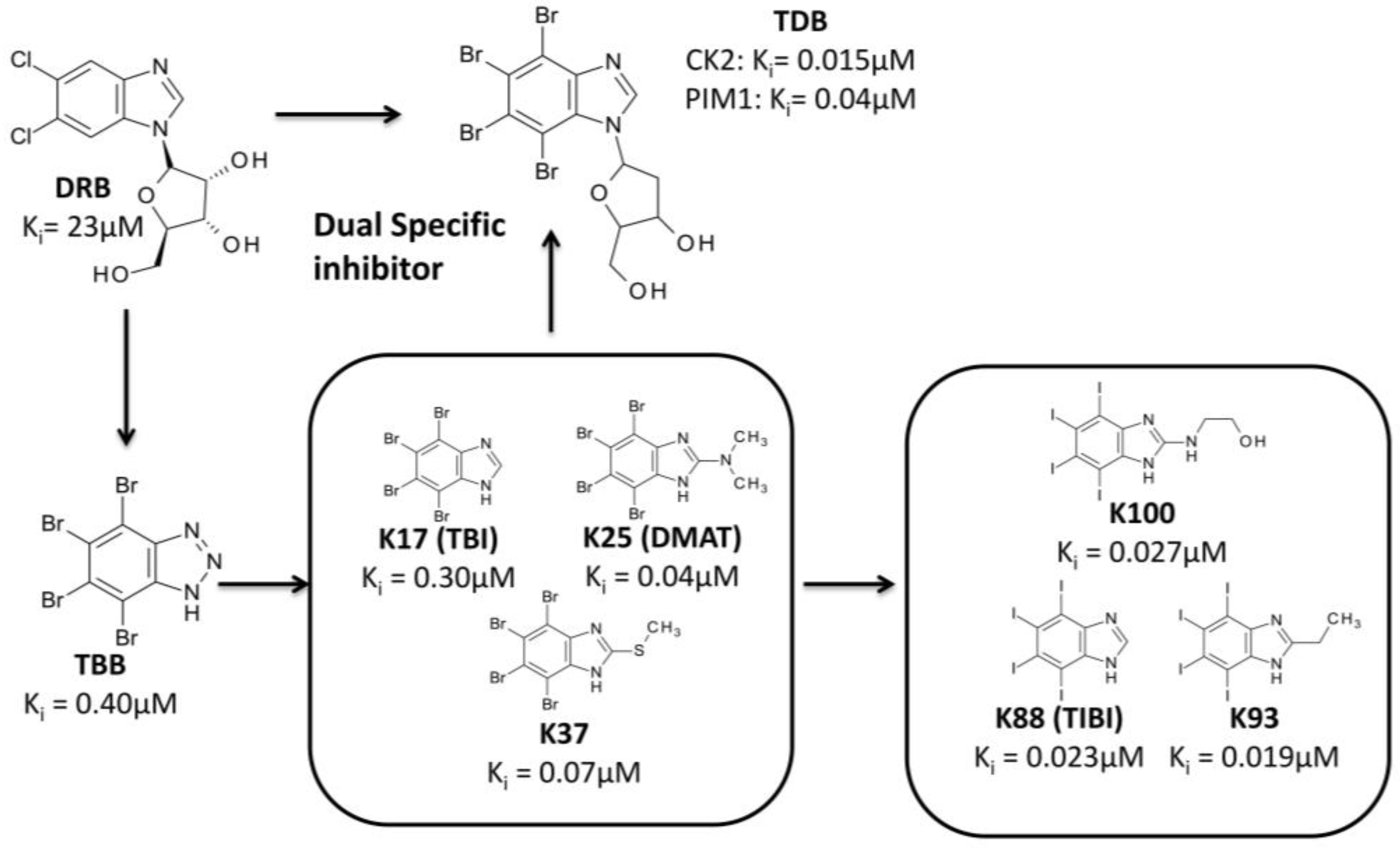
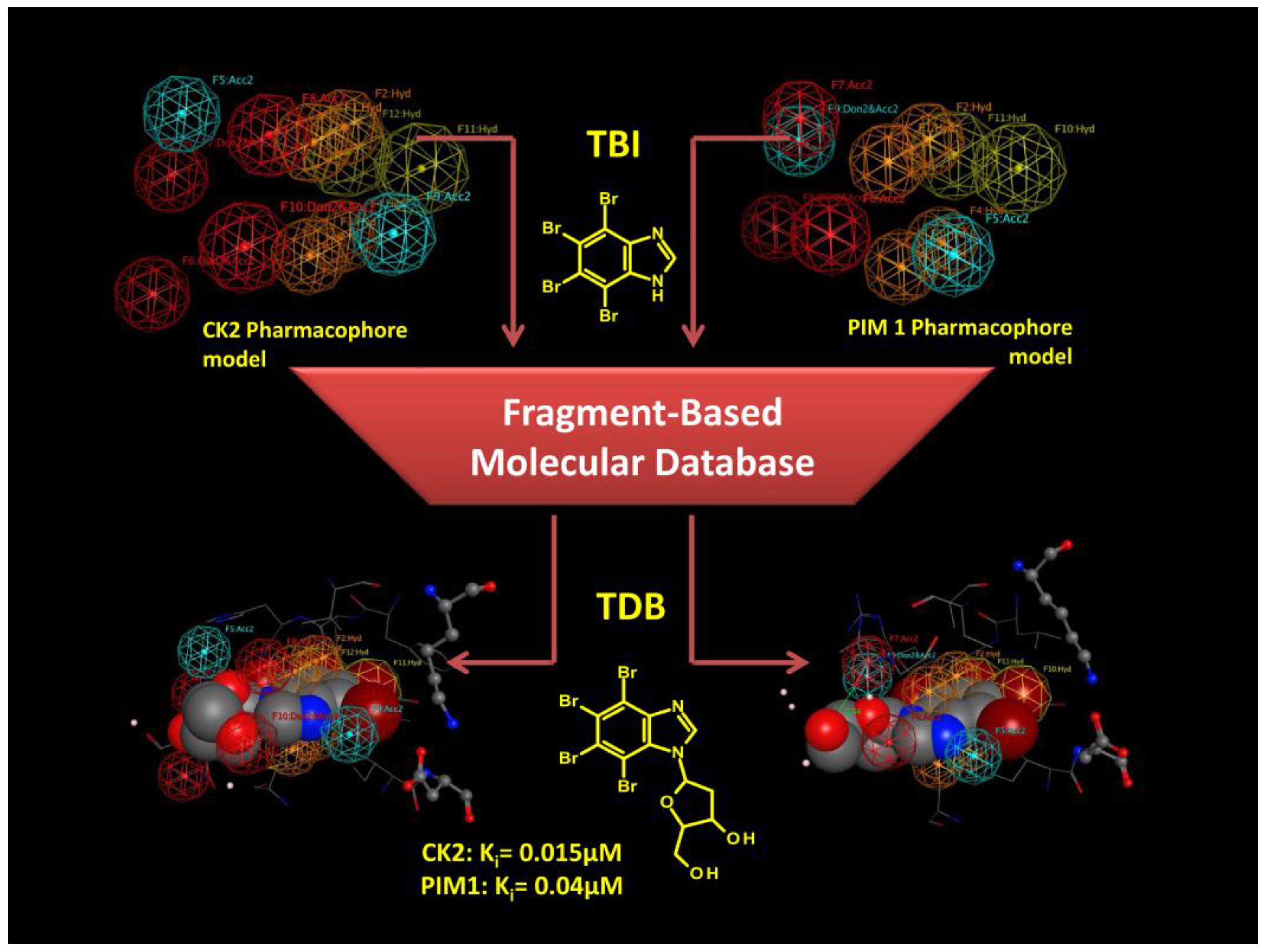
3.3.2. Hit to Lead: Benzimidazole Scaffold
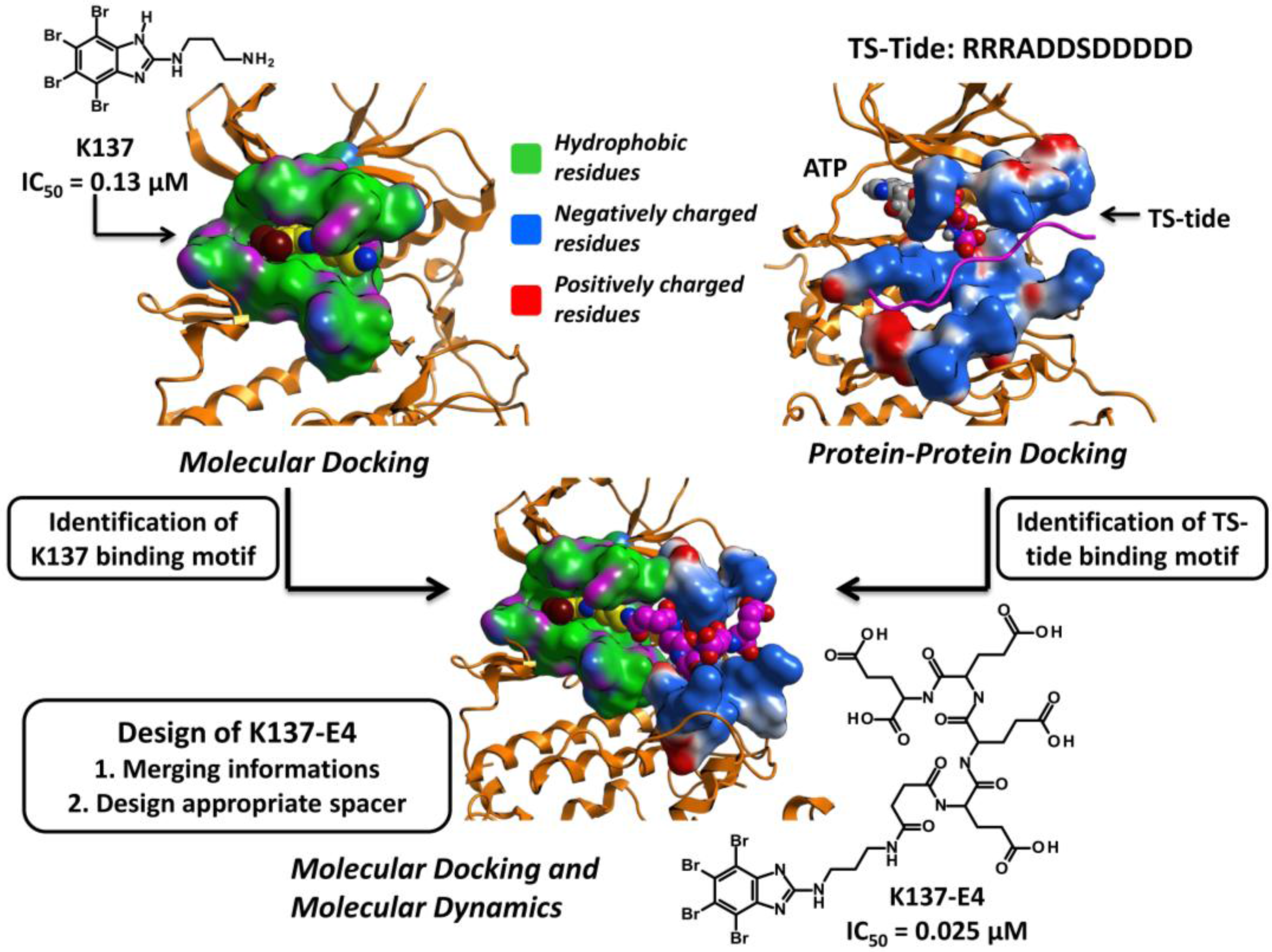
3.3.3. Hit to Lead: Other Examples
4. Rational Drug Design of CK2 Inhibitors: Ligand Based Drug Design
4.1. The Pharmacophore Approach
4.1.1. The Pharmacophore Approach: Applications
4.2. Quantitative Structure–Activity Relationship (QSAR)
4.2.1. The QSAR Approach: Applications
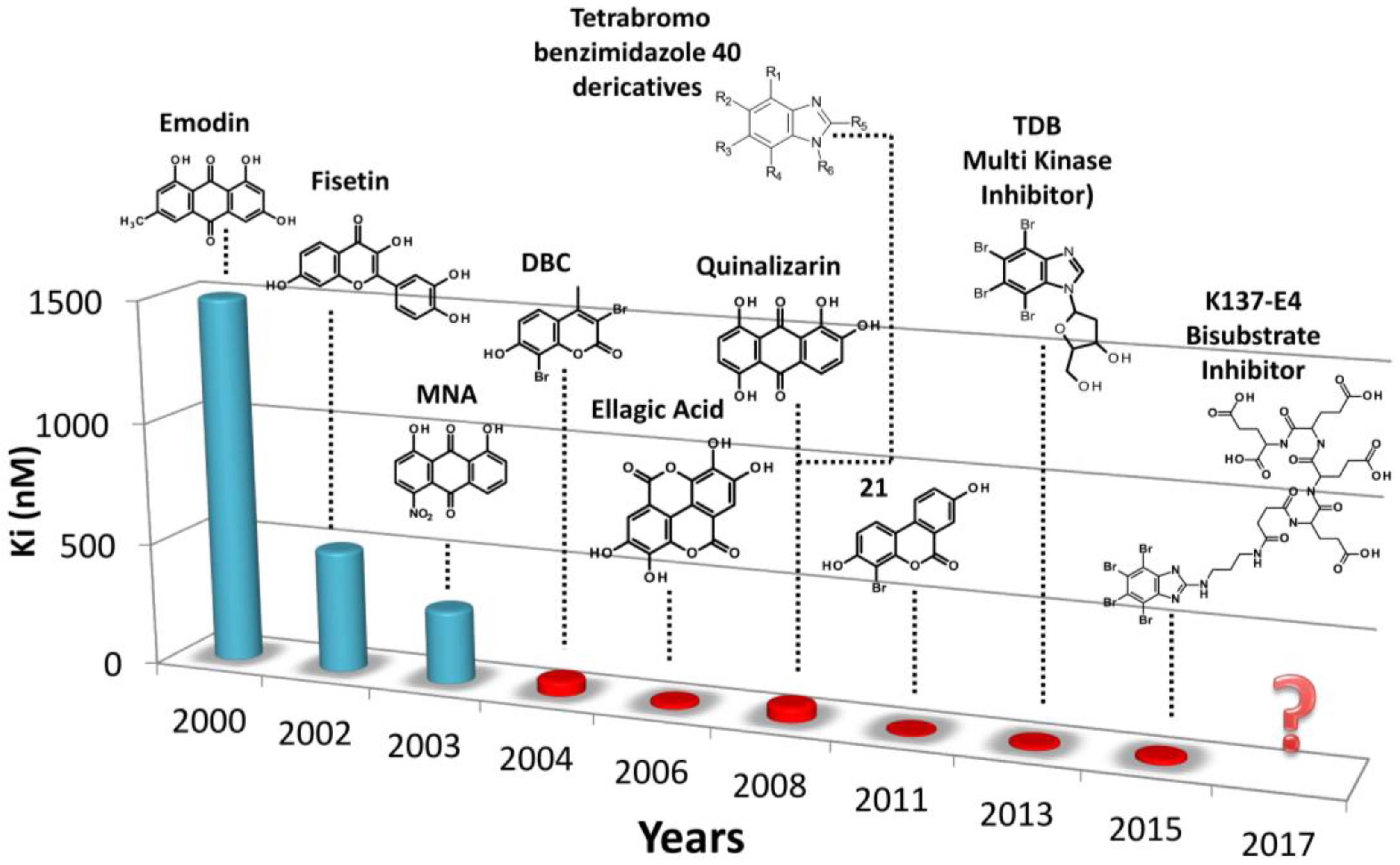
5. Discussion
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Burnett, G.; Kennedy, E.P. The enzymatic phosphorylation of proteins. J. Biol. Chem. 1954, 211, 969–980. [Google Scholar]
- Meggio, F.; Pinna, L.A. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 2003, 17, 349–368. [Google Scholar] [CrossRef]
- Salvi, M.; Sarno, S.; Cesaro, L.; Nakamura, H.; Pinna, L.A. Extraordinary pleiotropy of protein kinase CK2 revealed by weblogo phosphoproteome analysis. Biochim. Biophys. Acta 2009, 1793, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Sarno, S.; Ghisellini, P.; Pinna, L.A. Unique activation mechanism of protein kinase CK2. The n-terminal segment is essential for constitutive activity of the catalytic subunit but not of the holoenzyme. J. Biol. Chem. 2002, 277, 22509–22514. [Google Scholar] [CrossRef] [PubMed]
- Cristiani, A.; Costa, G.; Cozza, G.; Meggio, F.; Scapozza, L.; Moro, S. The role of the n-terminal domain in the regulation of the “constitutively active” conformation of protein kinase CK2alpha: Insight from a molecular dynamics investigation. ChemMedChem 2011, 6, 1207–1216. [Google Scholar] [CrossRef]
- Ortega, C.E.; Seidner, Y.; Dominguez, I. Mining CK2 in cancer. PLoS ONE 2014, 9, e115609. [Google Scholar] [CrossRef] [PubMed]
- Murtaza, I.; Wang, H.X.; Feng, X.; Alenina, N.; Bader, M.; Prabhakar, B.S.; Li, P.F. Down-regulation of catalase and oxidative modification of protein kinase CK2 lead to the failure of apoptosis repressor with caspase recruitment domain to inhibit cardiomyocyte hypertrophy. J. Biol. Chem. 2008, 283, 5996–6004. [Google Scholar] [CrossRef] [PubMed]
- Axtell, R.C.; Xu, L.; Barnum, S.R.; Raman, C. Cd5-CK2 binding/activation-deficient mice are resistant to experimental autoimmune encephalomyelitis: Protection is associated with diminished populations of il-17-expressing t cells in the central nervous system. J. Immunol. 2006, 177, 8542–8549. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, K.I.; Puustinen, P.; Gabrenaite, R.; Vihinen, H.; Ronnstrand, L.; Valmu, L.; Kalkkinen, N.; Makinen, K. Phosphorylation of the potyvirus capsid protein by protein kinase CK2 and its relevance for virus infection. Plant Cell 2003, 15, 2124–2139. [Google Scholar] [CrossRef] [PubMed]
- Foka, P.; Dimitriadis, A.; Kyratzopoulou, E.; Giannimaras, D.A.; Sarno, S.; Simos, G.; Georgopoulou, U.; Mamalaki, A. A complex signaling network involving protein kinase CK2 is required for hepatitis c virus core protein-mediated modulation of the iron-regulatory hepcidin gene expression. Cell. Mol. Life Sci. 2014, 71, 4243–4258. [Google Scholar] [CrossRef] [PubMed]
- Meggio, F.; Marin, O.; Boschetti, M.; Sarno, S.; Pinna, L.A. Hiv-1 rev transactivator: A beta-subunit directed substrate and effector of protein kinase CK2. Mol. Cell. Biochem. 2001, 227, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Pagano, M.A.; Arrigoni, G.; Marin, O.; Sarno, S.; Meggio, F.; Treharne, K.J.; Mehta, A.; Pinna, L.A. Modulation of protein kinase CK2 activity by fragments of CFTR encompassing f508 may reflect functional links with cystic fibrosis pathogenesis. Biochemistry 2008, 47, 7925–7936. [Google Scholar] [CrossRef] [PubMed]
