
DNA Barcode Contamination Screen (DBCscreen): A Pipeline to Rapidly Detect DNA Barcode Contamination for Biodiversity Research


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
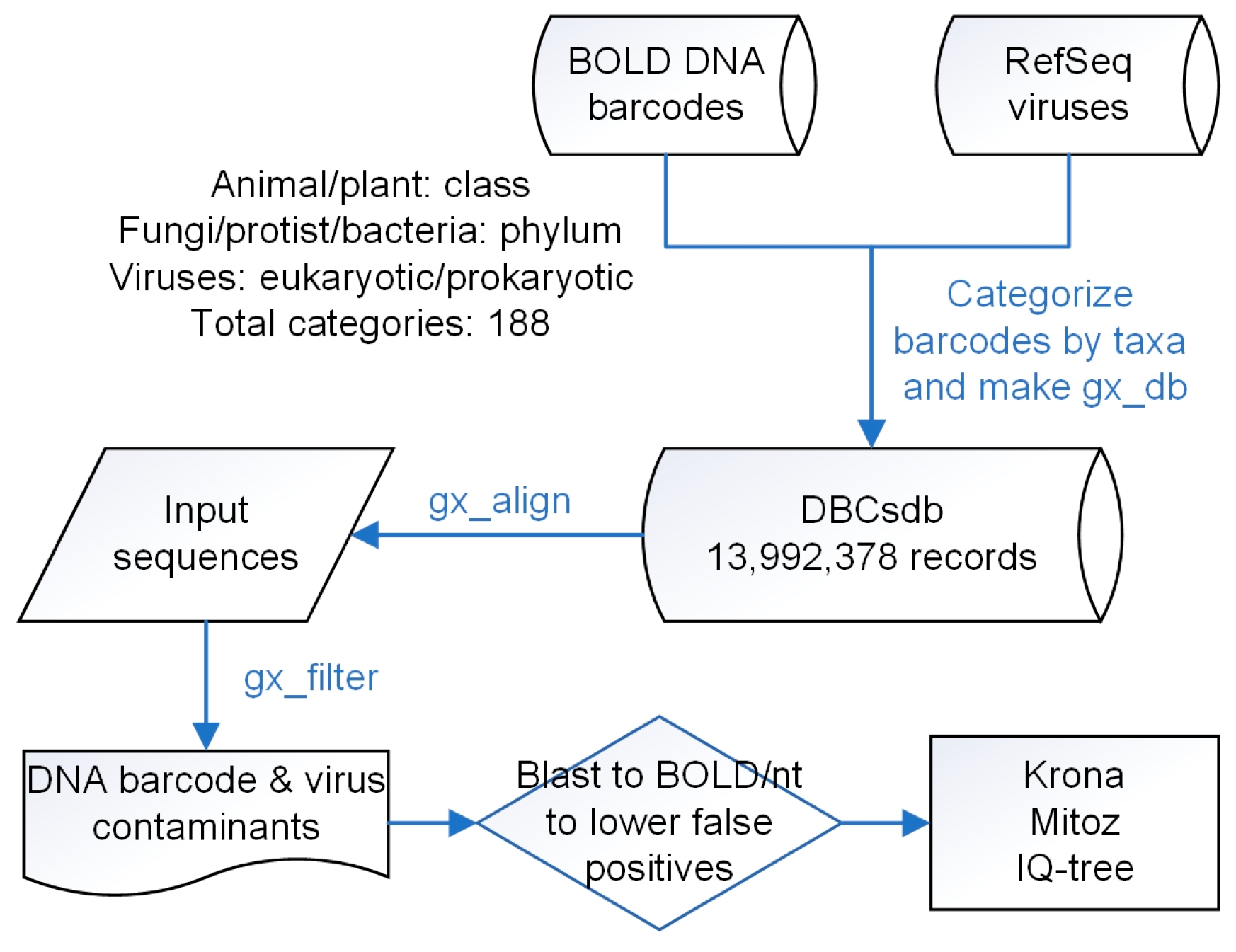
2. Materials and Methods
2.1. Database Retrieval
2.2. DBCscreen Database Construction
2.3. NCBI TSA/WGS Database Screening
2.4. Contamination Taxonomy Classification
2.5. Phylogenetic Analysis
3. Results
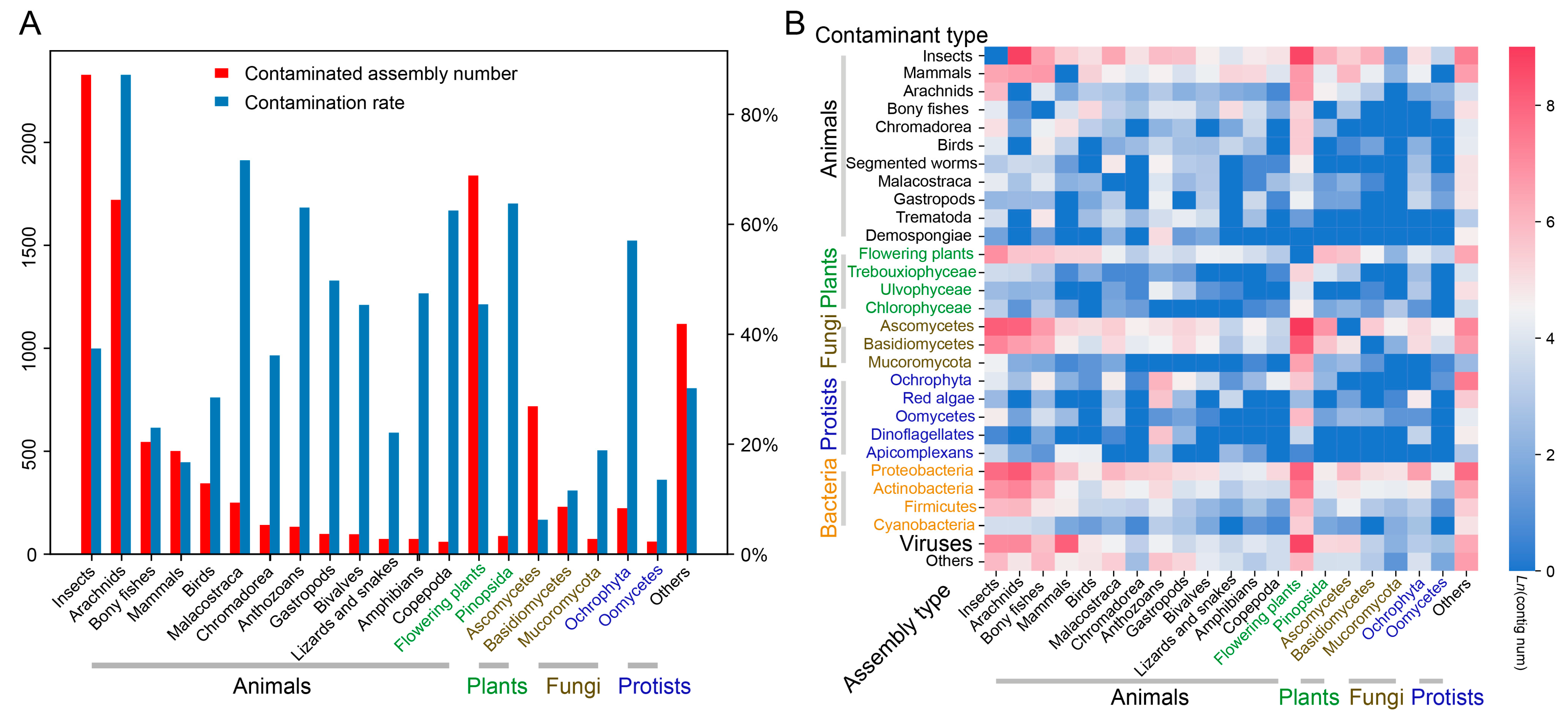
3.1. DBCscreen Detects a Vast Range of Contaminants in the TSA/WGS Database
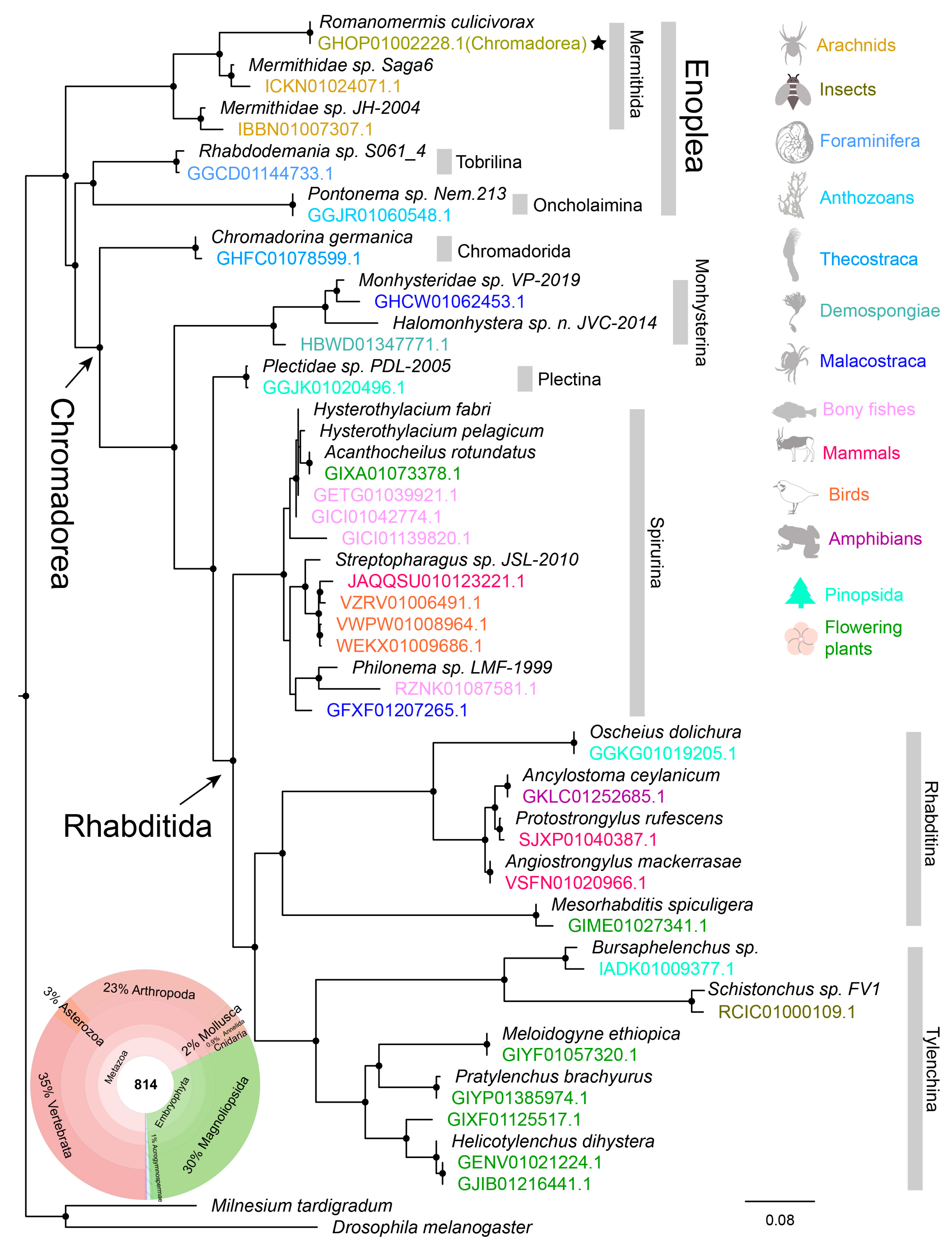
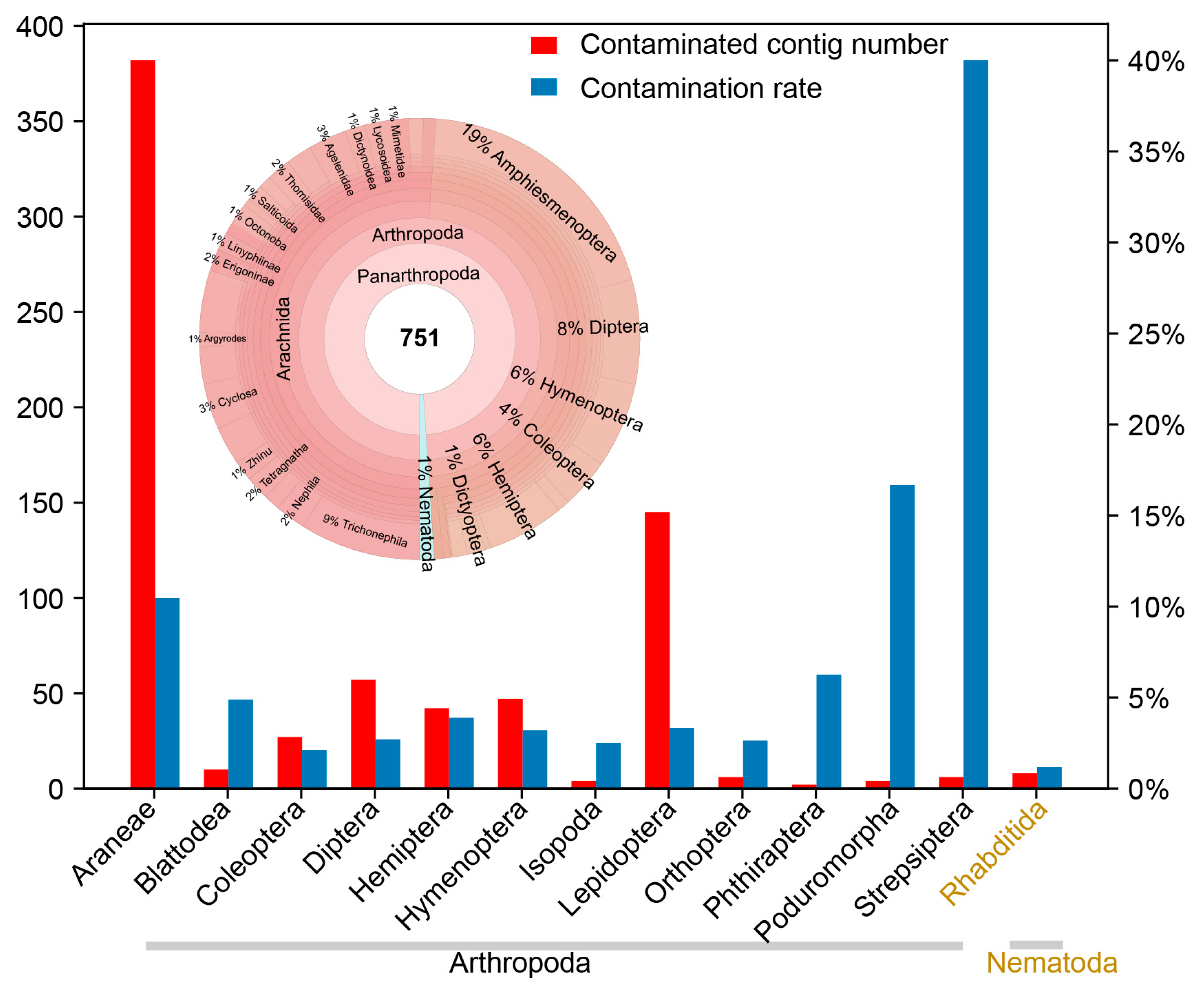
3.2. Application 1—Understanding the Diversity of Nematodes
3.3. Application 2—Exploring the Distribution of Wolbachia
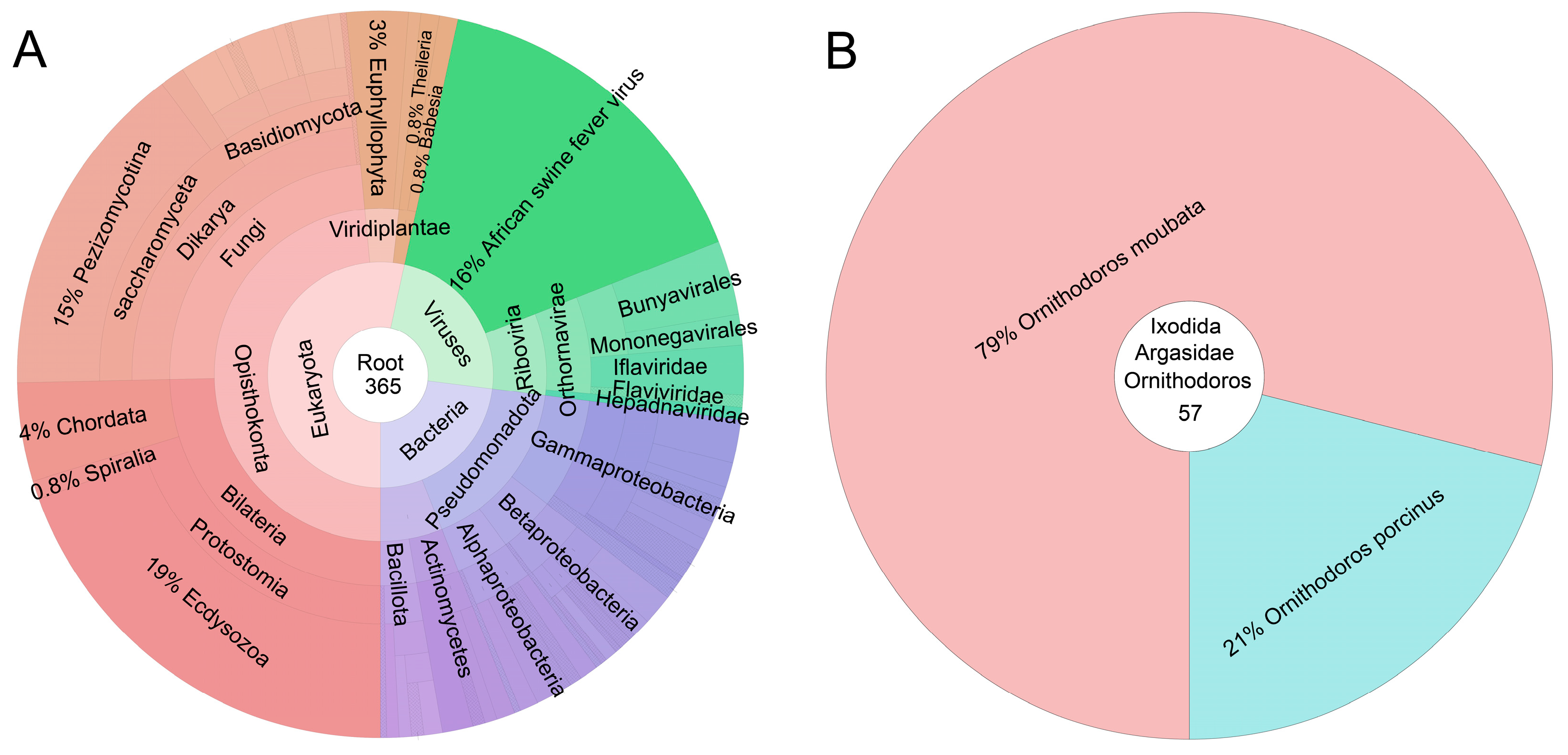
3.4. Application 3—Investigation of the Origins of Arboviruses
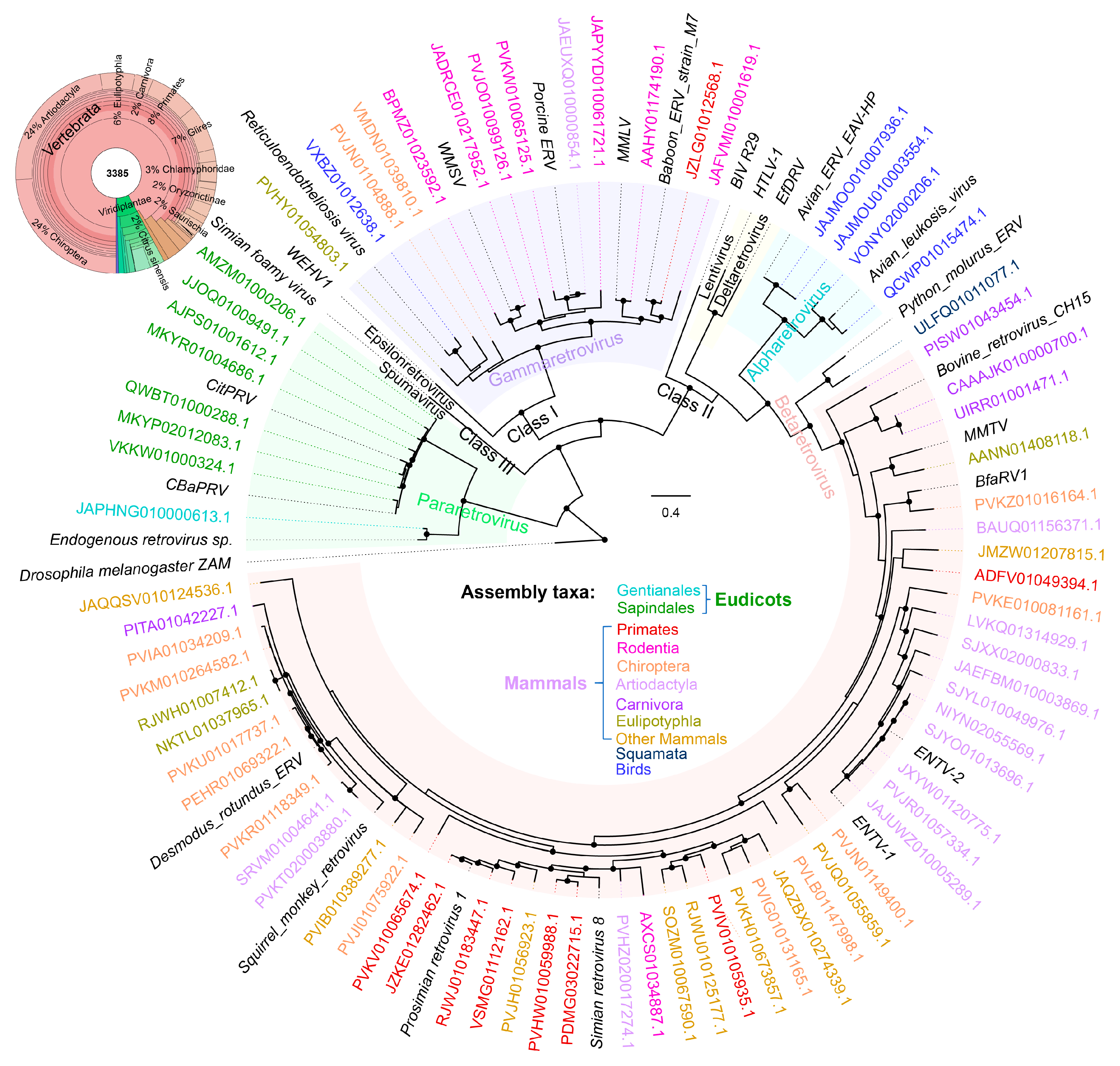
3.5. Application 4—Searching for Endogenous Retroviruses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HTLV-1 | Human T-cell leukemia virus type I |
