


Fecal Microbiota and Feeding Habitats of Nomadic Indigenous Animals (Deer, Yak, Sheep and Camel) in Baikal Siberia (Russia)

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Objects and Area
- Deer (Rangifer tarandus): As a breed of mountain taiga deer that lives in more northern latitudes, Tofalar deer graze freely in the valleys of high mountain rivers.
- Yaks (Bos grunniens): The Okinsky yak population exhibits a considerable degree of adaptation in order to survive in harsh ecological and geographical conditions, including year-round grazing on high alpine meadows of the Eastern Sayan and Small Khamar-Daban. This adaptation is evident in the yaks’ ecology and behavior, marked breeding seasonality, accelerated development of young animals in the warm season, and seasonal rotation of pastures.
- Sheep (Ovis aries): Buryat-native coarse-wool sheep were bred during the era of nomadic pastoralism. This breed shows exceptional adaptability to local conditions and is suited for year-round grazing.
- Camels (Camelus bactrianus): The ransbaikal camel is best adapted to life in a sharply continental climate with pronounced changes in habitat conditions both throughout the year and day. Thanks to their anatomical and physiological features, camels can tolerate unusually long periods without water and are satisfied with the coarsest and most nutritious food sources [29].
2.2. Sample Collection
2.3. Ethical Considerations
2.4. DNA Extraction
2.5. The 16S rRNA Gene Profiling
2.6. Data Analysis
3. Results
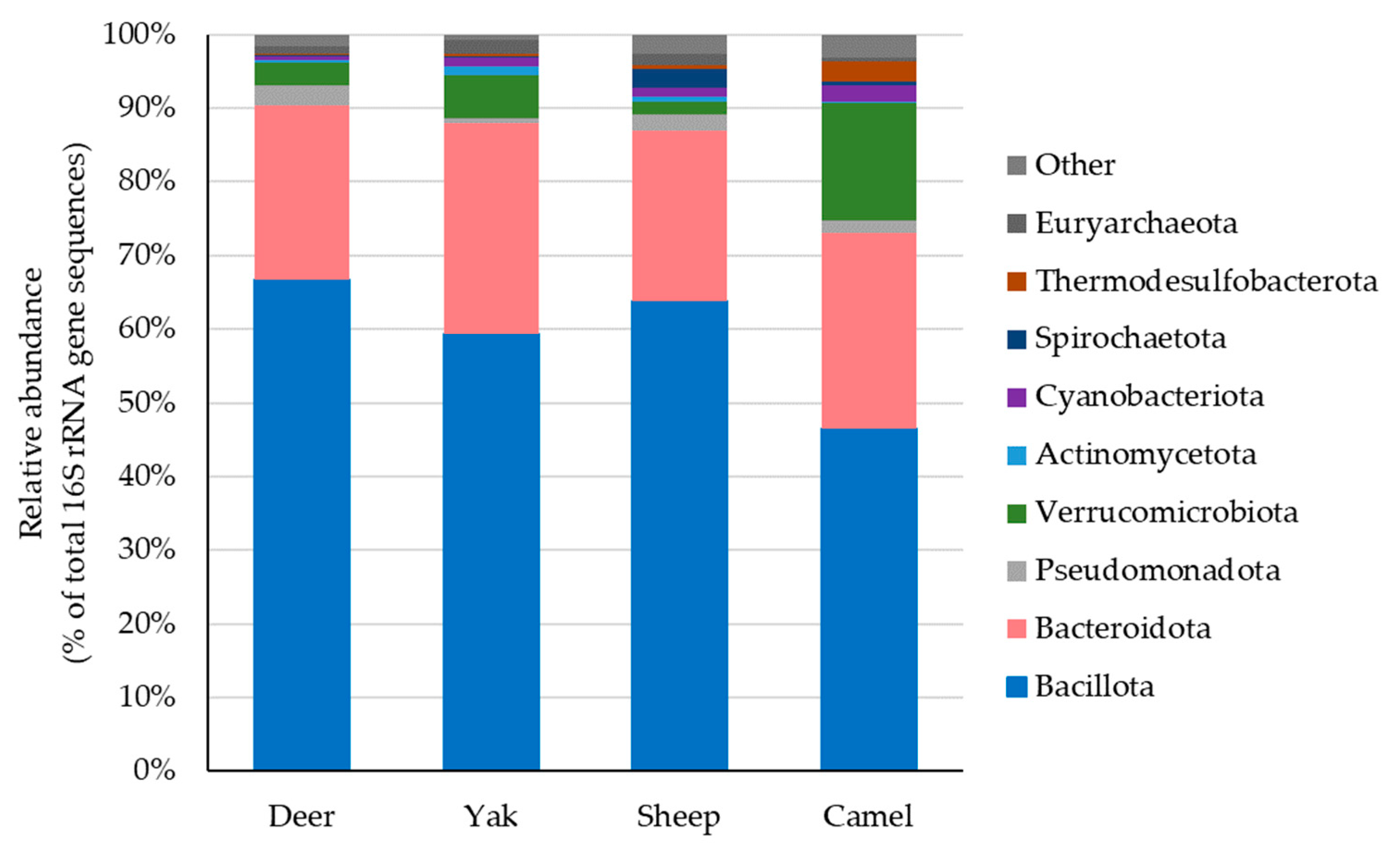
3.1. Fecal Microbiota of Four Animal Species
3.1.1. Fecal Microbiota of Deer
3.1.2. Fecal Microbiota of Yaks
3.1.3. Fecal Microbiota of Sheep
3.1.4. Fecal Microbiota of Camels
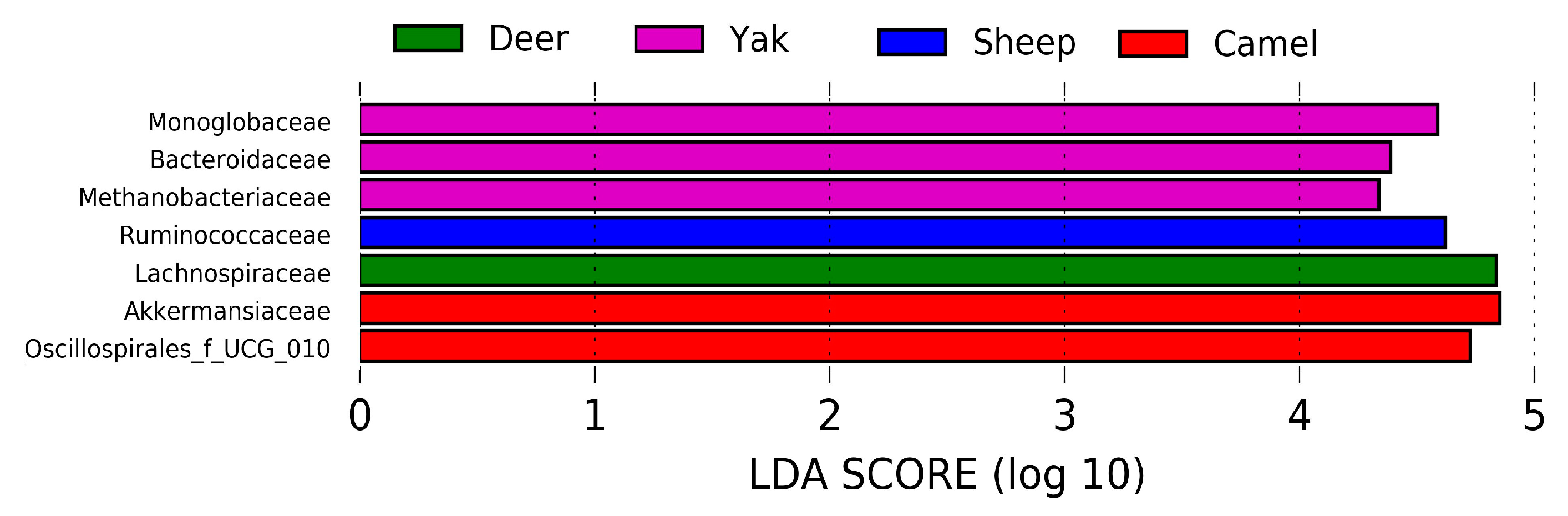
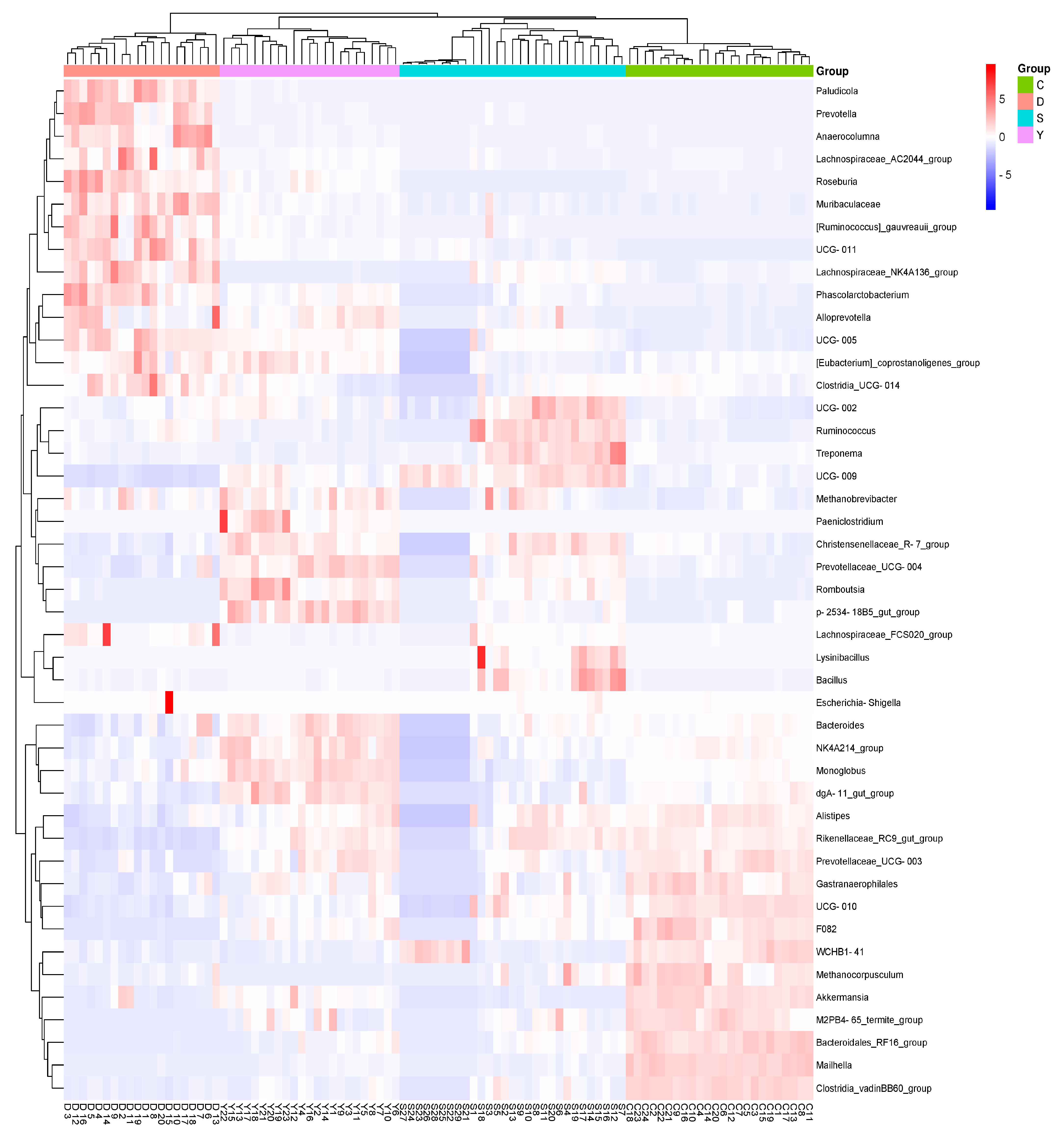
3.1.5. LEfSe Analysis
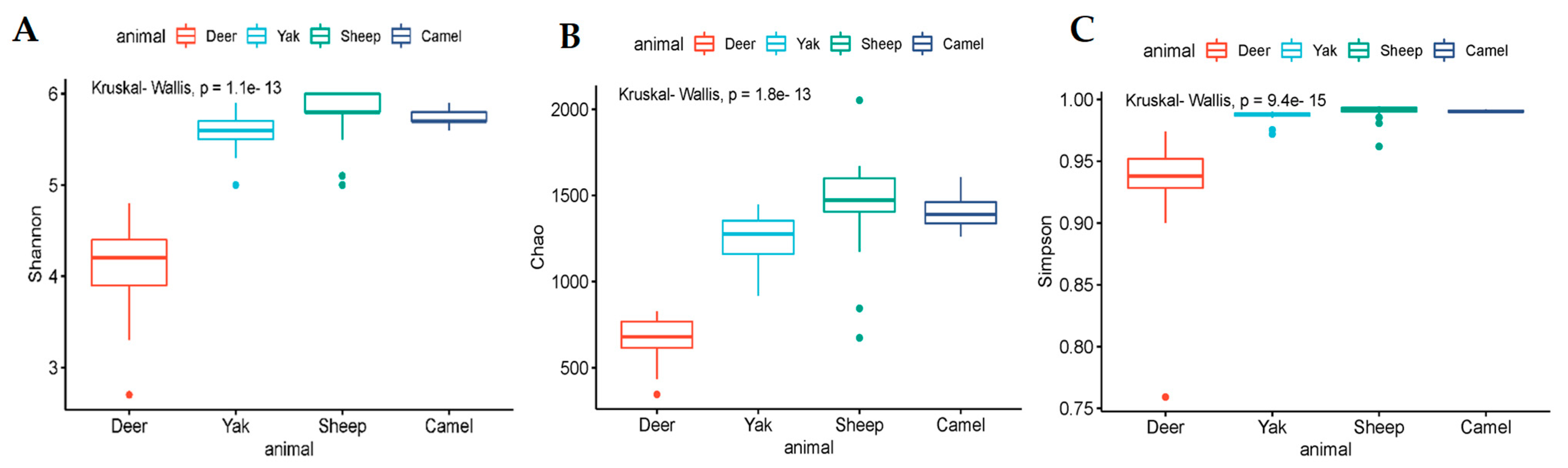
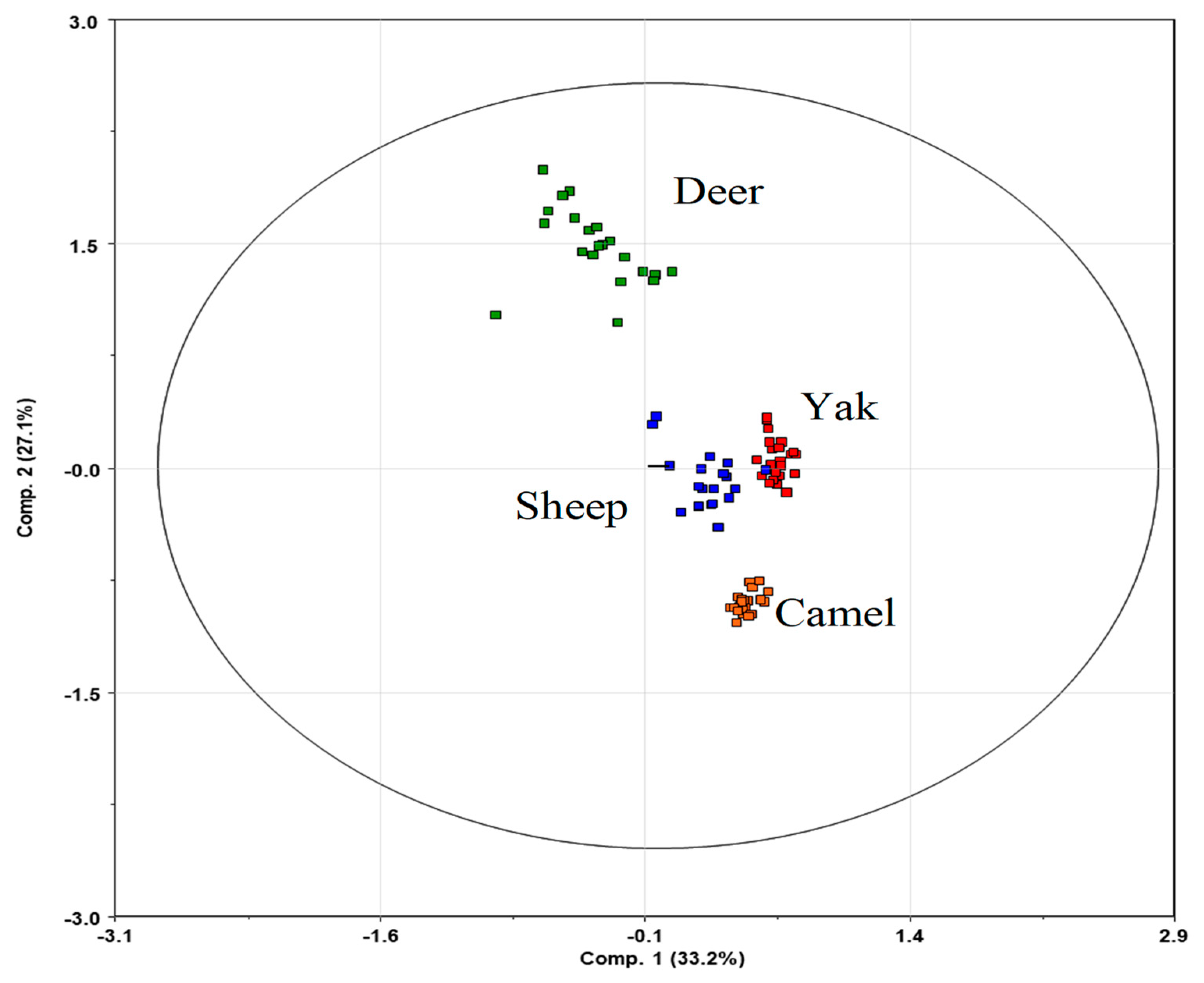
3.2. Diversity Analysis
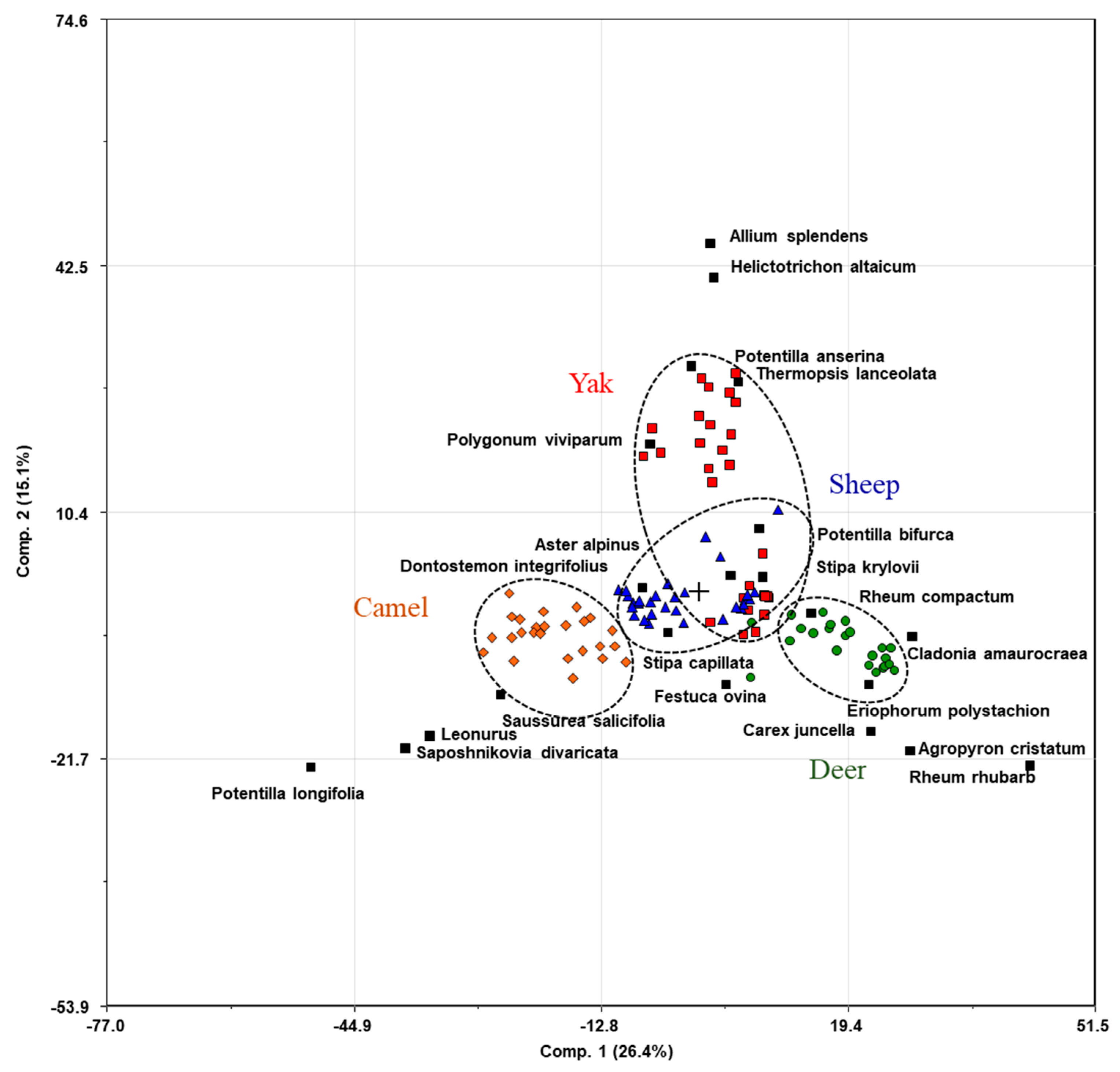
3.3. Diet
4. Discussion
4.1. The Core Fecal Microbiota of Nomadic Animals (Deer, Yaks, Sheep, and Camels) and Their Species Specificity
4.2. Diet Preferences Is an Important Factor That Affects Fecal Microbiota
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Aricha, H.; Simujide, H.; Wang, C.; Zhang, J.; Lv, W.; Jimisi, X.; Liu, B.; Chen, H.; Zhang, C.; He, L.; et al. Comparative analysis of fecal microbiota of grazing Mongolian cattle from different regions in Inner Mongolia, China. Animals 2021, 11, 1938. [Google Scholar] [CrossRef] [PubMed]
- Henderson, G.; Cox, F.; Ganesh, S.; Jonker, A.; Young, W.; Global Rumen Census Collaborators; Janssen, P.H. Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci. Rep. 2015, 5, 14567. [Google Scholar] [CrossRef]
- Delgado, M.L.; Singh, P.; Funk, J.A.; Moore, J.A.; Cannell, E.M.; Kanesfsky, J.; Manning, S.D.; Scribner, K.T. Intestinal microbial community dynamics of white-tailed deer (Odocoileus virginianus) in an agroecosystem. Microb. Ecol. 2017, 74, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Groussin, M.; Mazel, F.; Sanders, J.G.; Smillie, C.S.; Lavergne, S.; Thuiller, W.; Alm, E.J. Unraveling the processes shaping mammalian gut microbiomes over evolutionary time. Nat. Commun. 2017, 8, 14319. [Google Scholar] [CrossRef]