- Pagano, M.A.; Marin, O.; Cozza, G.; Sarno, S.; Meggio, F.; Treharne, K.J.; Mehta, A.; Pinna, L.A. Cystic fibrosis transmembrane regulator fragments with the phe508 deletion exert a dual allosteric control over the master kinase CK2. Biochem. J. 2010, 426, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Cesaro, L.; Marin, O.; Venerando, A.; Donella-Deana, A.; Pinna, L.A. Phosphorylation of cystic fibrosis transmembrane conductance regulator (CFTR) serine-511 by the combined action of tyrosine kinases and CK2: The implication of tyrosine-512 and phenylalanine-508. Amino Acids 2013, 45, 1423–1429. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui-Jain, A.; Drygin, D.; Streiner, N.; Chua, P.; Pierre, F.; O’Brien, S.E.; Bliesath, J.; Omori, M.; Huser, N.; Ho, C.; et al. Cx-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010, 70, 10288–10298. [Google Scholar] [CrossRef] [PubMed]
- Pierre, F.; Chua, P.C.; O’Brien, S.E.; Siddiqui-Jain, A.; Bourbon, P.; Haddach, M.; Michaux, J.; Nagasawa, J.; Schwaebe, M.K.; Stefan, E.; et al. Pre-clinical characterization of cx-4945, a potent and selective small molecule inhibitor of CK2 for the treatment of cancer. Mol. Cell. Biochem. 2011, 356, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, A.D.; Sheth, P.R.; Basso, A.D.; Paliwal, S.; Gray, K.; Fischmann, T.O.; Le, H.V. Structural basis of cx-4945 binding to human protein kinase CK2. FEBS Lett. 2011, 585, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Wilson, L.K.; Dhillon, N.; Thorner, J.; Martin, G.S. Casein kinase ii catalyzes tyrosine phosphorylation of the yeast nucleolar immunophilin fpr3. J. Biol. Chem. 1997, 272, 12961–12967. [Google Scholar] [CrossRef] [PubMed]
- Marin, O.; Meggio, F.; Sarno, S.; Cesaro, L.; Pagano, M.A.; Pinna, L.A. Tyrosine versus serine/threonine phosphorylation by protein kinase casein kinase-2. A study with peptide substrates derived from immunophilin FPR3. J. Biol. Chem. 1999, 274, 29260–29265. [Google Scholar] [CrossRef] [PubMed]
- Sarno, S.; Vaglio, P.; Meggio, F.; Issinger, O.G.; Pinna, L.A. Protein kinase CK2 mutants defective in substrate recognition. Purification and kinetic analysis. J. Biol. Chem. 1996, 271, 10595–10601. [Google Scholar] [PubMed]
- Marin, O.; Sarno, S.; Boschetti, M.; Pagano, M.A.; Meggio, F.; Ciminale, V.; D’Agostino, D.M.; Pinna, L.A. Unique features of hiv-1 rev protein phosphorylation by protein kinase CK2 ('casein kinase-2'). FEBS Lett. 2000, 481, 63–67. [Google Scholar] [CrossRef]
- Poletto, G.; Vilardell, J.; Marin, O.; Pagano, M.A.; Cozza, G.; Sarno, S.; Falques, A.; Itarte, E.; Pinna, L.A.; Meggio, F. The regulatory beta subunit of protein kinase CK2 contributes to the recognition of the substrate consensus sequence. A study with an EIF2 beta-derived peptide. Biochemistry 2008, 47, 8317–8325. [Google Scholar] [CrossRef] [PubMed]
- Meggio, F.; Boldyreff, B.; Marin, O.; Marchiori, F.; Perich, J.W.; Issinger, O.G.; Pinna, L.A. The effect of polylysine on casein-kinase-2 activity is influenced by both the structure of the protein/peptide substrates and the subunit composition of the enzyme. Eur. J. Biochem. 1992, 205, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Boldyreff, B.; James, P.; Staudenmann, W.; Issinger, O.G. Ser2 is the autophosphorylation site in the beta subunit from bicistronically expressed human casein kinase-2 and from native rat liver casein kinase-2 beta. Eur. J. Biochem. 1993, 218, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, D.W.; Lozeman, F.J.; Cicirelli, M.F.; Harrylock, M.; Ericsson, L.H.; Piening, C.J.; Krebs, E.G. Phosphorylation of the beta subunit of casein kinase ii in human a431 cells. Identification of the autophosphorylation site and a site phosphorylated by p34cdc2. J. Biol. Chem. 1991, 266, 20380–20389. [Google Scholar] [PubMed]
