
Molecular Taxonomy of South Africa’s Catsharks: How Far Have We Come?

 , , , , , ,
, , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction, Amplification, and Sequencing
2.3. Genetic Data Analyses
2.4. Species Delimitation Analyses
2.5. Specimen Assignment Analyses
3. Results
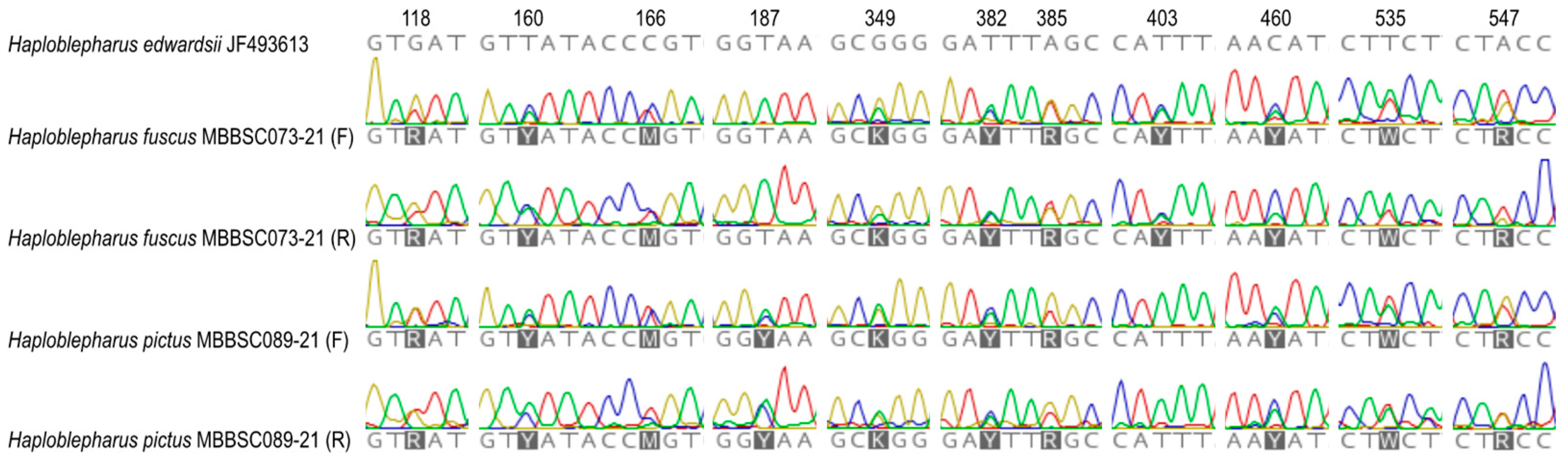
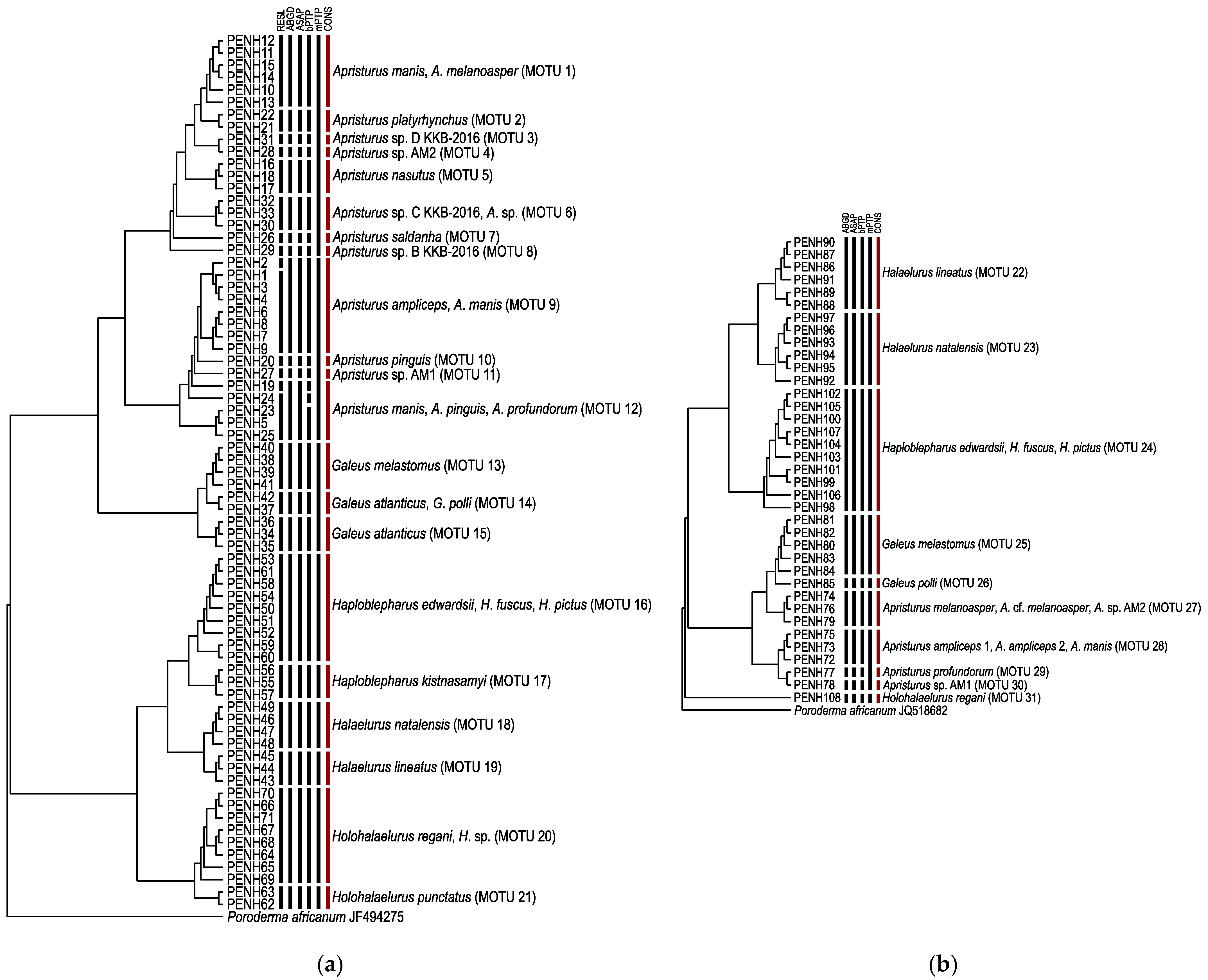
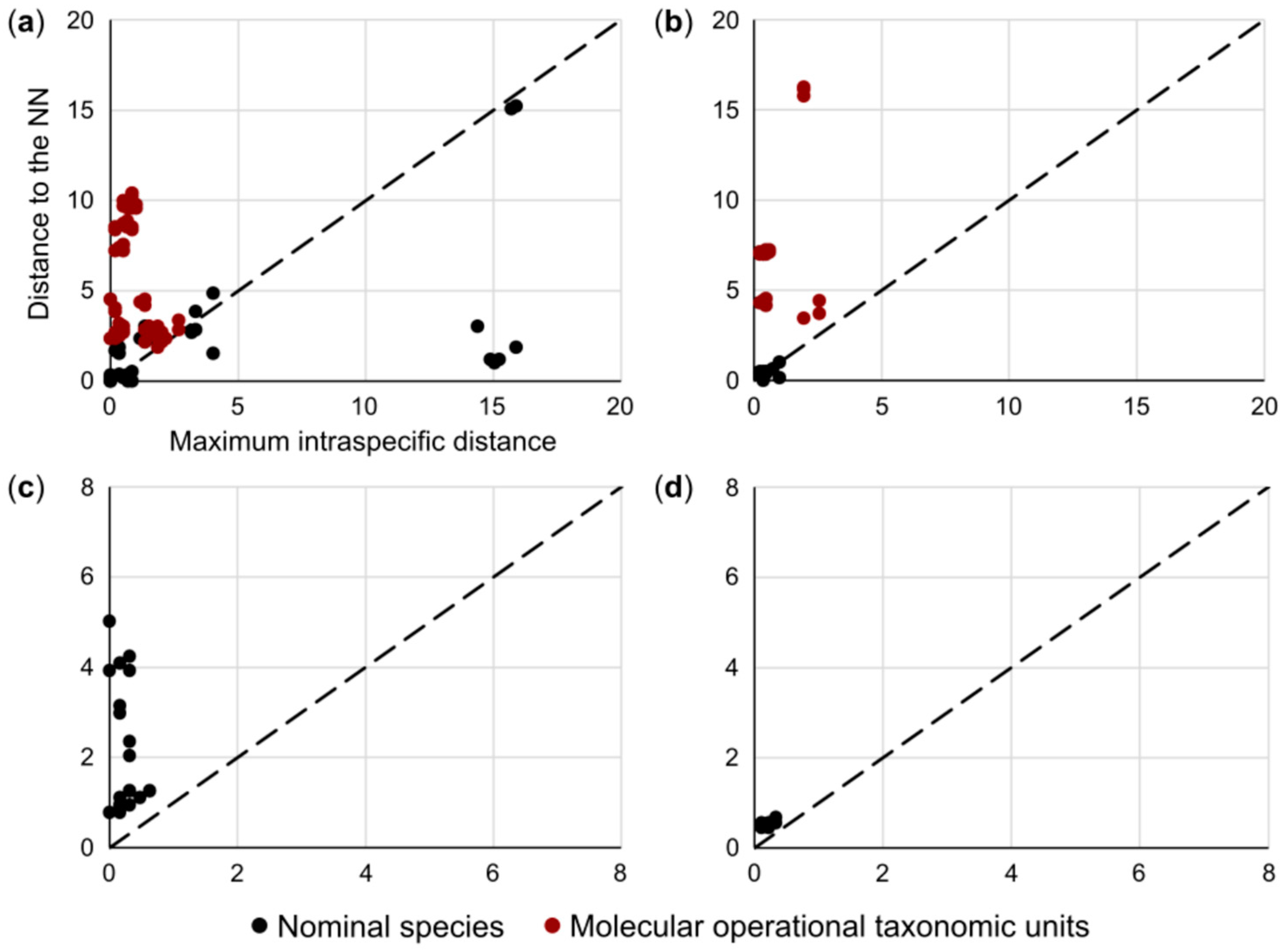
3.1. Species Delimitation and Specimen Assignment Analyses within the Family Pentanchidae
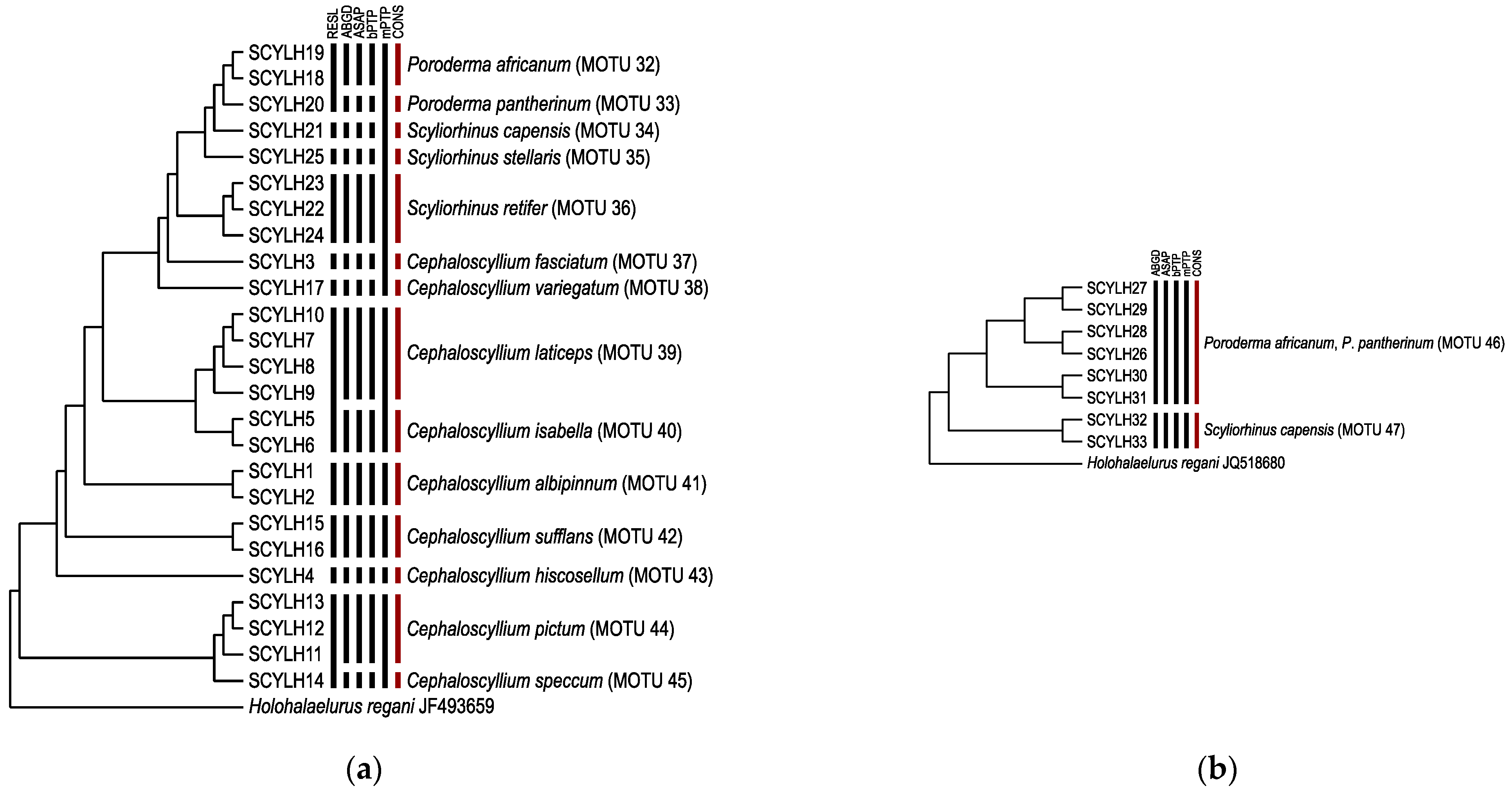
3.2. Species Delimitation and Specimen Assignment Analyses within the Family Scyliorhinidae
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weigmann, S. Annotated Checklist of the Living Sharks, Batoids and Chimaeras (Chondrichthyes) of the World, with a Focus on Biogeographical Diversity. J. Fish Biol. 2016, 88, 837–1037. [Google Scholar] [CrossRef] [PubMed]
- Ebert, D.A.; Dando, M.; Fowler, S. Sharks of the World: A Complete Guide; Princeton University Press: Princeton, NJ, USA, 2021; ISBN 978-0-691-20599-1. [Google Scholar]
