
Integrative Phylogeography Reveals Conservation Priorities for the Giant Anteater Myrmecophaga tridactyla in Brazil

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
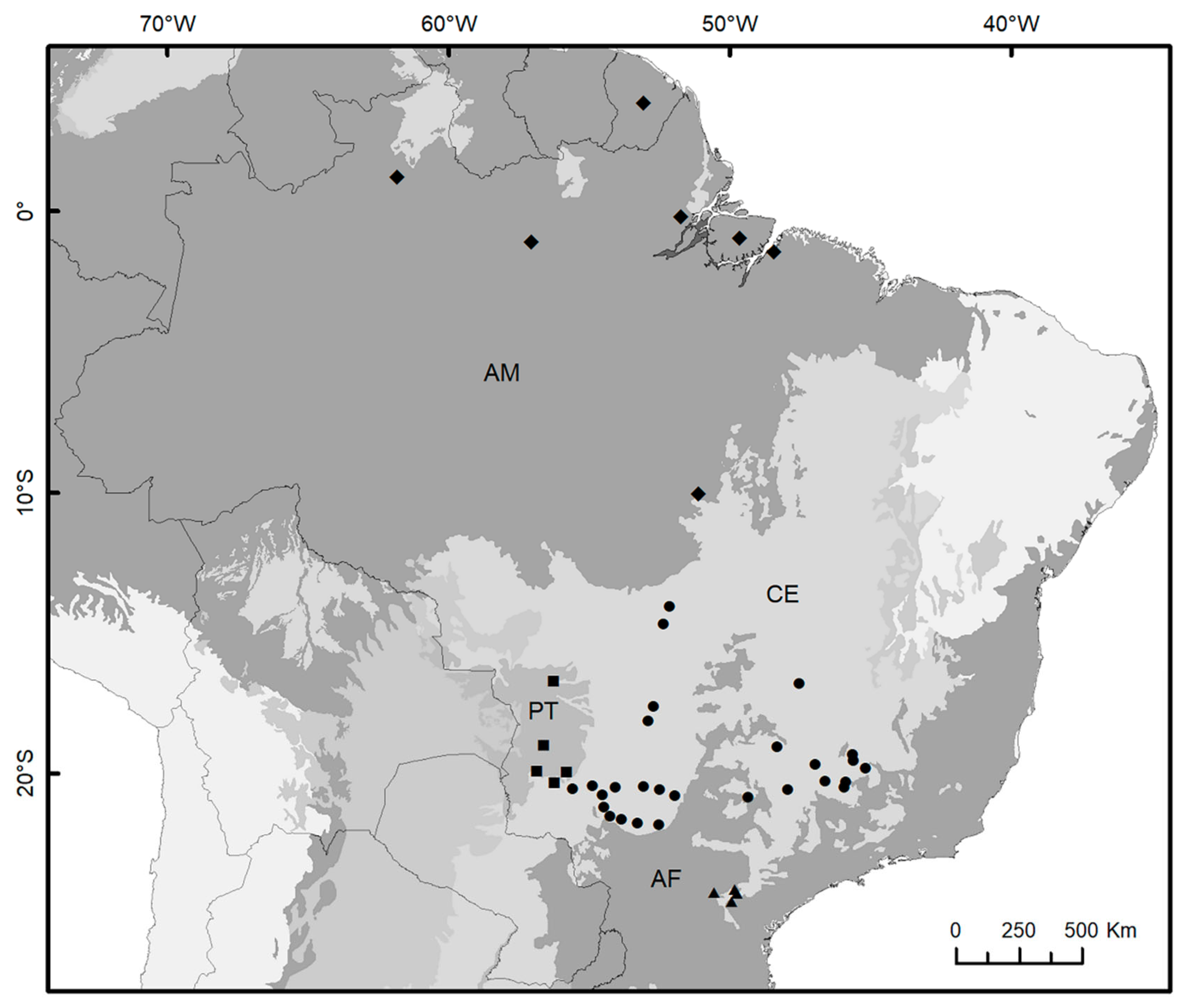
2.1. Biological Samples, DNA Data Obtention and Processing
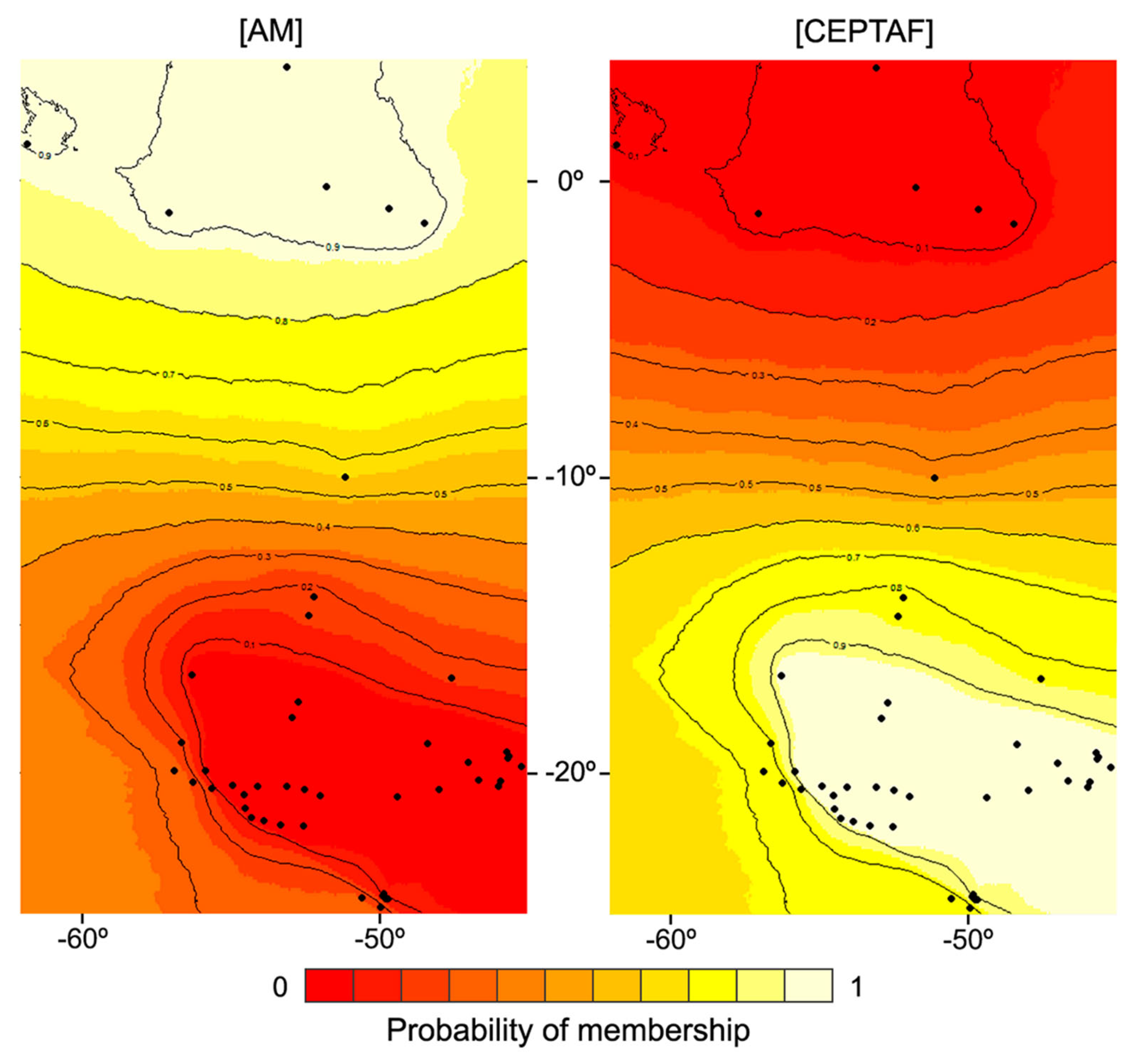
2.2. Assessing Spatial Genetic Patterns and Gene Flow
2.3. Historical Demography Reconstruction
2.4. Ecological Niche Models (ENMs)
3. Results
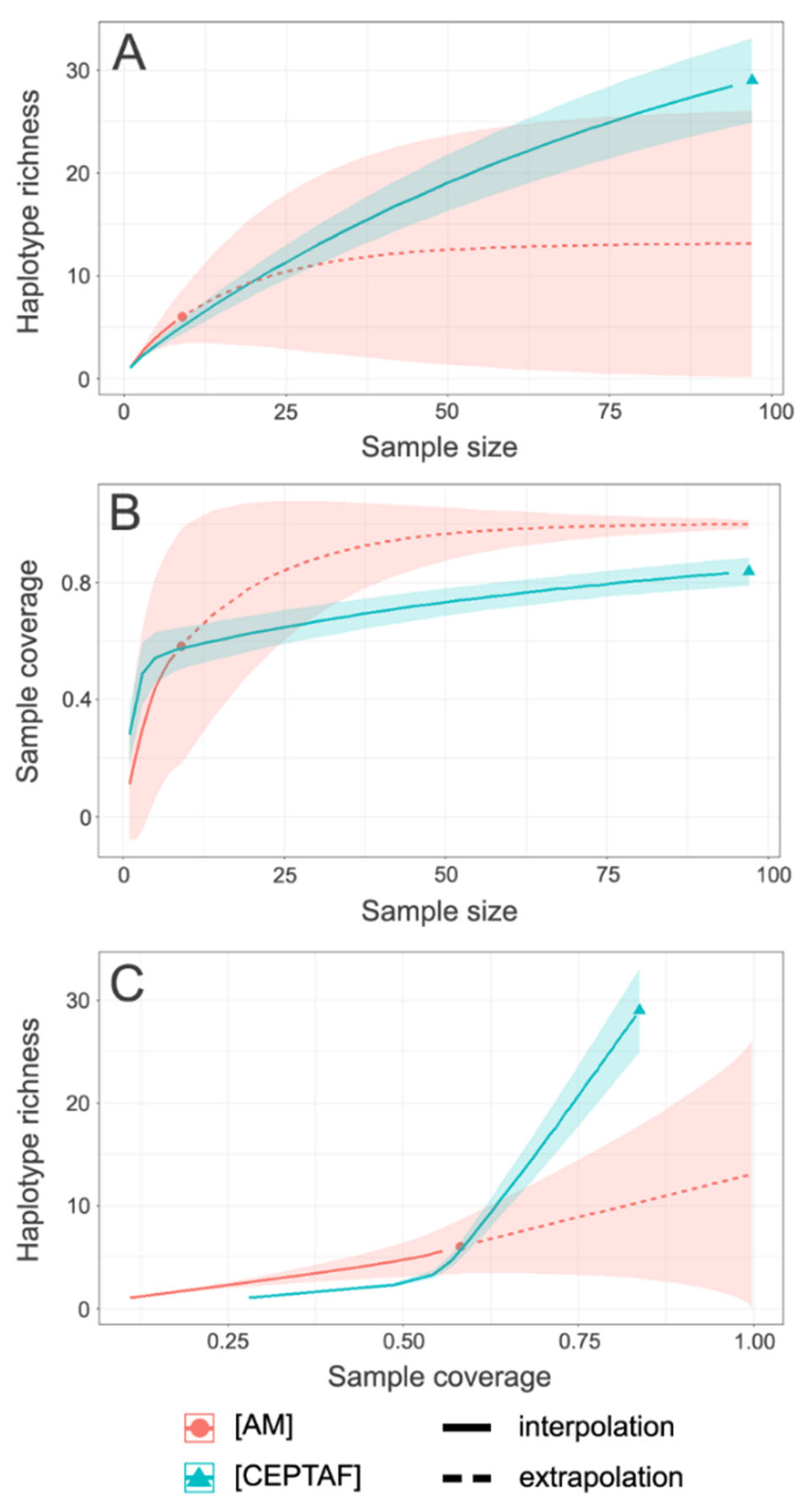
3.1. Sequencing and Haplotype Phasing
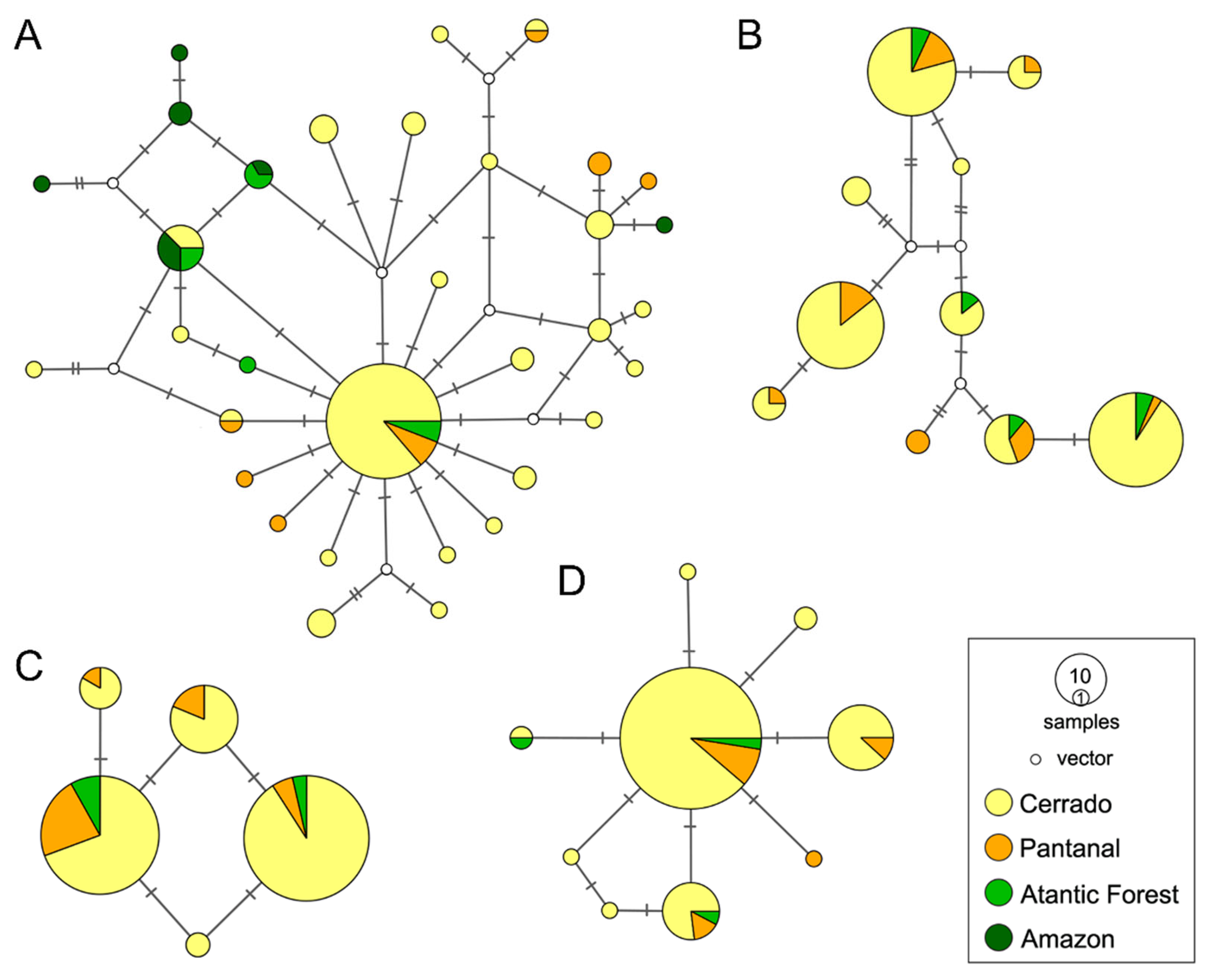
3.2. Spatial Patterns, Genetic Diversity Characterization, and Evidence for Unidirectional Gene Flow
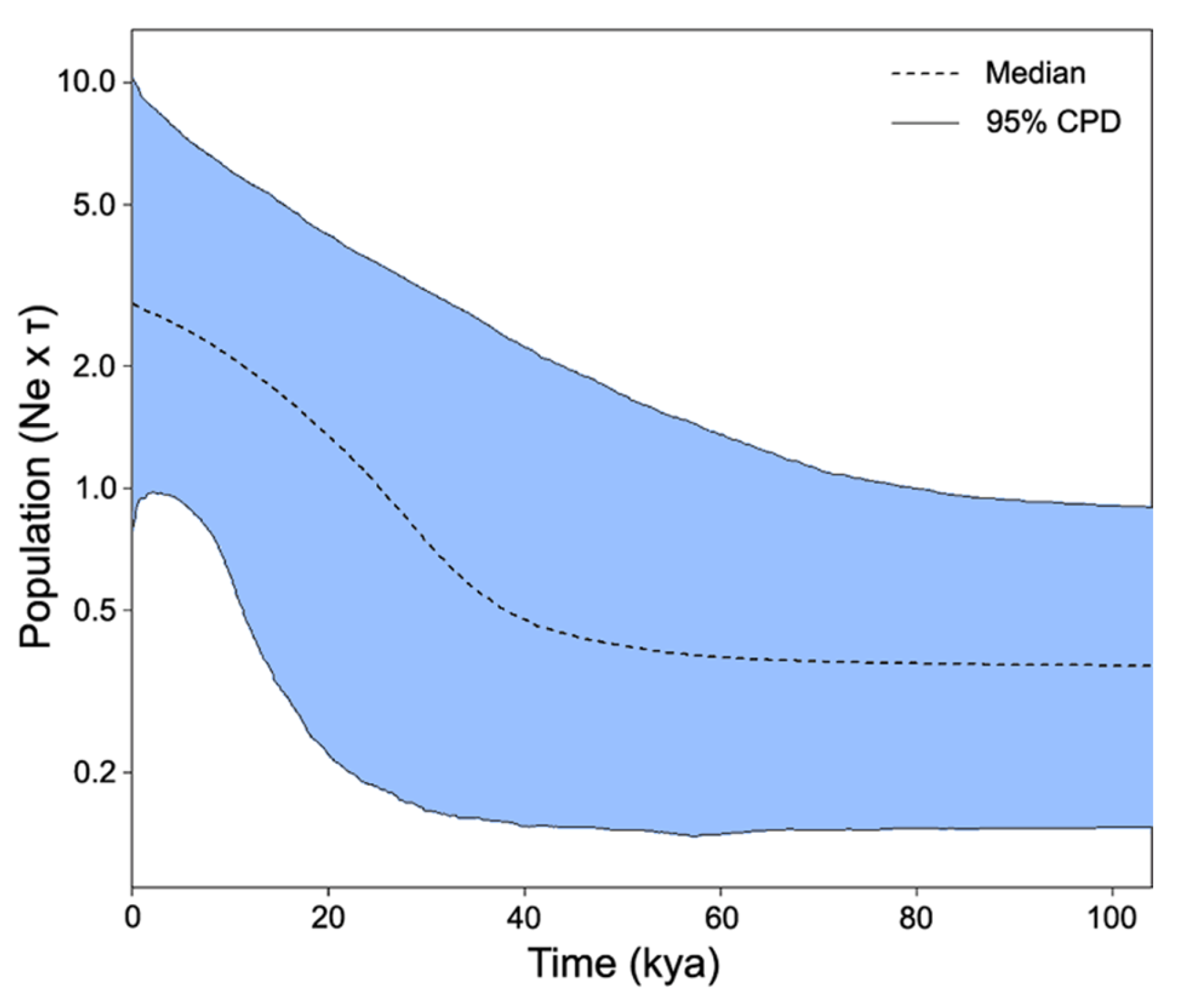
3.3. Historical Population Expansion
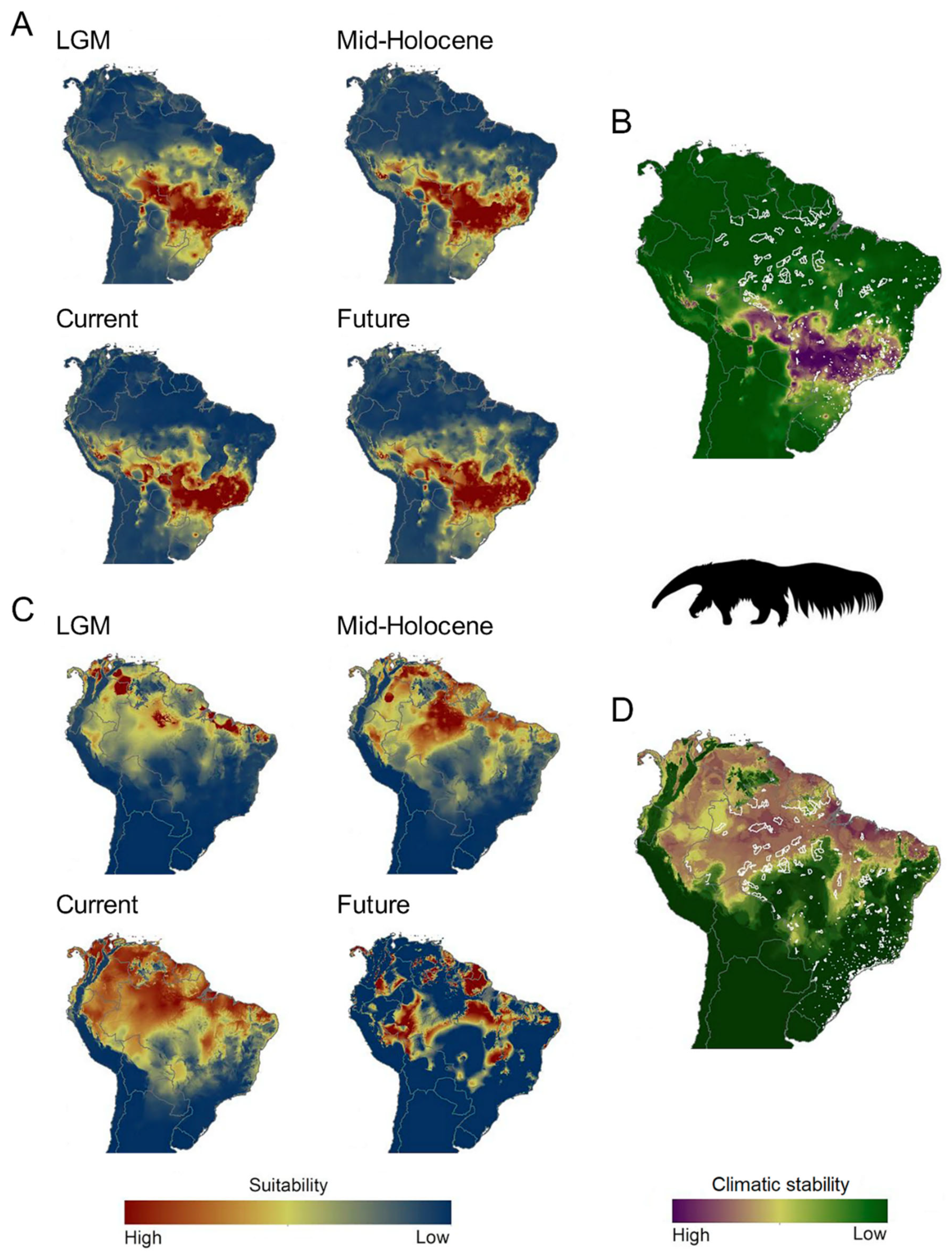
3.4. ENMs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Torres-Florez, J.P.; Johnson, W.E.; Nery, M.F.; Eizirik, E.; Oliveira-Miranda, M.A.; Galetti, P.M. The Coming of Age of Conservation Genetics in Latin America: What Has Been Achieved and What Needs to Be Done. Conserv. Genet. 2018, 19, 1–15. [Google Scholar] [CrossRef]
- Gutiérrez-Tapia, P.; Palma, R.E. Integrating Phylogeography and Species Distribution Models: Cryptic Distributional Responses to Past Climate Change in an Endemic Rodent from the Central Chile Hotspot. Divers. Distrib. 2016, 22, 638–650. [Google Scholar] [CrossRef] [PubMed]
- Lecocq, T.; Harpke, A.; Rasmont, P.; Schweiger, O. Integrating Intraspecific Differentiation in Species Distribution Models: Consequences on Projections of Current and Future Climatically Suitable Areas of Species. Divers. Distrib. 2019, 25, 1088–1100. [Google Scholar] [CrossRef]
- Forester, B.R.; DeChaine, E.G.; Bunn, A.G. Integrating Ensemble Species Distribution Modelling and Statistical Phylogeography to Inform Projections of Climate Change Impacts on Species Distributions. Divers. Distrib. 2013, 19, 1480–1495. [Google Scholar] [CrossRef]