| ZAM | Drosophila melanogaster ZAM |
| ENTV-1 | Ovine enzootic nasal tumor virus |
| ENTV-2 | Enzootic nasal tumor virus 2 |
| WEHV1 | Walleye epidermal hyperplasia virus 1 |
| CBaPRV | Citrus blight-associated pararetrovirus |
| CitPRV | Citrus endogenous pararetrovirus |
| BIV R29 | Bovine immunodeficiency virus R29 |
| EfDRV | Eptesicus fuscus deltaretrovirus |
| MMLV | Moloney murine leukemia virus |
| BfaRV1 | Bat faecal associated retrovirus 1 |
| MMTV | Mouse mammary tumor virus |
| WMSV | Woolly monkey sarcoma virus |
References
- Steinegger, M.; Salzberg, S.L. Terminating contamination: Large-scale search identifies more than 2,000,000 contaminated entries in GenBank. Genome Biol. 2020, 21, 115. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, X.; Wang, Y.; Liang, H.; Yang, Y.; Huang, X.; Deng, J. Contamination Survey of Insect Genomic and Transcriptomic Data. Animals 2024, 14, 3432. [Google Scholar] [CrossRef] [PubMed]
- Borner, J.; Burmester, T. Parasite infection of public databases: A data mining approach to identify apicomplexan contaminations in animal genome and transcriptome assemblies. BMC Genom. 2017, 18, 100. [Google Scholar] [CrossRef] [PubMed]
- Medina, P.; Russell, S.L.; Corbett-Detig, R. Deep data mining reveals variable abundance and distribution of microbial reproductive manipulators within and among diverse host species. PLoS ONE 2023, 18, e0288261. [Google Scholar] [CrossRef]
- Scholz, M.; Albanese, D.; Tuohy, K.; Donati, C.; Segata, N.; Rota-Stabelli, O. Large scale genome reconstructions illuminate Wolbachia evolution. Nat. Commun. 2020, 11, 5235. [Google Scholar] [CrossRef]
- Sangiovanni, M.; Granata, I.; Thind, A.S.; Guarracino, M.R. From trash to treasure: Detecting unexpected contamination in unmapped NGS data. BMC Bioinform. 2019, 20, 168. [Google Scholar] [CrossRef]
- Xie, J.; Tan, B.; Zhang, Y. A Large-Scale Study into Protist-Animal Interactions Based on Public Genomic Data Using DNA Barcodes. Animals 2023, 13, 2243. [Google Scholar] [CrossRef]