- Jize, Z.; Zhuoga, D.; Xiaoqing, Z.; Na, T.; Jiacuo, G.; Cuicheng, L.; Bandan, P. Different feeding strategies can affect growth performance and rumen functions in Gangba sheep as revealed by integrated transcriptome and microbiome analyses. Front. Microbiol. 2022, 13, 908326. [Google Scholar] [CrossRef]
- Glendinning, L.; Genç, B.; Wallace, R.J.; Watson, M. Metagenomic analysis of the cow, sheep, reindeer and red deer rumen. Sci. Rep. 2021, 11, 1990. [Google Scholar] [CrossRef]
- Fonty, G.; Joblin, K.; Chavarot, M.; Roux, R.; Naylor, G.; Michallon, F. Establishment and development of ruminal hydrogenotrophs in methanogen-free lambs. Appl. Environ. Microbiol. 2007, 73, 6391–6403. [Google Scholar] [CrossRef] [PubMed]
- Bolnick, D.I.; Snowberg, L.K.; Hirsch, P.E.; Lauber, C.L.; Org, E.; Parks, B.; Lusis, A.J.; Knight, R.; Caporaso, J.G.; Svanbäck, R. Individual diet has sex-dependent effects on vertebrate gut microbiota. Nat. Commun. 2014, 5, 4500. [Google Scholar] [CrossRef]
- Grond, K.; Sandercock, B.K.; Jumpponen, A.; Zeglin, L.H. The avian gut microbiota: Community, physiology and function in wild birds. J. Avian. Biol. 2018, 49, e01788. [Google Scholar] [CrossRef]
- Sundset, M.A.; Præsteng, K.E.; Cann, I.K.O.; Mathiesen, S.D.; Mackie, R.I. Novel rumen bacterial diversity in two geographically separated sub-species of reindeer. Microb. Ecol. 2007, 54, 424–438. [Google Scholar] [CrossRef]
- Crater, A.R.; Barboza, P.S.; Forster, R. Regulation of rumen fermentation during seasonal fluctuations in food intake of muskoxen. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2007, 146, 233–241. [Google Scholar] [CrossRef]
- Zaitseva, S.; Dagurova, O.; Radnagurueva, A.; Kozlova, A.; Izotova, A.; Krylova, A.; Noskov, S.; Begmatov, S.; Patutina, E.; Barkhutova, D. Fecal microbiota and diet composition of buryatian horses grazing warm- and cold-season grass pastures. Microorganisms 2023, 11, 1947. [Google Scholar] [CrossRef]
- Ilina, L.A.; Filippova, V.A.; Layshev, K.A.; Yildirim, E.A.; Dunyashev, T.P.; Brazhnik, E.A.; Dubrovin, A.V.; Sobolev, D.V.; Tiurina, D.G.; Novikova, N.I.; et al. Variation in the Russian arctic reindeer (Rangifer tarandus) rumen microbiome related to season change. Agr. Biol. 2020, 55, 697–713. [Google Scholar] [CrossRef]
- Rustomo, B.; AlZahal, O.; Odongo, N.; Duffield, T.F.; McBride, B.W. Effects of rumen acid load from feed and forage particle size on ruminal pH and dry matter intake in the lactating dairy cow. J. Dairy Sci. 2006, 89, 4758–4768. [Google Scholar] [CrossRef]
- Zhou, Z.; Fang, L.; Meng, Q.; Li, S.; Chai, S.; Liu, S.; Schonewille, J.T. Assessment of ruminal bacterial and archaeal community structure in yak (Bos grunniens). Front. Microbiol. 2017, 8, 179. [Google Scholar] [CrossRef]
- Ming, L.; Siriguleng, Y.L.; Hasi, S.; He, J.; Hai, L.; Wang, Z.; Guo, F.; Qiao, X.; Jirimutu. Comparative analysis of fecal microbial communities in cattle and Bactrian camels. PLoS ONE 2017, 12, e0173062. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, Z.; Li, B.; Hao, W.; Yin, W.; Ai, S.; Han, J.; Wang, R.; Duan, Z. Depicting fecal microbiota characteristic in yak, cattle, yak-cattle hybrid and Tibetan sheep in different eco-regions of Qinghai-Tibetan plateau. Microbiol. Spectr. 2022, 10, e00021-22. [Google Scholar] [CrossRef]
- Chen, S.; Luo, S.; Yan, C. Gut microbiota implications for health and welfare in farm animals: A Review. Animals 2021, 12, 93. [Google Scholar] [CrossRef]