- Ruiz-García, M.; Pinilla-Beltrán, D.; Murillo-García, O.E.; Pinto, C.M.; Brito, J.; Shostell, J.M. Comparative Mitogenome Phylogeography of Two Anteater Genera (Tamandua and Myrmecophaga; Myrmecophagidae, Xenarthra): Evidence of Discrepant Evolutionary Traits. Zool. Res. 2021, 42, 525–547. [Google Scholar] [CrossRef] [PubMed]
- Naples, V.L. Morphology, Evolution and Function of Feeding in the Giant Anteater (Myrmecophaga tridactyla). J. Zool. 1999, 249, 19–41. [Google Scholar] [CrossRef]
- Casali, D.M.; Martins-Santos, E.; Santos, A.L.Q.; Miranda, F.R.; Mahecha, G.A.B.; Perini, F.A. Morphology of the Tongue of Vermilingua (Xenarthra: Pilosa) and Evolutionary Considerations. J. Morphol. 2017, 278, 1380–1399. [Google Scholar] [CrossRef]
- Miranda, F.; Bertassoni, A.; Abba, A.M. Myrmecophaga tridactyla. The IUCN Red List of Threatened Species 2014: E.T14224A47441961; International Union for Conservation of Nature and Natural Resources: Gland, Switzerland, 2014. [Google Scholar]
- Figel, J.J.; Botero-Cañola, S.; Sánchez-Londoño, J.D.; Quintero-Ángel, A. Unexpected Documentation and Inter-Andean Range Expansion of a Vulnerable Large Mammal (Mammalia, Pilosa, Myrmecophaga tridactyla) in Colombia. Mammalia 2016, 80, 449–452. [Google Scholar] [CrossRef]
- Wetzel, R.M. Systematics, Distribution, Ecology, and Conservation of South American Edentates. In The Pymatuning Symposia in Ecology—Mammalian Biology in South America, Vol. 6; Mares, M.A., Genoways, H.H., Eds.; Pymatuning Laboratory of Ecology, University of Pittsburgh: Linesville, PA, USA, 1982; pp. 345–375. [Google Scholar]
- Medri, Í.M.; Mourão, G.M.; Rodrigues, F.H.G. Ordem Pilosa. In Mamíferos do Brasil; Reis, N.R., Peracchi, A.L., Pedro, W.A., Lima, I.P., Eds.; Nelio R. Dos Reis: Londrina, Brazil, 2011; pp. 91–106. [Google Scholar]