- Fricke, R.; Eschmeyer, W.N.; Van der Laan, R. Eschmeyer’s Catalog of Fishes: Genera, Species, References. Available online: https://researcharchive.calacademy.org/research/ichthyology/catalog/fishcatmain.asp (accessed on 14 June 2023).
- Compagno, L.J.V. Sharks of the Order Carcharhiniformes; Princeton University Press: Princeton, NJ, USA, 1988; ISBN 0-691-08453-X. [Google Scholar]
- De Carvalho, M.R. Higher-level Elasmobranch Phylogeny, Basal Squaleans, and Paraphyly. In Interrelationships of Fishes; Stiassny, M.L.J., Parenti, L.R., Johnson, G.D., Eds.; Academic Press: New York, NY, USA, 1996; pp. 35–62. ISBN 978-0-12-670950-6. [Google Scholar]
- Springer, S. A Revision of the Catsharks, Family Scyliorhinidae; NOAA Technical Report NMFS Circular; U.S. Department of Commerce: Washington, DC, USA, 1979; Volume 422, pp. 1–152.
- Soares, K.D.A.; Mathubara, K. Combined Phylogeny and New Classification of Catsharks (Chondrichthyes: Elasmobranchii: Carcharhiniformes). Zool. J. Linn. Soc. 2022, 195, 761–814. [Google Scholar] [CrossRef]
- Stein, R.W.; Mull, C.G.; Kuhn, T.S.; Aschliman, N.C.; Davidson, L.N.K.; Joy, J.B.; Smith, G.J.; Dulvy, N.K.; Mooers, A.O. Global Priorities for Conserving the Evolutionary History of Sharks, Rays and