- He, J.; Yi, L.; Hai, L.; Ming, L.; Gao, W.; Ji, R. Characterizing the bacterial microbiota in different gastrointestinal tract segments of the Bactrian camel. Sci. Rep. 2018, 8, 654. [Google Scholar] [CrossRef]
- He, J.; Hai, L.; Orgoldol, K.; Yi, L.; Ming, L.; Guo, F.; Li, G.; Ji, R. High-throughput sequencing reveals the gut microbiome of the Bactrian camel in different ages. Curr. Microbiol. 2019, 76, 810–817. [Google Scholar] [CrossRef]
- Fujio-Vejar, S.; Vasquez, Y.; Morales, P.; Magne, F.; Vera-Wolf, P.; Ugalde, J.A.; Navarrete, P.; Gotteland, M. The gut microbiota of healthy Chilean subjects reveals a high abundance of the phylum Verrucomicrobia. Front. Microbiol. 2017, 8, 1221. [Google Scholar] [CrossRef]
- Gharechahi, J.; Sarikhan, S.; Han, J.L.; Ding, X.Z.; Salekdeh, G.H. Functional and phylogenetic analyses of camel rumen microbiota associated with different lignocellulosic substrates. NPJ Biofilms Microbiomes 2022, 8, 46. [Google Scholar] [CrossRef] [PubMed]
- Kleen, J.L.; Hooijer, G.A.; Rehage, J.; Noordhuizen, J.P. Subacute ruminal acidosis (SARA): A review. J. Vet. Med. Ser. A 2003, 50, 406–414. [Google Scholar] [CrossRef] [PubMed]
- McEwan, N.R.; Abecia, L.; Regensbogenova, M.; Adam, C.L.; Findlay, P.A.; Newbold, C.J. Rumen microbial population dynamics in response to photoperiod. Lett. Appl. Microbiol. 2005, 41, 97–101. [Google Scholar] [CrossRef]
- Uyeno, Y.; Sekiguchi, Y.; Tajima, K.; Takenaka, A.; Kurihara, M.; Kamagata, Y. An rRNA-based analysis for evaluating the effect of heat stress on the rumen microbial composition of Holstein heifers. Anaerobe 2010, 16, 27–33. [Google Scholar] [CrossRef]
- Romero-Pérez, G.A.; Ominski, K.H.; McAllister, T.A.; Krause, D.O. Effect of environmental factors and influence of rumen and hindgut biogeography on bacterial communities in steers. Appl. Environ. Microbiol. 2011, 77, 258–268. [Google Scholar] [CrossRef]
- Petrov, K.A.; Dudareva, L.V.; Nokhsorov, V.V.; Stoyanov, K.N.; Makhutova, O.N. Fatty acid content and composition of the Yakutian horses and their main food source: Living in extreme winter conditions. Biomolecules 2020, 10, 315. [Google Scholar] [CrossRef]
- O’Donnell, M.M.; Harris, M.B.; Ross, R.P.; O’Toole, P.W. Core fecal microbiota of domesticated herbivorous ruminant, hindgut fermenters, and monogastric animals. Microbiol. Op. 2017, 6, e509. [Google Scholar]
- Taishin, V.A.; Lkhasaranov, B.B.; James, R. Atlas of Migratory Animals; Publishing House of the Siberian Branch of the Russian Academy of Sciences: Novosibirsk, Russia, 1999; p. 284. [Google Scholar]
- Taishin, V.A. Rare endangered species of agricultural animals in Buryatia. Agric. J. 2009, 2. [Google Scholar]
- Anenkhonov, O.A.; Pykhalova, T.D.; Osipov, K.I.; Sekulich, I.R.; Badmaeva, N.K.; Namzalov, B.B.; Krivobokov, L.V.; Munkueva, M.S.; Sutkin, A.V.; Tubshinova, D.B.; et al. Key to Identification of the Plants in Buryatia; JSC Republican Printing House: Ulan-Ude, Russia, 2001; p. 672. [Google Scholar]
- Magŏc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using Qiime 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Boil. 2011, 12, R60. [Google Scholar] [CrossRef]
- Fazlollahi, M.; Lee, T.D.; Andrade, J.; Oguntuyo, K.; Chun, Y.; Grishina, G.; Grishin, A.; Bunyavanich, S. The nasal microbiome in asthma. J. Allergy. Clin. Immunol. 2018, 142, 834–843.e2. [Google Scholar] [CrossRef] [PubMed]