- Mourão, G.; Medri, Í.M. Activity of a Specialized Insectivorous Mammal (Myrmecophaga tridactyla) in the Pantanal of Brazil. J. Zool. 2007, 271, 187–192. [Google Scholar] [CrossRef]
- Bertassoni, A.; Mourão, G.; Ribeiro, R.C.; Cesário, C.S.; Oliveira, J.P.; Bianchi, R.C. Movement Patterns and Space Use of the First Giant Anteater (Myrmecophaga tridactyla) Monitored in São Paulo State, Brazil. Stud. Neotrop. Fauna Environ. 2017, 52, 68–74. [Google Scholar] [CrossRef]
- Quiroga, V.A.; Noss, A.J.; Boaglio, G.I.; di Bitetti, M.S. Local and Continental Determinants of Giant Anteater (Myrmecophaga tridactyla) Abundance: Biome, Human and Jaguar Roles in Population Regulation. Mamm. Biol. 2016, 81, 274–280. [Google Scholar] [CrossRef]
- Di Blanco, Y.E.; Desbiez, A.L.J.; Jiménez-Pérez, I.; Kluyber, D.; Massocato, G.F.; di Bitetti, M.S. Habitat Selection and Home-Range Use by Resident and Reintroduced Giant Anteaters in 2 South American Wetlands. J. Mammal. 2017, 98, 1118–1128. [Google Scholar] [CrossRef]
- Garcia, J.E.; Vilas Boas, L.A.; Lemos, M.V.F.; Lemos, E.G.M.; Contel, E.P.B. Identification of Microsatellite DNA Markers for the Giant Anteater Myrmecophaga tridactyla. J. Hered. 2005, 96, 600–602. [Google Scholar] [CrossRef][Green Version]
- Collevatti, R.G.; Leite, K.C.E.; Miranda, G.H.B.; Rodrigues, F.H.G. Evidence of High Inbreeding in a Population of the Endangered Giant Anteater, Myrmecophaga tridactyla (Myrmecophagidae), from Emas National Park, Brazil. Genet. Mol. Biol. 2007, 30, 112–120. [Google Scholar] [CrossRef]
- Clozato, C.L.; Miranda, F.R.; Lara-Ruiz, P.; Collevatti, R.G.; Santos, F.R. Population Structure and Genetic Diversity of the Giant Anteater (Myrmecophaga tridactyla: Myrmecophagidae, Pilosa) in Brazil. Genet. Mol. Biol. 2017, 40, 50–60. [Google Scholar] [CrossRef]