- Astashyn, A.; Tvedte, E.S.; Sweeney, D.; Sapojnikov, V.; Bouk, N.; Joukov, V.; Mozes, E.; Strope, P.K.; Sylla, P.M.; Wagner, L.; et al. Rapid and sensitive detection of genome contamination at scale with FCS-GX. Genome Biol. 2024, 25, 60. [Google Scholar] [CrossRef]
- Ratnasingham, S.; Hebert, P.D.N. BOLD: The Barcode of Life Data System (www.barcodinglife.org). Mol. Ecol. Notes 2007, 7, 355–364. [Google Scholar] [CrossRef]
- Benson, D.A.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2009, 37, D26–D31. [Google Scholar] [CrossRef]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Espagne, E.; Lespinet, O.; Malagnac, F.; Da Silva, C.; Jaillon, O.; Porcel, B.M.; Couloux, A.; Aury, J.M.; Ségurens, B.; Poulain, J.; et al. The genome sequence of the model ascomycete fungus Podospora anserina. Genome Biol. 2008, 9, R77. [Google Scholar] [CrossRef]
- Xie, J.; Zhang, Y. Diversity and Distribution of Mites (ACARI) Revealed by Contamination Survey in Public Genomic Databases. Animals 2023, 13, 3172. [Google Scholar] [CrossRef]