Chimaeras. Nat. Ecol. Evol. 2018, 2, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Derrick, D.H.; Cheok, J.; Dulvy, N.K. Spatially Congruent Sites of Importance for Global Shark and Ray Biodiversity. PLoS ONE 2020, 15, e0235559. [Google Scholar] [CrossRef]
- Davidson, L.N.K.; Dulvy, N.K. Global Marine Protected Areas to Prevent Extinctions. Nat. Ecol. Evol. 2017, 1, 0040. [Google Scholar] [CrossRef]
- Davies, K.F.; Margules, C.R.; Lawrence, J.F. A Synergistic Effect Puts Rare, Specialized Species at a Greater Risk of Extinction. Ecology 2004, 85, 265–271. [Google Scholar] [CrossRef]
- Ebert, D.A.; Wintner, S.P.; Kyne, P.M. An Annotated Checklist of the Chondrichthyans of South Africa. Zootaxa 2021, 4947, 1–127. [Google Scholar] [CrossRef]
- da Silva, C.; Booth, A.J.; Dudley, S.F.J.; Kerwath, S.E.; Lamberth, S.J.; Leslie, R.W.; McCord, M.E.; Sauer, W.H.H.; Zweig, T. The Current Status and Management of South Africa’s Chondrichthyan Fisheries. Afr. J. Mar. Sci. 2015, 37, 233–248. [Google Scholar] [CrossRef]
- The IUCN Red List of Threatened Species. Version 2022-2. Available online: https://www.iucnredlist.org (accessed on 5 February 2023).