- Sartori, R.Q.; Lopes, A.G.; Aires, L.P.N.; Bianchi, R.D.C.; de Mattos, C.C.B.; Morales, A.C.; Castiglioni, L. Identifying Priority Giant Anteater (Myrmecophaga tridactyla) Populations for Conservation in São Paulo State, Brazil. Ecol. Evol. 2021, 11, 700–713. [Google Scholar] [CrossRef]
- Barragán-Ruiz, C.E.; Silva-Santos, R.; Saranholi, B.H.; Desbiez, A.L.J.; Galetti, P.M. Moderate Genetic Diversity and Demographic Reduction in the Threatened Giant Anteater, Myrmecophaga tridactyla. Front. Genet. 2021, 12, 669350. [Google Scholar] [CrossRef]
- Shaw, J.H.; Machado-Neto, J.; Carter, T.S. Behavior of Free-Living Giant Anteaters (Myrmecophaga tridactyla). Biotropica 1987, 19, 255–259. [Google Scholar] [CrossRef]
- Zimbres, B.; Furtado, M.M.; Jácomo, A.T.A.; Silveira, L.; Sollmann, R.; Tôrres, N.M.; Machado, R.B.; Marinho-Filho, J. The Impact of Habitat Fragmentation on the Ecology of Xenarthrans (Mammalia) in the Brazilian Cerrado. Landsc. Ecol. 2013, 28, 259–269. [Google Scholar] [CrossRef]
- Vynne, C.; Keim, J.L.; Machado, R.B.; Marinho-Filho, J.; Silveira, L.; Groom, M.J.; Wasser, S.K. Resource Selection and Its Implications for Wide-Ranging Mammals of the Brazilian Cerrado. PLoS ONE 2011, 6, e28939. [Google Scholar] [CrossRef]
- Di Blanco, Y.E.; Jiménez-Pérez, I.; di Bitetti, M.S. Habitat Selection in Reintroduced Giant Anteaters: The Critical Role of Conservation Areas. J. Mammal. 2015, 96, 1024–1035. [Google Scholar] [CrossRef]
- Leeuwenberg, F. Edentata as a Food Resource: Subsistence Hunting by Xavante Indians, Brazil. Edentata 1997, 3, 4–5. [Google Scholar]
- Merrit, D.A., Jr. Xenarthrans of the Paraguayan Chaco. In The Biology of the Xenarthra; Vizcaino, S.F., Loughry, W.J., Eds.; University Press of Florida: Gainesville, FL, USA, 2008; pp. 294–299. ISBN 978-0-8130-3718-9. [Google Scholar]