- Bennett, J.; Poulin, R.; Presswell, B. Large-scale genetic investigation of nematode diversity and their phylogenetic patterns in New Zealand’s marine animals. Parasitology 2022, 149, 1794–1809. [Google Scholar] [CrossRef]
- Bert, W.; Leliaert, F.; Vierstraete, A.R.; Vanfleteren, J.R.; Borgonie, G. Molecular phylogeny of the Tylenchina and evolution of the female gonoduct (Nematoda: Rhabditida). Mol. Phylogenet. Evol. 2008, 48, 728–744. [Google Scholar] [CrossRef]
- Wijová, M.; Moravec, F.; Horák, A.; Lukeš, J. Evolutionary relationships of Spirurina (Nematoda: Chromadorea: Rhabditida) with special emphasis on dracunculoid nematodes inferred from SSU rRNA gene sequences. Int. J. Parasitol. 2006, 36, 1067–1075. [Google Scholar] [CrossRef]
- Fang, H.; Poinar, G.O.; Wang, H.; Wang, B.; Luo, C. First spider-parasitized mermithid nematode from mid-Cretaceous Kachin amber of northern Myanmar. Cretac. Res. 2024, 158, 105866. [Google Scholar] [CrossRef]
- Kajtoch, Ł.; Kotásková, N. Current state of knowledge on Wolbachia infection among Coleoptera: A systematic review. PeerJ 2018, 6, e4471. [Google Scholar] [CrossRef] [PubMed]