- Dulvy, N.K.; Pacoureau, N.; Rigby, C.L.; Pollom, R.A.; Jabado, R.W.; Ebert, D.A.; Finucci, B.; Pollock, C.M.; Cheok, J.; Derrick, D.H.; et al. Overfishing Drives over One-Third of All Sharks and Rays toward a Global Extinction Crisis. Curr. Biol. 2021, 31, 4773–4787. [Google Scholar] [CrossRef] [PubMed]
- Human, B.A. A Taxonomic Revision of the Catshark Genus Haploblepharus Garman 1913 (Chondrichthyes: Carcharhiniformes: Scyliorhinidae). Zootaxa 2007, 1451, 1–40. [Google Scholar] [CrossRef]
- Human, B.A.; Compagno, L.J.V. Description of Haploblepharus kistnasamyi, a New Catshark (Chondrichthyes: Scyliorhinidae) from South Africa. Zootaxa 2006, 1318, 41–58. [Google Scholar] [CrossRef]
- Human, B.A. Size-Corrected Shape Variation Analysis and Quantitative Species Discrimination in a Morphologically Conservative Catshark Genus, Haploblepharus Garman, 1913 (Chondrichthyes: Carcharhiniformes: Scyliorhinidae). Afr. Nat. Hist. 2007, 3, 59–73. [Google Scholar]
- van Staden, M.; Gledhill, K.S.; Gennari, E.; McCord, M.E.; Parkinson, M.; Watson, R.G.A.; Rhode, C.; Bester-van der Merwe, A.E. Microsatellite Development and Detection of Admixture among Three Sympatric Haploblepharus Species (Carcharhiniformes: Scyliorhinidae). Aquat. Conserv. 2020, 30, 2336–2350. [Google Scholar] [CrossRef]
- Abercrombie, D.L.; Clarke, S.C.; Shivji, M.S. Global-Scale Genetic Identification of Hammerhead Sharks: Application to Assessment of the International Fin Trade and Law Enforcement. Conserv. Genet. 2005, 6, 775–788. [Google Scholar] [CrossRef]
- Dayrat, B. Towards Integrative Taxonomy. Biol. J. Linn. 2005, 85, 407–417. [Google Scholar] [CrossRef]
- Cerutti-Pereyra, F.; Meekan, M.G.; Wei, N.-W.V.; O’Shea, O.; Bradshaw, C.J.A.; Austin, C.M. Identification of Rays through DNA Barcoding: An Application for Ecologists. PLoS ONE 2012, 7, e36479. [Google Scholar] [CrossRef]
- Pank, M.; Stanhope, M.; Natanson, L.; Kohler, N.; Shivji, M. Rapid and Simultaneous Identification of Body Parts from the Morphologically Similar Sharks Carcharhinus obscurus and Carcharhinus plumbeus (Carcharhinidae) Using Multiplex PCR. Mar. Biotechnol. 2001, 3, 231–240. [Google Scholar] [CrossRef]
- Morgan, J.A.T.; Harry, A.V.; Welch, D.J.; Street, R.; White, J.; Geraghty, P.T.; Macbeth, W.G.; Tobin, A.; Simpfendorfer, C.A.; Ovenden, J.R. Detection of Interspecies Hybridisation in Chondrichthyes: Hybrids and Hybrid Offspring between Australian (Carcharhinus tilstoni) and Common (C. limbatus) Blacktip Shark Found in an Australian Fishery. Conserv. Genet. 2012, 13, 455–463. [Google Scholar] [CrossRef]
- Nakaya, K.; Kawauchi, J. A Review of the Genus Apristurus (Chondrichthyes: Carcharhiniformes: Scyliorhinidae) from Taiwanese Waters. Zootaxa 2013, 3752, 130–171. [Google Scholar] [CrossRef] [PubMed]
- Steinke, D.; Connell, A.D.; Hebert, P.D.N. Linking Adults and Immatures of South African Marine Fishes. Genome 2016, 59, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Blaxter, M. Counting Angels with DNA. Nature 2003, 421, 122–123. [Google Scholar] [CrossRef]
- Tautz, D.; Arctander, P.; Minelli, A.; Thomas, R.H.; Vogler, A.P. A Plea for DNA Taxonomy. Trends Ecol. Evol. 2003, 18, 70–74. [Google Scholar] [CrossRef]
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; deWaard, J.R. Biological Identifications through DNA Barcodes. Proc. R. Soc. B 2003, 270, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Hebert, P.D.N.; Stoeckle, M.Y.; Zemlak, T.S.; Francis, C.M. Identification of Birds through DNA Barcodes. PLoS Biol. 2004, 2, e312. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.D.; Hanner, R.; Hebert, P.D.N. The Campaign to DNA Barcode All Fishes, FISH-BOL. J. Fish Biol. 2009, 74, 329–356. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.B.M.; White, W.T.; Ward, R.D.; Naylor, G.J.P.; Peirce, R. Rediscovery and Redescription of the Smoothtooth Blacktip Shark, Carcharhinus leiodon (Carcharhinidae), from Kuwait, with Notes on Its Possible Conservation Status. Mar. Freshw. Res. 2011, 62, 528–539. [Google Scholar] [CrossRef]
- Hara, Y.; Yamaguchi, K.; Onimaru, K.; Kadota, M.; Koyanagi, M.; Keeley, S.D.; Tatsumi, K.; Tanaka, K.; Motone, F.; Kageyama, Y.; et al. Shark Genomes Provide Insights into Elasmobranch Evolution and the Origin of Vertebrates. Nat. Ecol. Evol. 2018, 2, 1761–1771. [Google Scholar] [CrossRef]