- Noss, A.J.; Cuéllar, R.L.; Cuéllar, E. Exploitation of Xenarthrans by the Guaraní-Isoseño Indigenous People of the Bolivian Chaco: Comparisons with Hunting by Other Indigenous Groups in Latin America, and Implications for Conservation. In The Biology of the Xenarthra; Vizcaino, S.F., Loughry, W.J., Eds.; University Press of Florida: Gainesville, FL, USA, 2008; pp. 244–254. ISBN 978-0-8130-3718-9. [Google Scholar]
- Harris, M.B.; Tomas, W.; Mourão, G.; Silva, C.J.; Guimarães, E.; Sonoda, F.; Fachim, E. Safeguarding the Pantanal Wetlands: Threats and Conservation Initiatives. Conserv. Biol. 2005, 19, 714–720. [Google Scholar] [CrossRef]
- Klink, C.A.; Machado, R.B. Conservation of the Brazilian Cerrado. Conserv. Biol. 2005, 19, 707–713. [Google Scholar] [CrossRef]
- Ribeiro, M.C.; Metzger, J.P.; Martensen, A.C.; Ponzoni, F.J.; Hirota, M.M. The Brazilian Atlantic Forest: How Much Is Left, and How Is the Remaining Forest Distributed? Implications for Conservation. Biol. Conserv. 2009, 142, 1141–1153. [Google Scholar] [CrossRef]
- Silveira, L.; Rodrigues, F.H.G.; Jácomo, A.T.A.; Diniz-Filho, J.A.F. Impact of Wildfires on the Megafauna of Emas National Park, Central Brazil. Oryx 1999, 33, 108–114. [Google Scholar] [CrossRef]
- Cáceres, N.C.; Hannibal, W.; Freitas, D.R.; Silva, E.L.; Roman, C.; Casella, J. Mammal Occurrence and Roadkill in Two Adjacent Ecoregions (Atlantic Forest and Cerrado) in South-Western Brazil. Zoologia 2010, 27, 709–717. [Google Scholar] [CrossRef]
- Cunha, H.F.; Moreira, F.G.A.; Silva, S.S. Roadkill of Wild Vertebrates along the GO-060 Road between Goiânia and Iporá, Goiás State, Brazil. Acta Sci. Biol. Sci. 2010, 32, 257–263. [Google Scholar] [CrossRef]
- Freitas, C.H.; Justino, C.S.; Setz, E.Z.F. Road-Kills of the Giant Anteater in South-Eastern Brazil: 10 Years Monitoring Spatial and Temporal Determinants. Wildl. Res. 2014, 41, 673–680. [Google Scholar] [CrossRef]