- Lolli, G.; Pinna, L.A.; Battistutta, R. Structural determinants of protein kinase CK2 regulation by autoinhibitory polymerization. ACS Chem. Biol. 2012, 7, 1158–1163. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Pinna, L.A.; Moro, S. Kinase CK2 inhibition: An update. Curr. Med. Chem. 2013, 20, 671–693. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Pinna, L.A. Casein kinases as potential therapeutic targets. Expert Opin. Ther. Targets 2015, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Hubert, A.; Paris, S.; Piret, J.P.; Ninane, N.; Raes, M.; Michiels, C. Casein Kinase 2 inhibition decreases hypoxia-inducible factor-1 activity under hypoxia through elevated p53 protein level. J. Cell Sci. 2006, 119, 3351–3362. [Google Scholar] [CrossRef] [PubMed]
- Hupp, T.R.; Meek, D.W.; Midgley, C.A.; Lane, D.P. Regulation of the specific DNA binding function of p53. Cell 1992, 71, 875–886. [Google Scholar] [CrossRef]
- Mottet, D.; Ruys, S.P.; Demazy, C.; Raes, M.; Michiels, C. Role for casein kinase 2 in the regulation of HIF-1 activity. Int. J. Cancer 2005, 117, 764–774. [Google Scholar] [PubMed]
- Pluemsampant, S.; Safronova, O.S.; Nakahama, K.; Morita, I. Protein kinase CK2 is a key activator of histone deacetylase in hypoxia-associated tumors. Int. J. Cancer 2008, 122, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Charo, I.F.; Taubman, M.B. Chemokines in the pathogenesis of vascular disease. Circ. Res. 2004, 95, 858–866. [Google Scholar] [CrossRef] [PubMed]
- Harvey, E.J.; Li, N.; Ramji, D.P. Critical role for casein kinase 2 and phosphoinositide-3-kinase in the interferon-gamma-induced expression of monocyte chemoattractant protein-1 and other key genes implicated in atherosclerosis. Arter. Thromb. Vasc. Biol. 2007, 27, 806–812. [Google Scholar] [CrossRef]
- Okochi, M.; Walter, J.; Koyama, A.; Nakajo, S.; Baba, M.; Iwatsubo, T.; Meijer, L.; Kahle, P.J.; Haass, C. Constitutive phosphorylation of the parkinson's disease associated alpha-synuclein. J. Biol. Chem. 2000, 275, 390–397. [Google Scholar] [CrossRef]
- Lee, G.; Tanaka, M.; Park, K.; Lee, S.S.; Kim, Y.M.; Junn, E.; Lee, S.H.; Mouradian, M.M. Casein Kinase II-mediated phosphorylation regulates alpha-synuclein/synphilin-1 interaction and inclusion body formation. J. Biol. Chem. 2004, 279, 6834–6839. [Google Scholar] [CrossRef] [PubMed]
- Ishii, A.; Nonaka, T.; Taniguchi, S.; Saito, T.; Arai, T.; Mann, D.; Iwatsubo, T.; Hisanaga, S.; Goedert, M.; Hasegawa, M. Casein kinase 2 is the major enzyme in brain that phosphorylates ser129 of human alpha-synuclein: Implication for alpha-synucleinopathies. FEBS Lett. 2007, 581, 4711–4717. [Google Scholar] [CrossRef] [PubMed]
- Iimoto, D.S.; Masliah, E.; DeTeresa, R.; Terry, R.D.; Saitoh, T. Aberrant casein kinase II in alzheimer's disease. Brain Res. 1990, 507, 273–280. [Google Scholar] [CrossRef]
- Aksenova, M.V.; Burbaeva, G.S.; Kandror, K.V.; Kapkov, D.V.; Stepanov, A.S. The decreased level of casein kinase 2 in brain cortex of schizophrenic and alzheimer's disease patients. FEBS Lett. 1991, 279, 55–57. [Google Scholar] [CrossRef]