- Holechek, J.L.; Vavra, M.; Pieper, R.D. Botanical composition determination of range herbivore diets: A review. J. Range Manag. 1982, 35, 309–315. [Google Scholar] [CrossRef]
- Ley, R.E.; Hamady, M.; Lozupone, C.; Turnbaugh, P.; Ramey, R.R.; Bircher, J.S.; Schlegel, M.L.; Tucker, T.A.; Schrenzel, M.D.; Knight, R.; et al. Evolution of mammals and their gut microbes. Science 2008, 320, 1647–1651. [Google Scholar] [CrossRef] [PubMed]
- Jonge, N.; Carlsen, B.; Christensen, M.H.; Pertoldi, C.; Nielsen, L.J. The gut microbiome of 54 mammalian species. Front. Microbiol. 2022, 13, 886252. [Google Scholar] [CrossRef]
- Karnachuk, O.V.; Panova, I.A.; Panov, V.L.; Ikkert, O.P.; Kadnikov, V.V.; Rusanov, I.I.; Avakyan, M.R.; Glukhova, L.B.; Lukina, A.P.; Rakitin, A.V.; et al. Active sulfate-reducing bacterial community in the camel gut. Microorganisms 2023, 11, 401. [Google Scholar] [CrossRef]
- Pope, P.B.; Mackenzie, A.K.; Gregor, I.; Smith, W.; Sundset, M.A.; McHardy, A.C.; Morrison, M.; Eijsink, V.G.H. Metagenomics of the Svalbard reindeer rumen microbiome reveals abundance of polysaccharide utilization loci. PLoS ONE 2012, 7, e38571. [Google Scholar] [CrossRef]
- Li, B.; Jia, G.; Wen, D.; Zhao, X.; Zhang, J.; Xu, Q.; Zhao, X.; Jiang, N.; Liu, Z.; Wang, Y. Rumen microbiota of indigenous and introduced ruminants and their adaptation to the Qinghai–Tibetan plateau. Front. Microbiol. 2022, 13, 1027138. [Google Scholar] [CrossRef]
- Xin, J.; Chai, Z.; Zhang, C.; Zhang, Q.; Zhu, Y.; Cao, H.; Zhong, J.; Ji, Q. Comparing the microbial community in four stomach of dairy cattle, yellow cattle and three yak herds in Qinghai-Tibetan Plateau. Front. Microbiol. 2019, 10, 1547. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hu, X.; Yang, S.; Zhou, J.; Qi, L.; Sun, X.; Fan, M.; Xu, S.; Cha, M.; Zhang, M.; et al. Comparison between the fecal bacterial microbiota of healthy and diarrheic captive musk deer. Front. Microbiol. 2018, 9, 300. [Google Scholar] [CrossRef]
- Kibegwa, F.M.; Bett, R.C.; Gachuiri, C.K.; Machuka, E.; Stomeo, F.; Mujibi, F.D. Diversity and functional analysis of rumen and fecal microbial communities associated with dietary changes in crossbreed dairy cattle. PLoS ONE 2023, 18, e0274371. [Google Scholar] [CrossRef] [PubMed]
- Shah, T.; Ding, L.; Din, A.U.; Hassan, F.; Ahmad, A.A.; Wei, H.; Wang, X.; Yan, Q.; Ishaq, M.; Ali, N.; et al. Differential effects of natural grazing and feedlot feeding on yak fecal microbiota. Front. Vet. Sci. 2022, 9, 791245. [Google Scholar] [CrossRef] [PubMed]
- Jami, E.; White, B.A.; Mizrahi, I. Potential role of the bovine rumen microbiome in modulating milk composition and feed efficiency. PLoS ONE 2014, 9, e85423. [Google Scholar] [CrossRef]
- Koliada, A.; Syzenko, G.; Moseiko, V.; Budovska, L.; Puchkov, K.; Perederiy, V.; Gavalko, Y.; Dorofeyev, A.; Romanenko, M.; Tkach, S.; et al. Association between body mass index and Firmicutes/Bacteroidetes ratio in an adult Ukrainian population. BMC Microbiol. 2017, 17, 120. [Google Scholar] [CrossRef]
- Haworth, S.E.; White, K.S.; Côté, S.D.; Shafer, A.B.A. Space, time and captivity: Quantifying the factors influencing the fecal microbiome of an alpine ungulate. FEMS Microbiol. Ecol. 2019, 95, fiz095. [Google Scholar] [CrossRef]