- Ward, R.D.; Holmes, B.H.; White, W.T.; Last, P.R. DNA Barcoding Australasian Chondrichthyans: Results and Potential Uses in Conservation. Mar. Freshw. Res. 2008, 59, 57–71. [Google Scholar] [CrossRef]
- Ferrari, A.; Di Crescenzo, S.; Cariani, A.; Crobe, V.; Benvenuto, A.; Piattoni, F.; Mancusi, C.; Bonnici, L.; Bonello, J.J.; Schembri, P.J.; et al. Puzzling over Spurdogs: Molecular Taxonomy Assessment of the Squalus Species in the Strait of Sicily. Eur. Zool. J. 2021, 88, 181–190. [Google Scholar] [CrossRef]
- Naylor, G.J.P.; Caira, J.N.; Jensen, K.; Rosana, K.A.M.; White, W.T.; Last, P.R. A DNA Sequence—Based Approach to the Identification of Shark and Ray Species and Its Implications for Global Elasmobranch Diversity and Parasitology. Bull. Am. Mus. Nat. Hist. 2012, 367, 1–262. [Google Scholar] [CrossRef]
- Giles, J.L.; Riginos, C.; Naylor, G.J.P.; Dharmadi; Ovenden, J.R. Genetic and Phenotypic Diversity in the Wedgefish Rhynchobatus australiae, a Threatened Ray of High Value in the Shark Fin Trade. Mar. Ecol. Prog. Ser. 2016, 548, 165–180. [Google Scholar] [CrossRef]
- Henderson, A.C.; Reeve, A.J.; Jabado, R.W.; Naylor, G.J.P. Taxonomic Assessment of Sharks, Rays and Guitarfishes (Chondrichthyes: Elasmobranchii) from South-Eastern Arabia, Using the NADH Dehydrogenase Subunit 2 (NADH2) Gene. Zool. J. Linn. Soc. 2016, 176, 399–442. [Google Scholar] [CrossRef]
- Fernando, D.; Bown, R.M.K.; Tanna, A.; Gobiraj, R.; Ralicki, H.; Jockusch, E.L.; Ebert, D.A.; Jensen, K.; Caira, J.N. New Insights into the Identities of the Elasmobranch Fauna of Sri Lanka. Zootaxa 2019, 4585, 201–238. [Google Scholar] [CrossRef] [PubMed]
- Petean, F.F.; Naylor, G.J.P.; Lima, S.M.Q. Integrative Taxonomy Identifies a New Stingray Species of the Genus Hypanus Rafinesque, 1818 (Dasyatidae, Myliobatiformes), from the Tropical Southwestern Atlantic. J. Fish Biol. 2020, 97, 1120–1142. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.D.J.; Collins, R.A.; Boyer, S.; Lefort, M.-C.; Malumbres-Olarte, J.; Vink, C.J.; Cruickshank, R.H. SPIDER: An R Package for the Analysis of Species Identity and Evolution, with Particular Reference to DNA Barcoding. Mol. Ecol. Resour. 2012, 12, 562–565. [Google Scholar] [CrossRef]
- Puillandre, N.; Lambert, A.; Brouillet, S.; Achaz, G. ABGD, Automatic Barcode Gap Discovery for Primary Species Delimitation. Mol. Ecol. 2012, 21, 1864–1877. [Google Scholar] [CrossRef]
- Ratnasingham, S.; Hebert, P.D.N. A DNA-Based Registry for All Animal Species: The Barcode Index Number (BIN) System. PLoS ONE 2013, 8, e66213. [Google Scholar] [CrossRef]
- Dellicour, S.; Flot, J.-F. The Hitchhiker’s Guide to Single-Locus Species Delimitation. Mol. Ecol. Resour. 2018, 18, 1234–1246. [Google Scholar] [CrossRef]
- Zhang, A.-B.; Muster, C.; Liang, H.-B.; Zhu, C.-D.; Crozier, R.; Wan, P.; Feng, J.; Ward, R.D. A Fuzzy-Set-Theory-Based Approach to Analyse Species Membership in DNA Barcoding. Mol. Ecol. 2012, 21, 1848–1863. [Google Scholar] [CrossRef]
- Ratnasingham, S.; Hebert, P.D.N. BOLD: The Barcode of Life Data System (http://www.barcodinglife.org). Mol. Ecol. Notes 2007, 7, 355–364. [Google Scholar] [CrossRef]
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Pruitt, K.D.; Schoch, C.L.; Sherry, S.T.; Karsch-Mizrachi, I. GenBank. Nucleic Acids Res. 2022, 50, D161–D164. [Google Scholar] [CrossRef]
- Hubert, N.; Delrieu-Trottin, E.; Irisson, J.-O.; Meyer, C.; Planes, S. Identifying Coral Reef Fish Larvae through DNA Barcoding: A Test Case with the Families Acanthuridae and Holocentridae. Mol. Phylogenet. Evol. 2010, 55, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Hubert, N.; Hanner, R. DNA Barcoding, Species Delineation and Taxonomy: A Historical Perspective. DNA Barcodes 2015, 3, 44–58. [Google Scholar] [CrossRef]
- Delrieu-Trottin, E.; Durand, J.-D.; Limmon, G.; Sukmono, T.; Kadarusman; Sugeha, H.Y.; Chen, W.-J.; Busson, F.; Borsa, P.; Dahruddin, H.; et al. Biodiversity Inventory of the Grey Mullets (Actinopterygii: Mugilidae) of the Indo-Australian Archipelago through the Iterative Use of DNA-based Species Delimitation and Specimen Assignment Methods. Evol. Appl. 2020, 13, 1451–1467. [Google Scholar] [CrossRef] [PubMed]