- Masliah, E.; Iimoto, D.S.; Mallory, M.; Albright, T.; Hansen, L.; Saitoh, T. Casein kinase ii alteration precedes tau accumulation in tangle formation. Am. J. Pathol. 1992, 140, 263–268. [Google Scholar] [PubMed]
- Greenwood, J.A.; Scott, C.W.; Spreen, R.C.; Caputo, C.B.; Johnson, G.V. Casein kinase ii preferentially phosphorylates human tau isoforms containing an amino-terminal insert. Identification of threonine 39 as the primary phosphate acceptor. J. Biol. Chem. 1994, 269, 4373–4380. [Google Scholar] [PubMed]
- Yamada, M.; Katsuma, S.; Adachi, T.; Hirasawa, A.; Shiojima, S.; Kadowaki, T.; Okuno, Y.; Koshimizu, T.A.; Fujii, S.; Sekiya, Y.; et al. Inhibition of protein kinase CK2 prevents the progression of glomerulonephritis. Proc. Natl. Acad. Sci. USA 2005, 102, 7736–7741. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, T.; Kosuge, S.; Karino, A.; Okano, T.; Ito, J.; Munakata, H.; Ohtsuki, K. Biochemical characterization of 60s acidic ribosomal p proteins from porcine liver and the inhibition of their immunocomplex formation with sera from systemic lupus erythematosus (sle) patients by glycyrrhizin in vitro. Biol. Pharm. Bull. 2000, 23, 27–32. [Google Scholar]
- Caponi, L.; Anzilotti, C.; Longombardo, G.; Migliorini, P. Antibodies directed against ribosomal p proteins cross-react with phospholipids. Clin. Exp. Immunol. 2007, 150, 140–143. [Google Scholar] [CrossRef] [PubMed]
- Hauck, L.; Harms, C.; An, J.; Rohne, J.; Gertz, K.; Dietz, R.; Endres, M.; von Harsdorf, R. Protein kinase CK2 links extracellular growth factor signaling with the control of p27(kip1) stability in the heart. Nat. Med. 2008, 14, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Tapia, J.C.; Bolanos-Garcia, V.M.; Sayed, M.; Allende, C.C.; Allende, J.E. Cell cycle regulatory protein p27kip1 is a substrate and interacts with the protein kinase CK2. J. Cell. Biochem. 2004, 91, 865–879. [Google Scholar] [CrossRef] [PubMed]
- De Stefano, D.; Villella, V.R.; Esposito, S.; Tosco, A.; Sepe, A.; De Gregorio, F.; Salvadori, L.; Grassia, R.; Leone, C.A.; De Rosa, G.; et al. Restoration of CFTR function in patients with cystic fibrosis carrying the f508del-CFTR mutation. Autophagy 2014, 10, 2053–2074. [Google Scholar] [CrossRef] [PubMed]
- Ruzzene, M.; Pinna, L.A. Addiction to protein kinase CK2: A common denominator of diverse cancer cells? Biochim. Biophys. Acta Proteins Proteom. 2010, 1804, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Mazzorana, M.; Papinutto, E.; Bain, J.; Elliott, M.; di Maira, G.; Gianoncelli, A.; Pagano, M.A.; Sarno, S.; Ruzzene, M.; et al. Quinalizarin as a potent, selective and cell-permeable inhibitor of protein kinase CK2. Biochem. J. 2009, 421, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Bonvini, P.; Zorzi, E.; Poletto, G.; Pagano, M.A.; Sarno, S.; Donella-Deana, A.; Zagotto, G.; Rosolen, A.; Pinna, L.A.; et al. Identification of ellagic acid as potent inhibitor of protein kinase CK2: A successful example of a virtual screening application. J. Med. Chem. 2006, 49, 2363–2366. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Brooks, C.L., 3rd; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. Charmm: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Merck molecular force field. 1. Basis, form, scope, parameterization, and performance of mmff94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.J.; Reboul, M.; Xiang, J.Y.; Wang, L.L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. Opls3: A force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Gianoncelli, A.; Bonvini, P.; Zorzi, E.; Pasquale, R.; Rosolen, A.; Pinna, L.A.; Meggio, F.; Zagotto, G.; Moro, S. Urolithin as a converging scaffold linking ellagic acid and coumarin analogues: Design of potent protein kinase CK2 inhibitors. ChemMedChem 2011, 6, 2273–2286. [Google Scholar] [CrossRef] [PubMed]