- Lindenberg, F.; Krych, L.; Fielden, J.; Kot, W.; Frøkiær, H.; van Galen, G.; Nielsen, D.S.; Hansen, A.K. Expression of immune regulatory genes correlate with the abundance of specific Clostridiales and Verrucomicrobia species in the equine ileum and cecum. Sci. Rep. 2019, 9, 12674. [Google Scholar] [CrossRef]
- Gharechahi, J.; Vahidi, M.F.; Sharifi, G.; Ariaeenejad, S.; Ding, X.-Z.; Han, J.-L.; Salekdeh, G.H. Lignocellulose degradation by rumen bacterial communities: New insights from metagenome analyses. Environ. Res. 2023, 229, 115925. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, G.; Xu, H.; Xin, H.; Zhang, Y. Metagenomic analyses of microbial and carbohydrate-active enzymes in the rumen of holstein cows fed different forage-to-concentrate ratios. Front. Microbiol. 2019, 10, 649. [Google Scholar] [CrossRef]
- Gharechahi, J.; Vahidi, M.F.; Bahram, M.; Han, J.-L.; Ding, X.-Z.; Salekdeh, G.H. Metagenomic analysis reveals a dynamic microbiome with diversified adaptive functions to utilize high lignocellulosic forages in the cattle rumen. ISME J. 2021, 15, 1108–1120. [Google Scholar] [CrossRef] [PubMed]
- Welch, C.B.; Lourenco, J.M.; Krause, T.R.; Seidel, D.S.; Fluharty, F.L.; Pringle, T.D.; Callaway, T.R. Evaluation of fecal bacterial communities of angus bulls with varying feeding efficiency throughout life from weaning to slaughter. Front. Vet. Sci. 2021, 29, 597405. [Google Scholar] [CrossRef] [PubMed]
- Andrade, B.G.N.; Bressani, F.A.; Cuadrat, R.R.C.; Tizioto, P.C.; De Oliveira, P.S.N.; Mourão, G.B.; Coutinho, L.L.; Reecy, J.M.; Koltes, J.E.; Walsh, P.; et al. The structure of microbial populations in Nelore GIT reveals inter-dependency of methanogens in feces and rumen. J. Anim. Sci. Biotechnol. 2020, 11, 6. [Google Scholar] [CrossRef]
- Boukerb, A.M.; Noël, C.; Quenot, E.; Cadiou, B.; Chevé, J.; Quintric, L.; Cormier, A.; Dantan, L.; Gourmelon, M. Comparative analysis of fecal microbiomes from wild waterbirds to poultry, cattle, pigs, and wastewater treatment plants for a microbial source tracking approach. Front. Microbiol. 2021, 12, 697553. [Google Scholar] [CrossRef]
- Guo, J.; Li, P.; Zhang, K.; Zhang, L.; Wang, X.; Li, L.; Zhang, H. Distinct stage changes in early-life colonization and acquisition of the gut microbiota and its correlations with volatile fatty acids in goat kids. Front. Microbiol. 2020, 11, 584742. [Google Scholar] [CrossRef]
- Li, Y.; Hu, X.; Yang, S.; Zhou, J.; Zhang, T.; Qi, L.; Sun, X.; Fan, M.; Xu, S.; Cha, M.; et al. Comparative analysis of the gut microbiota composition between captive and wild forest musk deer. Front. Microbiol. 2017, 8, 1705. [Google Scholar] [CrossRef] [PubMed]
- Zoelzer, F.; Burger, A.L.; Dierkes, P.W. Unraveling differences in fecal microbiota stability in mammals: From high variable carnivores and consistently stable herbivores. Anim. Microbiome 2021, 3, 77. [Google Scholar] [CrossRef]
- Youngblut, N.D.; Reischer, G.H.; Walters, W.; Schuster, N.; Walzer, C.; Stalder, G.; Ley, R.E.; Farnleitner, A.H. Host diet and evolutionary history explain different aspects of gut microbiome diversity among vertebrate clades. Nat. Commun. 2019, 10, 2200. [Google Scholar] [CrossRef]
- Li, Z.; Wright, A.D.; Liu, H.; Fan, Z.; Yang, F.; Zhang, Z.; Li, G. Response of the rumen microbiota of sika deer (Cervus nippon) fed different concentrations of tannin rich plants. PLoS ONE 2015, 10, e0123481. [Google Scholar] [CrossRef]