- Carugati, L.; Melis, R.; Cariani, A.; Cau, A.; Crobe, V.; Ferrari, A.; Follesa, M.C.; Geraci, M.L.; Iglésias, S.P.; Pesci, P.; et al. Combined COI Barcode-based Methods to Avoid Mislabeling of Threatened Species of Deep-sea Skates. Anim. Conserv. 2022, 25, 38–52. [Google Scholar] [CrossRef]
- Piemontese, L.; Giovannini, I.; Guidetti, R.; Pellegri, G.; Dioli, P.; Maistrello, L.; Rebecchi, L.; Cesari, M. The Species Identification Problem in Mirids (Hemiptera: Heteroptera) Highlighted by DNA Barcoding and Species Delimitation Studies. Eur. Zool. J. 2020, 87, 310–324. [Google Scholar] [CrossRef]
- Bellodi, A.; Benvenuto, A.; Melis, R.; Mulas, A.; Barone, M.; Barría, C.; Cariani, A.; Carugati, L.; Chatzispyrou, A.; Desrochers, M.; et al. Call Me by My Name: Unravelling the Taxonomy of the Gulper Shark Genus Centrophorus in the Mediterranean Sea through an Integrated Taxonomic Approach. Zool. J. Linn. Soc. 2022, 195, 815–840. [Google Scholar] [CrossRef]
- Compagno, L.J.V.; Ebert, D.A.; Smale, M.J. Guide to the Sharks and Rays of Southern Africa; Struik: Cape Town, South Africa, 1989; ISBN 978-0-86977-880-7. [Google Scholar]
- Dunlop, S.W.; Mann, B.Q.; Van Der Elst, R.P. A Review of the Oceanographic Research Institute’s Cooperative Fish Tagging Project: 27 Years down the Line. Afr. J. Mar. Sci. 2013, 35, 209–221. [Google Scholar] [CrossRef]
- Sambrook, J.F.; Russell, D.W. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001; ISBN 978-0-87969-577-4. [Google Scholar]
- Ward, R.D.; Zemlak, T.S.; Innes, B.H.; Last, P.R.; Hebert, P.D.N. DNA Barcoding Australia’s Fish Species. Philos. Trans. R. Soc. B 2005, 360, 1847–1857. [Google Scholar] [CrossRef]
- Ivanova, N.V.; Zemlak, T.S.; Hanner, R.H.; Hebert, P.D.N. Universal Primer Cocktails for Fish DNA Barcoding. Mol. Ecol. Notes 2007, 7, 544–548. [Google Scholar] [CrossRef]
- Naylor, G.J.P.; Ryburn, J.A.; Fedrigo, O.; Lopez, A. Phylogenetic Relationships among the Major Lineages of Modern Elasmobranchs. In Reproductive Biology and Phylogeny of Chondrichthyes: Sharks, Batoids, and Chimaeras; Hamlett, W.C., Ed.; CRC Press: Boca Raton, FL, USA, 2005; pp. 1–25. ISBN 978-0-429-06290-2. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Jukes, T.H.; Cantor, C.R. Evolution of Protein Molecules. In Mammalian Protein Metabolism; Munro, H.N., Ed.; Academic Press: New York, NY, USA, 1969; pp. 21–132. ISBN 978-1-4832-3211-9. [Google Scholar]
- Collins, R.A.; Cruickshank, R.H. The Seven Deadly Sins of DNA Barcoding. Mol. Ecol. Resour. 2013, 13, 969–975. [Google Scholar] [CrossRef] [PubMed]
- Moritz, C. Defining ‘Evolutionarily Significant Units’ for Conservation. Trends Ecol. Evol. 1994, 9, 373–375. [Google Scholar] [CrossRef]
- Vogler, A.P.; Desalle, R. Diagnosing Units of Conservation Management. Conserv. Biol. 1994, 8, 354–363. [Google Scholar] [CrossRef]
- Puillandre, N.; Brouillet, S.; Achaz, G. ASAP: Assemble Species by Automatic Partitioning. Mol. Ecol. Resour. 2021, 21, 609–620. [Google Scholar] [CrossRef]
- Zhang, J.; Kapli, P.; Pavlidis, P.; Stamatakis, A. A General Species Delimitation Method with Applications to Phylogenetic Placements. Bioinformatics 2013, 29, 2869–2876. [Google Scholar] [CrossRef]
- Kapli, P.; Lutteropp, S.; Zhang, J.; Kobert, K.; Pavlidis, P.; Stamatakis, A.; Flouri, T. Multi-Rate Poisson Tree Processes for Single-Locus Species Delimitation under Maximum Likelihood and Markov Chain Monte Carlo. Bioinformatics 2017, 33, 1630–1638. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Lefort, V.; Longueville, J.-E.; Gascuel, O. SMS: Smart Model Selection in PhyML. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef]
- Meier, R.; Shiyang, K.; Vaidya, G.; Ng, P.K.L. DNA Barcoding and Taxonomy in Diptera: A Tale of High Intraspecific Variability and Low Identification Success. Syst. Biol. 2006, 55, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.P.; Paulay, G. DNA Barcoding: Error Rates Based on Comprehensive Sampling. PLoS Biol. 2005, 3, e422. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.B.; Sikes, D.S.; Muster, C.; Li, S.Q. Inferring Species Membership Using DNA Sequences with Back-Propagation Neural Networks. Syst. Biol. 2008, 57, 202–215. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.-B.; Hao, M.-D.; Yang, C.-Q.; Shi, Z.-Y. BarcodingR: An Integrated R Package for Species Identification Using DNA Barcodes. Methods Ecol. Evol. 2017, 8, 627–634. [Google Scholar] [CrossRef]