- Sarno, S.; Moro, S.; Meggio, F.; Zagotto, G.; Dal Ben, D.; Ghisellini, P.; Battistutta, R.; Zanotti, G.; Pinna, L.A. Toward the rational design of protein kinase casein kinase-2 inhibitors. Pharmacol. Ther. 2002, 93, 159–168. [Google Scholar] [CrossRef]
- Pagano, M.A.; Sarno, S.; Poletto, G.; Cozza, G.; Pinna, L.A.; Meggio, F. Autophosphorylation at the regulatory beta subunit reflects the supramolecular organization of protein kinase CK2. Mol. Cell. Biochem. 2005, 274, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, R.T. Structure-based drug design: Docking and scoring. Curr. Protein Pept. Sci. 2007, 8, 312–328. [Google Scholar] [CrossRef] [PubMed]
- Cavasotto, C.N.; Orry, A.J. Ligand docking and structure-based virtual screening in drug discovery. Curr. Top. Med. Chem. 2007, 7, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and autodocktools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal chemistry and the molecular operating environment (moe): Application of qsar and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef] [PubMed]
- Allen, W.J.; Balius, T.E.; Mukherjee, S.; Brozell, S.R.; Moustakas, D.T.; Lang, P.T.; Case, D.A.; Kuntz, I.D.; Rizzo, R.C. Dock 6: Impact of new features and current docking performance. J. Comput. Chem. 2015, 36, 1132–1156. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- McGann, M. Fred pose prediction and virtual screening accuracy. J. Chem. Inf. Model. 2011, 51, 578–596. [Google Scholar] [CrossRef] [PubMed]
- Zsoldos, Z.; Reid, D.; Simon, A.; Sadjad, S.B.; Johnson, A.P. Ehits: A new fast, exhaustive flexible ligand docking system. J. Mol. Graph. Model. 2007, 26, 198–212. [Google Scholar] [CrossRef] [PubMed]
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar] [CrossRef] [PubMed]
- Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, C.J.; McLauchlan, H.; Klevernic, I.; Arthur, J.S.; Alessi, D.R.; Cohen, P. The selectivity of protein kinase inhibitors: A further update. Biochem. J. 2007, 408, 297–315. [Google Scholar] [CrossRef] [PubMed]
- De Moliner, E.; Moro, S.; Sarno, S.; Zagotto, G.; Zanotti, G.; Pinna, L.A.; Battistutta, R. Inhibition of protein kinase CK2 by anthraquinone-related compounds. A structural insight. J. Biol. Chem. 2003, 278, 1831–1836. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Venerando, A.; Sarno, S.; Pinna, L.A. The selectivity of CK2 inhibitor quinalizarin: A reevaluation. Biomed. Res. Int. 2015, 2015, 734127. [Google Scholar] [CrossRef] [PubMed]
- Franchin, C.; Salvi, M.; Arrigoni, G.; Pinna, L.A. Proteomics perturbations promoted by the protein kinase CK2 inhibitor quinalizarin. Biochim. Biophys. Acta 2015. [Google Scholar] [CrossRef] [PubMed]
- Vangrevelinghe, E.; Zimmermann, K.; Schoepfer, J.; Portmann, R.; Fabbro, D.; Furet, P. Discovery of a potent and selective protein kinase CK2 inhibitor by high-throughput docking. J. Med. Chem. 2003, 46, 2656–2662. [Google Scholar] [CrossRef] [PubMed]