- Li, Z.P.; Liu, H.L.; Li, G.Y.; Bao, K.; Wang, K.Y.; Xu, C.; Wright, A.-D.G. Molecular diversity of rumen bacterial communities from tannin-rich and fiber-rich forage fed domestic Sika deer (Cervus nippon) in China. BMC Microbiol. 2013, 13, 151. [Google Scholar] [CrossRef]
- Betancur-Murillo, C.L.; Aguilar-Marín, S.B.; Jovel, J. Prevotella: A key player in ruminal metabolism. Microorganisms 2023, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Zielińska, S.; Kidawa, D.; Stempniewicz, L.; Łoś, M.; Łoś, J.M. New insights into the microbiota of the Svalbard reindeer Rangifer tarandus platyrhynchus. Front. Microbiol. 2016, 7, 170. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, E.; Ilina, L.; Laptev, G.; Filippova, V.; Brazhnik, E.; Dunyashev, T.; Dubrovin, A.; Novikova, N.; Tiurina, D.; Tarlavin, N.; et al. The structure and functional profile of ruminal microbiota in young and adult reindeers (Rangifer tarandus) consuming natural winter-spring and summer-autumn seasonal diets. PeerJ 2021, 9, e12389. [Google Scholar] [CrossRef]
- Liu, W.; Wang, Q.; Song, J.; Xin, J.; Zhang, S.; Lei, Y.; Yang, Y.; Xie, P.; Suo, H. Comparison of gut microbiota of yaks from different geographical regions. Front. Microbiol. 2021, 12, 666940. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Zhou, Z.; Guan, J.; Xia, B.; Luo, X.; Yang, Y.; Fu, Y.; Sun, Q. Dynamic changes of yak (Bos grunniens) gut microbiota during growth revealed by polymerase chain reaction-denaturing gradient gel electrophoresis and metagenomics. Asian-Australas. J. Anim. Sci. (AJAS) 2017, 30, 957–966. [Google Scholar] [CrossRef][Green Version]
- Su, Y.; Su, J.; Li, F.; Tian, X.; Liu, Z.; Ding, G.; Bai, J.; Li, Z.; Ma, Z.; Peppelenbosch, M.P. Yak gut microbiota: A systematic review and meta-analysis. Front. Vet. Sci. 2022, 9, 889594. [Google Scholar] [CrossRef] [PubMed]
- Mtshali, K.; Khumalo, Z.T.H.; Kwenda, S.; Arshad, I.; Thekisoe, O.M.M. Exploration and comparison of bacterial communities present in bovine faeces, milk and blood using 16S rRNA metagenomic sequencing. PLoS ONE 2022, 17, e0273799. [Google Scholar] [CrossRef]
- Wu, Z.-L.; Wei, R.; Tan, X.; Yang, D.; Liu, D.; Zhang, J.; Wang, W. Characterization of gut microbiota dysbiosis of diarrheic adult yaks through 16S rRNA gene sequences. Front. Vet. Sci. 2022, 9, 946906. [Google Scholar] [CrossRef]
- Shang, Q.; Shan, X.; Cai, C.; Hao, J.; Li, G.; Yu, G. Dietary fucoidan modulates the gut microbiota in mice by increasing the abundance of Lactobacillus and Ruminococcaceae. Food Funct. 2016, 7, 3224–3232. [Google Scholar] [CrossRef]
- Tanca, A.; Fraumene, C.; Manghina, V.; Palomba, A.; Abbondio, M.; Deligios, M.; Pagnozzi, D.; Addis, M.F.; Uzzau, S. Diversity and functions of the sheep faecal microbiota: A multi-omic characterization. Microb. Biotechnol. 2017, 10, 541–554. [Google Scholar] [CrossRef]
- Chang, J.; Yao, X.; Zuo, C.; Qi, Y.; Chen, D.; Ma, W. The gut bacterial diversity of sheep associated with different breeds in Qinghai province. BMC Vet. Res. 2020, 16, 254. [Google Scholar] [CrossRef]
- Peng, S.; Yin, J.; Liu, X.; Jia, B.; Chang, Z.; Lu, H.; Jiang, N.; Chen, Q. First insights into the microbial diversity in the omasum and reticulum of bovine using Illumina sequencing. J. Appl. Genet. 2015, 56, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Tomšič, B.; Simončič, B.; Orel, B.; Vilčnik, A.; Spreizer, H. Biodegradability of cellulose fabric modified by imidazolidinone. Carbohyd. Polym. 2007, 69, 478–488. [Google Scholar] [CrossRef]