- Zurell, D.; Franklin, J.; König, C.; Bouchet, P.J.; Dormann, C.F.; Elith, J.; Fandos, G.; Feng, X.; Guillera-Arroita, G.; Guisan, A.; et al. A Standard Protocol for Reporting Species Distribution Models. Ecography 2020, 43, 1261–1277. [Google Scholar] [CrossRef]
- Peterson, A.T.; Soberón, J.; Pearson, R.G.; Anderson, R.P.; Martínez-Meyer, E.; Nakamura, M.; Araújo, M.B. Ecological Niches and Geographic Distributions—Monographs in Population Biology, No. 49; Princeton University Press: Princeton, NJ, USA, 2011; ISBN 9780691136868. [Google Scholar]
- Diniz-Filho, J.A.F.; Souza, K.S.; Bini, L.M.; Loyola, R.; Dobrovolski, R.; Rodrigues, J.F.M.; Lima-Ribeiro Matheus de, S.; Terribile, L.C.; Rangel, T.F.; Bione, I.; et al. A Macroecological Approach to Evolutionary Rescue and Adaptation to Climate Change. Ecography 2019, 42, 1124–1141. [Google Scholar] [CrossRef]
- Terribile, L.C.; Lima-Ribeiro, M.S.; Araújo, M.B.; Bizão, N.; Collevatti, R.G.; Dobrovolski, R.; Franco, A.A.; Guilhaumon, F.; Lima, J.S.; Murakami, D.M.; et al. Areas of Climate Stability of Species Ranges in the Brazilian Cerrado: Disentangling Uncertainties through Time. Nat. Conserv. 2012, 10, 152–159. [Google Scholar] [CrossRef]
- Ribeiro, B.R.; Sales, L.P.; Loyola, R. Strategies for Mammal Conservation under Climate Change in the Amazon. Biodivers. Conserv. 2018, 27, 1943–1959. [Google Scholar] [CrossRef]
- Hewitt, G. The Genetic Legacy of the Quaternary Ice Ages. Nature 2000, 405, 907–913. [Google Scholar] [CrossRef]
- Wang, I.J.; Bradburd, G.S. Isolation by Environment. Mol. Ecol. 2014, 23, 5649–5662. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the Sensitivity of Progressive Multiple Sequence Alignment through Sequence Weighting, Position-Specific Gap Penalties and Weight Matrix Choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Flot, J.-F. SEQPHASE: A Web Tool for Interconverting PHASE Input/Output Files and FASTA Sequence Alignments. Mol. Ecol. Resour. 2010, 10, 162–166. [Google Scholar] [CrossRef]