- Labuda, M.; Nuttall, P.A. Tick-borne viruses. Parasitology 2004, 129 (Suppl. 1), S221–S245. [Google Scholar] [CrossRef]
- Brites-Neto, J.; Duarte, K.M.; Martins, T.F. Tick-borne infections in human and animal population worldwide. Vet. World 2015, 8, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Jori, F.; Bastos, A.; Boinas, F.; Van Heerden, J.; Heath, L.; Jourdan-Pineau, H.; Martinez-Lopez, B.; Pereira de Oliveira, R.; Pollet, T.; Quembo, C.; et al. An Updated Review of Ornithodoros Ticks as Reservoirs of African Swine Fever in Sub-Saharan Africa and Madagascar. Pathogens 2023, 12, 469. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Wei, Y.; Han, G.-Z. The diversity and evolution of retroviruses: Perspectives from viral “fossils”. Virol. Sin. 2022, 37, 11–18. [Google Scholar] [CrossRef]
- Farkašová, H.; Hron, T.; Pačes, J.; Hulva, P.; Benda, P.; Gifford, R.J.; Elleder, D. Discovery of an endogenous Deltaretrovirus in the genome of long-fingered bats (Chiroptera: Miniopteridae). Proc. Natl. Acad. Sci. USA 2017, 114, 3145–3150. [Google Scholar] [CrossRef]