- Shabana, I.I.; Albakri, N.N.; Bouqellah, N.A. Metagenomic investigation of faecal microbiota in sheep and goats of the same ages. J. Taibah Univ. Sci. 2021, 15, 1–9. [Google Scholar] [CrossRef]
- Xiao, H.; Yan, H.; Tian, P.; Ji, S.; Zhao, W.; Lu, C.; Zhang, Y.; Liu, Y. The effect of early colonized gut microbiota on the growth performance of suckling lambs. Front. Microbiol. 2023, 14, 1273444. [Google Scholar] [CrossRef] [PubMed]
- Derrien, M.; Vaughan, E.E.; Plugge, C.M.; de Vos, W.M. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int. J. Syst. Evol. Microbiol. 2004, 54, 1469–1476. [Google Scholar] [CrossRef]
- Derrien, M.; Van Baarlen, P.; Hooiveld, G.; Norin, E.; Müller, M.; de Vos, W.M. Modulation of mucosal immune response, tolerance, and proliferation in mice colonized by the mucin-degrader Akkermansia muciniphila. Front. Microbiol. 2011, 1, 166. [Google Scholar] [CrossRef]
- Youngblut, N.D.; Reischer, G.H.; Dauser, S.; Maisch, S.; Walzer, C.; Stalder, G.; Farnleitner, A.H.; Ley, R.E. Vertebrate host phylogeny influences gut archaeal diversity. Nat. Microbiol. 2021, 6, 1443–1454. [Google Scholar] [CrossRef]
- Carmody, R.N.; Gerber, G.K.; Luevano, J.M., Jr.; Gatti, D.M.; Somes, L.; Svenson, K.L.; Turnbaugh, P.J. Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe 2015, 17, 72–84. [Google Scholar] [CrossRef]
- Pérez-Cobas, A.E.; Maiques, E.; Angelova, A.; Carrasco, P.; Moya, A.; Latorre, A. Diet shapes the gut microbiota of the omnivorous cockroach Blattella germanica. FEMS Microbiol. Ecol. 2015, 91, fiv022. [Google Scholar] [CrossRef]
- Tap, J.; Furet, J.P.; Bensaada, M.; Philippe, C.; Roth, H.; Rabot, S.; Lakhdari, O.; Lombard, V.; Henrissat, B.; Corthier, G.; et al. Gut microbiota richness promotes its stability upon increased dietary fibre intake in healthy adults. Environ. Microbiol. 2015, 17, 4954–4964. [Google Scholar] [CrossRef]
- Zhang, C.; Li, S.; Yang, L.; Huang, P.; Li, W.; Wang, S.; Zhao, G.; Zhang, M.; Pang, X.; Yan, Z.; et al. Structural modulation of gut microbiota in life-long calorie-restricted mice. Nat. Commun. 2013, 4, 2163. [Google Scholar] [CrossRef]
- Gebert, C.; Verheyden-Tixier, H. Variations of diet composition of Red Deer (Cervus elaphus L.) in Europe. Mammal Rev. 2001, 31, 189–201. [Google Scholar] [CrossRef]
- Bjørkvoll, E.; Pedersen, B.; Hytteborn, H.; Jónsdóttir, I.S.; Langvatn, R. Seasonal and interannual dietary variation during winter in female Svalbard reindeer (Rangifer tarandus platyrhynchus). Arct. Antarct. Alp. Res. 2009, 41, 88–96. [Google Scholar] [CrossRef]
- Hu, C.; Ding, L.; Jiang, C.; Ma, C.; Liu, B.; Li, D.; Degen, A.A. Effects of management, dietary intake, and genotype on rumen morphology, fermentation, and microbiota, and on meat quality in yaks and cattle. Front. Nutr. 2021, 8, 755255. [Google Scholar] [CrossRef]
- Tsevegemed, M.; Norovsambuu, T.; Jordan, G.; Schlecht, E. Feed intake of small ruminants on spring and summer pastures in the Mngolian Altai mountains. Sustainability 2019, 11, 5759. [Google Scholar] [CrossRef]
- Kazmin, V.; Abaturov, B.; Demina, O.; Kolesnikov, M. Food resources and nutrition of semi-free bison (Bison Bison) on the steppe pasture of the Western Manych valley. Zool. J. 2016, 95, 234–244. [Google Scholar]