- Stephens, M.; Scheet, P. Accounting for Decay of Linkage Disequilibrium in Haplotype Inference and Missing-Data Imputation. Am. J. Hum. Genet. 2005, 76, 449–462. [Google Scholar] [CrossRef]
- Stephens, M.; Smith, N.J.; Donnelly, P. A New Statistical Method for Haplotype Reconstruction from Population Data. Am. J. Hum. Genet. 2001, 68, 978–989. [Google Scholar] [CrossRef]
- Guillot, G.; Estoup, A.; Mortier, F.; Cosson, J.F. A Spatial Statistical Model for Landscape Genetics. Genetics 2005, 170, 1261–1280. [Google Scholar] [CrossRef]
- Guillot, G.; Mortier, F.; Estoup, A. Geneland: A Computer Package for Landscape Genetics. Mol. Ecol. Notes 2005, 5, 712–715. [Google Scholar] [CrossRef]
- Guillot, G. Inference of Structure in Subdivided Populations at Low Levels of Genetic Differentiation—The Correlated Allele Frequencies Model Revisited. Bioinformatics 2008, 24, 2222–2228. [Google Scholar] [CrossRef]
- Montgomery, G.G.; Lubin, Y.D. Prey Influences on Movements of Neotropical Anteaters. In Proceedings of the 1975 Predator Symposium; Phillips, R.L., Jonkel, C., Eds.; Montana Forest and Conservation Experiment Station, University of Montana: Missoula, MT, USA, 1977; pp. 103–131. [Google Scholar]
- Excoffier, L.; Lischer, H.E.L. Arlequin Suite Ver 3.5: A New Series of Programs to Perform Population Genetics Analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of Molecular Variance Inferred from Metric Distances among DNA Haplotypes: Application to Human Mitochondrial DNA Restriction Data. Genetics 1992, 131, 479–491. [Google Scholar] [CrossRef]