- Zhang, A.-B.; Feng, J.; Ward, R.D.; Wan, P.; Gao, Q.; Wu, J.; Zhao, W.-Z. A New Method for Species Identification via Protein-Coding and Non-Coding DNA Barcodes by Combining Machine Learning with Bioinformatic Methods. PLoS ONE 2012, 7, e30986. [Google Scholar] [CrossRef] [PubMed]
- White, D.J.; Wolff, J.N.; Pierson, M.; Gemmell, N.J. Revealing the Hidden Complexities of MtDNA Inheritance. Mol. Ecol. 2008, 17, 4925–4942. [Google Scholar] [CrossRef]
- Iglésias, S.P.; Nakaya, K.; Stehmann, M. Apristurus melanoasper, a New Species of Deep-Water Catshark from the North Atlantic (Chondrichthyes: Carcharhiniformes: Scyliorhinidae). Cybium 2004, 28, 345–356. [Google Scholar] [CrossRef]
- De Astarloa, J.M.D.; Mabragaña, E.; Hanner, R.; Figueroa, D.E. Morphological and Molecular Evidence for a New Species of Longnose Skate (Rajiformes: Rajidae: Dipturus) from Argentinean Waters Based on DNA Barcoding. Zootaxa 2008, 1921, 35–46. [Google Scholar] [CrossRef]
- Ebert, D.A.; White, W.T.; Goldman, K.J.; Compagno, L.J.V.; Daly-Engel, T.S.; Ward, R.D. Resurrection and Redescription of Squalus suckleyi (Girard, 1854) from the North Pacific, with Comments on the Squalus acanthias Subgroup (Squaliformes: Squalidae). Zootaxa 2010, 2612, 22–40. [Google Scholar] [CrossRef]
- Viana, S.T.F.L.; De Carvalho, M.R. Resurrection and Redescription of the Southern Dogfish Squalus probatovi (Squalidae), a Valid Species from Angola. J. Ichthyol. 2018, 58, 617–632. [Google Scholar] [CrossRef]
- Cariani, A.; Messinetti, S.; Ferrari, A.; Arculeo, M.; Bonello, J.J.; Bonnici, L.; Cannas, R.; Carbonara, P.; Cau, A.; Charilaou, C.; et al. Improving the Conservation of Mediterranean Chondrichthyans: The ELASMOMED DNA Barcode Reference Library. PLoS ONE 2017, 12, e0170244. [Google Scholar] [CrossRef]
- Nakaya, K.; Sato, K.; Iglésias, S.P. Occurrence of Apristurus melanoasper from the South Pacific, Indian and South Atlantic Oceans (Carcharhiniformes: Scyliorhinidae). In Descriptions of New Australian Chondrichthyans; CSIRO Marine & Atmospheric Research Paper; CSIRO: Hobart, Australia, 2008; pp. 61–74. [Google Scholar]
- Ebert, D.A.; Fowler, S.L.; Compagno, L.J.V. Sharks of the World: A Fully Illustrated Guide; Wild Nature Press: Plymouth, UK, 2013; ISBN 978-0-9573946-0-5. [Google Scholar]
- Ahrens, D.; Fujisawa, T.; Krammer, H.-J.; Eberle, J.; Fabrizi, S.; Vogler, A.P. Rarity and Incomplete Sampling in DNA-Based Species Delimitation. Syst. Biol. 2016, 65, 478–494. [Google Scholar] [CrossRef]
- Human, B.A. A Taxonomic Revision of the Catshark Genus Poroderma Smith, 1837 (Chondrichthyes: Carcharhiniformes: Scyliorhinidae). Zootaxa 2006, 1229, 1–32. [Google Scholar] [CrossRef]
- Marino, I.A.M.; Finotto, L.; Colloca, F.; Di Lorenzo, M.; Gristina, M.; Farrell, E.D.; Zane, L.; Mazzoldi, C. Resolving the Ambiguities in the Identification of Two Smooth-Hound Sharks (Mustelus mustelus and Mustelus punctulatus) Using Genetics and Morphology. Mar. Biodivers. 2018, 48, 1551–1562. [Google Scholar] [CrossRef]
- Kashiwagi, T.; Marshall, A.D.; Bennett, M.B.; Ovenden, J.R. The Genetic Signature of Recent Speciation in Manta Rays (Manta alfredi and M. birostris). Mol. Phylogenet. Evol. 2012, 64, 212–218. [Google Scholar] [CrossRef]
- Dudu, A.; Georgescu, S.E.; Berrebi, P.; Costache, M. Site Heteroplasmy in the Mitochondrial Cytochrome b Gene of the Sterlet Sturgeon Acipenser ruthenus. Genet. Mol. Biol. 2012, 35, 886–891. [Google Scholar] [CrossRef] [PubMed]
- Just, R.S.; Irwin, J.A.; Parson, W. Mitochondrial DNA Heteroplasmy in the Emerging Field of Massively Parallel Sequencing. Forensic Sci. Int. Genet. 2015, 18, 131–139. [Google Scholar] [CrossRef]
- Cree, L.M.; Samuels, D.C.; De Sousa Lopes, S.C.; Rajasimha, H.K.; Wonnapinij, P.; Mann, J.R.; Dahl, H.-H.M.; Chinnery, P.F. A Reduction of Mitochondrial DNA Molecules during Embryogenesis Explains the Rapid Segregation of Genotypes. Nat. Genet. 2008, 40, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Khrapko, K. Two Ways to Make an MtDNA Bottleneck. Nat. Genet. 2008, 40, 134–135. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Liu, S.; Liu, Y. Evidence for Recombination of Mitochondrial DNA in Triploid Crucian Carp. Genetics 2006, 172, 1745–1749. [Google Scholar] [CrossRef] [PubMed]