- Sarno, S.; de Moliner, E.; Ruzzene, M.; Pagano, M.A.; Battistutta, R.; Bain, J.; Fabbro, D.; Schoepfer, J.; Elliott, M.; Furet, P.; et al. Biochemical and three-dimensional-structural study of the specific inhibition of protein kinase CK2 by [5-oxo-5,6-dihydroindolo-(1,2-a)quinazolin-7-yl]acetic acid (iqa). Biochem. J. 2003, 374, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Xu, X.; Wu, X.; Zhang, X.; Liu, F.; Jia, J.; Guo, X.; Huang, J.; Jiang, Z.; Feng, T.; et al. Discovery and design of tricyclic scaffolds as protein kinase CK2 (CK2) inhibitors through a combination of shape-based virtual screening and structure-based molecular modification. J. Chem. Inf. Model. 2013, 53, 2093–2102. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Wu, X.; Xu, X.; Jiang, Z.; Liu, Z.; You, Q. Discovery of novel CK2 leads by cross-docking based virtual screening. Med. Chem. 2014, 10, 628–639. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The mm/pbsa and mm/gbsa methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, J.; Boukharta, L.; Aqvist, J. Combining docking, molecular dynamics and the linear interaction energy method to predict binding modes and affinities for non-nucleoside inhibitors to hiv-1 reverse transcriptase. J. Med. Chem. 2008, 51, 2648–2656. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, Y.; Nakaniwa, T.; Kinoshita, T.; Nakanishi, I.; Kitaura, K.; Hirasawa, A.; Tsujimoto, G.; Tada, T. Structural insight into human CK2alpha in complex with the potent inhibitor ellagic acid. Bioorg. Med. Chem. Lett. 2009, 19, 2920–2923. [Google Scholar] [CrossRef] [PubMed]
- Chilin, A.; Battistutta, R.; Bortolato, A.; Cozza, G.; Zanatta, S.; Poletto, G.; Mazzorana, M.; Zagotto, G.; Uriarte, E.; Guiotto, A.; et al. Coumarin as attractive casein kinase 2 (CK2) inhibitor scaffold: An integrate approach to elucidate the putative binding motif and explain structure-activity relationships. J. Med. Chem. 2008, 51, 752–759. [Google Scholar] [CrossRef] [PubMed]
- Zandomeni, R.; Zandomeni, M.C.; Shugar, D.; Weinmann, R. Casein kinase type ii is involved in the inhibition by 5,6-dichloro-1-beta-d-ribofuranosylbenzimidazole of specific rna polymerase ii transcription. J. Biol. Chem. 1986, 261, 3414–3419. [Google Scholar] [PubMed]
- Ruzzene, M.; Penzo, D.; Pinna, L.A. Protein kinase CK2 inhibitor 4,5,6,7-tetrabromobenzotriazole (TBB) induces apoptosis and caspase-dependent degradation of haematopoietic lineage cell-specific protein 1 (hs1) in jurkat cells. Biochem. J. 2002, 364, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Pagano, M.A.; Meggio, F.; Ruzzene, M.; Andrzejewska, M.; Kazimierczuk, Z.; Pinna, L.A. 2-dimethylamino-4,5,6,7-tetrabromo-1h-benzimidazole: A novel powerful and selective inhibitor of protein kinase CK2. Biochem. Biophys. Res. Commun. 2004, 321, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Gianoncelli, A.; Cozza, G.; Orzeszko, A.; Meggio, F.; Kazimierczuk, Z.; Pinna, L.A. Tetraiodobenzimidazoles are potent inhibitors of protein kinase CK2. Bioorg. Med. Chem. 2009, 17, 7281–7289. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Girardi, C.; Ranchio, A.; Lolli, G.; Sarno, S.; Orzeszko, A.; Kazimierczuk, Z.; Battistutta, R.; Ruzzene, M.; Pinna, L.A. Cell-permeable dual inhibitors of protein kinases CK2 and pim-1: Structural features and pharmacological potential. Cell. Mol. Life Sci. 2014, 71, 3173–3185. [Google Scholar] [CrossRef] [PubMed]
- Lorusso, P.M.; Eder, J.P. Therapeutic potential of novel selective-spectrum kinase inhibitors in oncology. Expert Opin. Investig. Drugs 2008, 17, 1013–1028. [Google Scholar] [CrossRef] [PubMed]