- Simpson, J.Z.; Kozak Christine, A.; Boso, G. Evolutionary conservation of an ancient retroviral gagpol gene in Artiodactyla. J. Virol. 2023, 97, e00535-23. [Google Scholar] [CrossRef]
- Keremane, M.; Singh, K.; Ramadugu, C.; Krueger, R.R.; Skaggs, T.H. Next Generation Sequencing, and Development of a Pipeline as a Tool for the Detection and Discovery of Citrus Pathogens to Facilitate Safer Germplasm Exchange. Plants 2024, 13, 411. [Google Scholar] [CrossRef]
- Swain, T.D.; Wulff, J.L. Diversity and specificity of Caribbean sponge–zoanthid symbioses: A foundation for understanding the adaptive significance of symbioses and generating hypotheses about higher-order systematics. Biol. J. Linn. Soc. 2007, 92, 695–711. [Google Scholar] [CrossRef]
- Smythe, A.B.; Holovachov, O.; Kocot, K.M. Improved phylogenomic sampling of free-living nematodes enhances resolution of higher-level nematode phylogeny. BMC Evol. Biol. 2019, 19, 121. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, J.; Zhang, Y.; Wang, L.; Deng, Y. DNA Barcode Contamination Screen (DBCscreen): A Pipeline to Rapidly Detect DNA Barcode Contamination for Biodiversity Research. Diversity 2025, 17, 186. https://doi.org/10.3390/d17030186
Xie J, Zhang Y, Wang L, Deng Y. DNA Barcode Contamination Screen (DBCscreen): A Pipeline to Rapidly Detect DNA Barcode Contamination for Biodiversity Research. Diversity. 2025; 17(3):186. https://doi.org/10.3390/d17030186
Chicago/Turabian StyleXie, Jiazheng, Yu Zhang, Lina Wang, and Yuting Deng. 2025. "DNA Barcode Contamination Screen (DBCscreen): A Pipeline to Rapidly Detect DNA Barcode Contamination for Biodiversity Research" Diversity 17, no. 3: 186. https://doi.org/10.3390/d17030186
APA StyleXie, J., Zhang, Y., Wang, L., & Deng, Y. (2025). DNA Barcode Contamination Screen (DBCscreen): A Pipeline to Rapidly Detect DNA Barcode Contamination for Biodiversity Research. Diversity, 17(3), 186. https://doi.org/10.3390/d17030186