- DeSalle, R.; Egan, M.G.; Siddall, M. The Unholy Trinity: Taxonomy, Species Delimitation and DNA Barcoding. Philos. Trans. R. Soc. B 2005, 360, 1905–1916. [Google Scholar] [CrossRef]
- Ebach, M.C.; Holdrege, C. DNA Barcoding Is No Substitute for Taxonomy. Nature 2005, 434, 697. [Google Scholar] [CrossRef]
- Blair, C.; Bryson, R.W. Cryptic Diversity and Discordance in Single-Locus Species Delimitation Methods within Horned Lizards (Phrynosomatidae: Phrynosoma). Mol. Ecol. Resour. 2017, 17, 1168–1182. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.H.; Shivji, M.S.; Hanner, R.H. Identifying Sharks with DNA Barcodes: Assessing the Utility of a Nucleotide diagnostic approach. Mol. Ecol. Resour. 2009, 9, 243–256. [Google Scholar] [CrossRef] [PubMed]
- van der Bank, H. DNA Barcoding Results for Some Southern African Elephantfish, Guitarfish, Rattails, Rays, Sharks and Skates. Int. J. Oceanogr. Aquac. 2019, 3, 000163. [Google Scholar] [CrossRef]
- McCusker, M.R.; Denti, D.; Van Guelpen, L.; Kenchington, E.; Bentzen, P. Barcoding Atlantic Canada’s Commonly Encountered Marine Fishes. Mol. Ecol. Resour. 2013, 13, 177–188. [Google Scholar] [CrossRef]
- Costa, F.O.; Landi, M.; Martins, R.; Costa, M.H.; Costa, M.E.; Carneiro, M.; Alves, M.J.; Steinke, D.; Carvalho, G.R. A Ranking System for Reference Libraries of DNA Barcodes: Application to Marine Fish Species from Portugal. PLoS ONE 2012, 7, e35858. [Google Scholar] [CrossRef]
- Landi, M.; Dimech, M.; Arculeo, M.; Biondo, G.; Martins, R.; Carneiro, M.; Carvalho, G.R.; Lo Brutto, S.; Costa, F.O. DNA Barcoding for Species Assignment: The Case of Mediterranean Marine Fishes. PLoS ONE 2014, 9, e106135. [Google Scholar] [CrossRef]
- Castilho, R.; Freitas, M.; Silva, G.; Fernandez-Carvalho, J.; Coelho, R. Morphological and Mitochondrial DNA Divergence Validates Blackmouth, Galeus melastomus, and Atlantic Sawtail Catsharks, Galeus atlanticus, as Separate Species. J. Fish Biol. 2007, 70, 346–358. [Google Scholar] [CrossRef]
- Kousteni, V.; Mazzoleni, S.; Vasileiadou, K.; Rovatsos, M. Complete Mitochondrial Genome of Nine Species of Sharks and Rays and Their Phylogenetic Placement among Modern Elasmobranchs. Genes 2021, 12, 324. [Google Scholar] [CrossRef]
- Stoeckle, M.Y.; Das Mishu, M.; Charlop-Powers, Z. Improved Environmental DNA Reference Library Detects Overlooked Marine Fishes in New Jersey, United States. Front. Mar. Sci. 2020, 7, 226. [Google Scholar] [CrossRef]
- Moftah, M.; Abdel Aziz, S.H.; Elramah, S.; Favereaux, A. Classification of Sharks in the Egyptian Mediterranean Waters Using Morphological and DNA Barcoding Approaches. PLoS ONE 2011, 6, e27001. [Google Scholar] [CrossRef] [PubMed]
- López, J.A.; Ryburn, J.A.; Fedrigo, O.; Naylor, G.J.P. Phylogeny of Sharks of the Family Triakidae (Carcharhiniformes) and its Implications for the Evolution of Carcharhiniform Placental Viviparity. Mol. Phylogenet. Evol. 2006, 40, 50–60. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Staden, M.; Ebert, D.A.; Gennari, E.; Leslie, R.W.; McCord, M.E.; Parkinson, M.; Watson, R.G.A.; Wintner, S.; da Silva, C.; Bester-van der Merwe, A.E. Molecular Taxonomy of South Africa’s Catsharks: How Far Have We Come? Diversity 2023, 15, 828. https://doi.org/10.3390/d15070828
van Staden M, Ebert DA, Gennari E, Leslie RW, McCord ME, Parkinson M, Watson RGA, Wintner S, da Silva C, Bester-van der Merwe AE. Molecular Taxonomy of South Africa’s Catsharks: How Far Have We Come? Diversity. 2023; 15(7):828. https://doi.org/10.3390/d15070828
Chicago/Turabian Stylevan Staden, Michaela, David A. Ebert, Enrico Gennari, Rob W. Leslie, Meaghen E. McCord, Matthew Parkinson, Ralph G. A. Watson, Sabine Wintner, Charlene da Silva, and Aletta E. Bester-van der Merwe. 2023. "Molecular Taxonomy of South Africa’s Catsharks: How Far Have We Come?" Diversity 15, no. 7: 828. https://doi.org/10.3390/d15070828
APA Stylevan Staden, M., Ebert, D. A., Gennari, E., Leslie, R. W., McCord, M. E., Parkinson, M., Watson, R. G. A., Wintner, S., da Silva, C., & Bester-van der Merwe, A. E. (2023). Molecular Taxonomy of South Africa’s Catsharks: How Far Have We Come? Diversity, 15(7), 828. https://doi.org/10.3390/d15070828