- Lackey, K.E. Lessons from the drug discovery of lapatinib, a dual erbb1/2 tyrosine kinase inhibitor. Curr. Top. Med. Chem. 2006, 6, 435–460. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.M.; Cockerill, S.; Guntrip, S.B.; Rusnak, D.; Smith, K.; Vanderwall, D.; Wood, E.; Lackey, K. Synthesis and sar of potent egfr/erbb2 dual inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Brault, L.; Gasser, C.; Bracher, F.; Huber, K.; Knapp, S.; Schwaller, J. Pim serine/threonine kinases in the pathogenesis and therapy of hematologic malignancies and solid cancers. Haematologica 2010, 95, 1004–1015. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Zanin, S.; Sarno, S.; Costa, E.; Girardi, C.; Ribaudo, G.; Salvi, M.; Zagotto, G.; Ruzzene, M.; Pinna, L.A. Design, validation and efficacy of bisubstrate inhibitors specifically affecting ecto-CK2 kinase activity. Biochem. J. 2015, 471, 415–430. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Brenke, R.; Comeau, S.R.; Vajda, S. Piper: An fft-based protein docking program with pairwise potentials. Proteins 2006, 65, 392–406. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with namd. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Golub, A.G.; Bdzhola, V.G.; Briukhovetska, N.V.; Balanda, A.O.; Kukharenko, O.P.; Kotey, I.M.; Ostrynska, O.V.; Yarmoluk, S.M. Synthesis and biological evaluation of substituted (thieno[2,3-d]pyrimidin-4-ylthio)carboxylic acids as inhibitors of human protein kinase CK2. Eur. J. Med. Chem. 2011, 46, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Ostrynska, O.V.; Balanda, A.O.; Bdzhola, V.G.; Golub, A.G.; Kotey, I.M.; Kukharenko, O.P.; Gryshchenko, A.A.; Briukhovetska, N.V.; Yarmoluk, S.M. Design and synthesis of novel protein kinase CK2 inhibitors on the base of 4-aminothieno[2,3-d]pyrimidines. Eur. J. Med. Chem. 2016, 115, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Borowiecki, P.; Wawro, A.M.; Winska, P.; Wielechowska, M.; Bretner, M. Synthesis of novel chiral tbbt derivatives with hydroxyl moiety. Studies on inhibition of human protein kinase CK2alpha and cytotoxicity properties. Eur. J. Med. Chem. 2014, 84, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Hochscherf, J.; Lindenblatt, D.; Steinkruger, M.; Yoo, E.; Ulucan, O.; Herzig, S.; Issinger, O.G.; Helms, V.; Gotz, C.; Neundorf, I.; et al. Development of a high-throughput screening-compatible assay to identify inhibitors of the CK2alpha/CK2beta interaction. Anal. Biochem. 2015, 468, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Li, X.; Zhang, N.; Zhong, R. Structural basis for low-affinity binding of non-r2 carboxylate-substituted tricyclic quinoline analogs to CK2alpha: Comparative molecular dynamics simulation studies. Chem. Biol. Drug Des. 2015, 85, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Retegan, M.; Milet, A.; Jamet, H. Exploring the binding of inhibitors derived from tetrabromobenzimidazole to the CK2 protein using a qm/mm-pb/sa approach. J. Chem. Inf. Model. 2009, 49, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Langer, T.; Hoffmann, R.D. Pharmacophores and pharmacophore searches; Wiley-VCH ; [Chichester : John Wiley, distributor]: Weinheim, 2006. [Google Scholar]
- Chirico, N.; Gramatica, P. Real external predictivity of qsar models. Part 2. New intercomparable thresholds for different validation criteria and the need for scatter plot inspection. J. Chem. Inf. Model. 2012, 52, 2044–2058. [Google Scholar] [CrossRef] [PubMed]