- Wei, X.; Dong, Z.; Cheng, F.; Shi, H.; Zhou, X.; Li, B.; Wang, L.; Wang, W.; Zhang, J. Seasonal diets supersede host species in shaping the distal gut microbiota of Yaks and Tibetan sheep. Sci. Rep. 2021, 11, 22626. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Animal | Sampling Place | GPS Location N/E | Altitude (m) | Habitat | Monthly Average Temperature, March (°C) | Number of Individuals (N) * | Age (Years) | Diet Preference (Geobotanical Descriptions) [31] |
|---|---|---|---|---|---|---|---|---|
| Deer | Russia, Republic of Buryatia, Onot area, deer camp | 52.1217 N 101.2553 E | 1762 | Mountain taiga | −10.2 | 20 | 1–9 | Rheum rhubarb, Cetraria islandica (L.) Ach., Cetraria laevigata Rassad., Cladonia amaurocraea (Florke) Schaer, Cladonia arbuscula (Wallr.) Flot., Carex juncella (E. Fries) T. Fries, Agropyron cristatum, Eriophorum polystachion L. |
| Yak | Russia, Republic of Buryatia, Bokson village, livestock camp | 52.0939 N 100.9503 E | 1360 | High mountainous areas and the valleys | −11.4 | 23 | 2–7 | Aster alpinus, Thermopsis lanceolata, Potentilla anserina L., Helictotrichon altaicum Tzvelev, Allium splendens Willd. ex Schultes et Schultes fil., Polygonum viviparum L., Potentilla bifurca L. |
| Sheep | Russia, Republic of Buryatia, Dabatui area | 50.7888 N 107.9373 E | 749 | Forest-steppe | −7.4 | 29 | 2–3 | Aster alpinus L, Stipa capillata L., Stipa krylovii, Dontostemon integrifolius, Potentilla bifurca L., Carex duriuscula, Pulsatilla turczaninovii Krylov et Serg. |
| Camel | Russia, Zabaikalsky Krai, Khapshur area | 51.4356 N 115.3508 E | 687 | Steppe | −8.1 | 24 | 2–10 | Allium anisopodium, Saposhnikovia divaricata, Aster alpinus, Saussurea salicifolia. Stipa krylovii Roshev, Carex duriuscula C.A.Meyer, Leonurus L. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lavrentyeva, E.; Banzaraktsaeva, T.; Kozyreva, L.; Danilova, E.; Tsyrenova, D.; Dambaev, V.; Buryukhaev, S.; Abidueva, E.; Begmatov, S.; Mardanov, A.; et al. Fecal Microbiota and Feeding Habitats of Nomadic Indigenous Animals (Deer, Yak, Sheep and Camel) in Baikal Siberia (Russia). Diversity 2024, 16, 52. https://doi.org/10.3390/d16010052
Lavrentyeva E, Banzaraktsaeva T, Kozyreva L, Danilova E, Tsyrenova D, Dambaev V, Buryukhaev S, Abidueva E, Begmatov S, Mardanov A, et al. Fecal Microbiota and Feeding Habitats of Nomadic Indigenous Animals (Deer, Yak, Sheep and Camel) in Baikal Siberia (Russia). Diversity. 2024; 16(1):52. https://doi.org/10.3390/d16010052
Chicago/Turabian StyleLavrentyeva, Elena, Tuyana Banzaraktsaeva, Lyudmila Kozyreva, Erzhena Danilova, Dulma Tsyrenova, Vyacheslav Dambaev, Savelii Buryukhaev, Elena Abidueva, Shahjahon Begmatov, Andrey Mardanov, and et al. 2024. "Fecal Microbiota and Feeding Habitats of Nomadic Indigenous Animals (Deer, Yak, Sheep and Camel) in Baikal Siberia (Russia)" Diversity 16, no. 1: 52. https://doi.org/10.3390/d16010052
APA StyleLavrentyeva, E., Banzaraktsaeva, T., Kozyreva, L., Danilova, E., Tsyrenova, D., Dambaev, V., Buryukhaev, S., Abidueva, E., Begmatov, S., Mardanov, A., & Barkhutova, D. D. (2024). Fecal Microbiota and Feeding Habitats of Nomadic Indigenous Animals (Deer, Yak, Sheep and Camel) in Baikal Siberia (Russia). Diversity, 16(1), 52. https://doi.org/10.3390/d16010052