- Chirico, N.; Gramatica, P. Real external predictivity of qsar models: How to evaluate it? Comparison of different validation criteria and proposal of using the concordance correlation coefficient. J. Chem. Inf. Model. 2011, 51, 2320–2335. [Google Scholar] [CrossRef]
- Di-wu, L.; Li, L.L.; Wang, W.J.; Xie, H.Z.; Yang, J.; Zhang, C.H.; Huang, Q.; Zhong, L.; Feng, S.; Yang, S.Y. Identification of CK2 inhibitors with new scaffolds by a hybrid virtual screening approach based on bayesian model; pharmacophore hypothesis and molecular docking. J. Mol. Graph. Model. 2012, 36, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.; Akthar, S.; Sharma, R.; Mishra, S. Identification of ellagic acid analogues as potent inhibitor of protein kinase CK2:A chemopreventive role in oral cancer. Bioinformation 2015, 11, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Morshed, M.N.; Muddassar, M.; Pasha, F.A.; Cho, S.J. Pharmacophore identification and validation study of CK2 inhibitors using comfa/comsia. Chem. Biol. Drug Des. 2009, 74, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Zhong, R. Docking and 3d-qsar studies of 7-hydroxycoumarin derivatives as CK2 inhibitors. Eur J. Med. Chem. 2010, 45, 292–297. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kinase | % Activity | Kinase | % Activity | Kinase | % Activity |
|---|---|---|---|---|---|
| PRAK | 3 | PDK1 | 66 | Lck | 101 |
| SRPK1 | 9 | PKC zeta | 67 | HIPK3 | 102 |
| DYRK3 | 9 | MSK1 | 68 | ERK1 | 103 |
| DYRK2 | 12 | S6K1 | 68 | PRK2 | 105 |
| CK2 | 13 | ROCK 2 | 68 | JNK3 | 106 |
| PAK4 | 14 | PKA | 69 | P38b MAPK | 107 |
| MELK | 17 | PKBb | 71 | EFK2 | 111 |
| BRSK2 | 18 | CSK | 73 | CHK2 | 117 |
| PIM3 | 21 | PLK1 | 76 | CAMKKa | 118 |
| DYRK1A | 26 | Src | 76 | SmMLCK | 127 |
| MAPKAP-K2 | 26 | JNK1 | 78 | ||
| PAK5 | 29 | SGK1 | 80 | ||
| CAMKKb | 29 | PKBa | 80 | ||
| GSK3b | 30 | PKCa | 81 | ||
| IKKb | 36 | CHK1 | 81 | ||
| MAPKAP-K3 | 39 | NEK7 | 82 | ||
| PIM1 | 40 | PHK | 82 | ||
| PAK6 | 42 | CAMK1 | 82 | ||
| ERK8 | 46 | ERK2 | 84 | ||
| AURORA C | 46 | AMPK | 86 | ||
| MARK3 | 47 | JNK2 | 87 | ||
| PKD1 | 48 | HIPK2 | 89 | ||
| PLK1 | 51 | NEK6 | 91 | ||
| RSK2 | 51 | MNK2 | 94 | ||
| CK1 | 58 | p38s MAPK | 98 | ||
| RSK1 | 58 | p38a MAPK | 100 | ||
| MKK1 | 62 | MNK1 | 100 | ||
| AURORA B | 64 | p38g MAPK | 101 | ||
| NEK2a | 65 | MST2 | 101 | ||
| PIM2 | 65 | CDK2-Cyclin A | 101 |
| CK2 | PIM1 | ||
|---|---|---|---|
| Training Set | Test Set | Training Set | Test Set |
| 3KXM | 3KXN | 4ENY | 3UIX |
| 3KXH | 3KXG | 4A7C | 3T9I |
| 3PVG | 3PWD | 3R00 | 3R01 |
| 3NGA | 3Q9Y | 3R02 | 3R04 |
| 3AMY | 3OWK | 3XJ1 | 3XJ2 |
| 4DGN | 3MB7 | 3JPV | 3DCV |
| 3OWL | 3OWJ | 3C4E | 3BGP |
| 3MB6 | 3RPS | 3BGQ | 3BGZ |
| 1ZOH | 1ZOG | 3UMX | 4ENX |
| 1M2R | 1M2Q | 4ALW | 3UMW |
| 2OXD | 2OXY | 4ALU | 4ALV |
| 1OM1 | 1M2P | 4K18 | 4K1B |
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cozza, G. The Development of CK2 Inhibitors: From Traditional Pharmacology to in Silico Rational Drug Design. Pharmaceuticals 2017, 10, 26. https://doi.org/10.3390/ph10010026
Cozza G. The Development of CK2 Inhibitors: From Traditional Pharmacology to in Silico Rational Drug Design. Pharmaceuticals. 2017; 10(1):26. https://doi.org/10.3390/ph10010026
Chicago/Turabian StyleCozza, Giorgio. 2017. "The Development of CK2 Inhibitors: From Traditional Pharmacology to in Silico Rational Drug Design" Pharmaceuticals 10, no. 1: 26. https://doi.org/10.3390/ph10010026
APA StyleCozza, G. (2017). The Development of CK2 Inhibitors: From Traditional Pharmacology to in Silico Rational Drug Design. Pharmaceuticals, 10(1), 26. https://doi.org/10.3390/ph10010026