- Kimura, M. A Simple Method for Estimating Evolutionary Rates of Base Substitutions through Comparative Studies of Nucleotide Sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Chao, A.; Gotelli, N.J.; Hsieh, T.C.; Sander, E.L.; Ma, K.H.; Colwell, R.K.; Ellison, A.M. Rarefaction and Extrapolation with Hill Numbers: A Framework for Sampling and Estimation in Species Diversity Studies. Ecol. Monogr. 2014, 84, 45–67. [Google Scholar] [CrossRef]
- Hsieh, T.C.; Ma, K.H.; Chao, A. INEXT: An R Package for Rarefaction and Extrapolation of Species Diversity (Hill Numbers). Methods Ecol. Evol. 2016, 7, 1451–1456. [Google Scholar] [CrossRef]
- Leigh, J.W.; Bryant, D. POPART: Full-feature Software for Haplotype Network Construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Forster, P.; Rohl, A. Median-Joining Networks for Inferring Intraspecific Phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Lichstein, J.W. Multiple Regression on Distance Matrices: A Multivariate Spatial Analysis Tool. Plant Ecol. 2007, 188, 117–131. [Google Scholar] [CrossRef]
- Goslee, S.C.; Urban, D.L. The Ecodist Package for Dissimilarity-Based Analysis of Ecological Data. J. Stat. Softw. 2007, 22, 1–19. [Google Scholar] [CrossRef]
- Paradis, E.; Schliep, K. Ape 5.0: An Environment for Modern Phylogenetics and Evolutionary Analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package, R Package Version 2.4-6. Available online: https://cran.r-project.org/package=vegan (accessed on 24 January 2018).
- Magalhães, R.F.; Lemes, P.; Camargo, A.; Oliveira, U.; Brandão, R.A.; Thomassen, H.; Garcia, P.C.A.; Leite, F.S.F.; Santos, F.R. Evolutionarily Significant Units of the Critically Endangered Leaf Frog Pithecopus ayeaye (Anura, Phyllomedusidae) Are Not Effectively Preserved by the Brazilian Protected Areas Network. Ecol. Evol. 2017, 7, 8812–8828. [Google Scholar] [CrossRef]
- Beerli, P. Comparison of Bayesian and Maximum-Likelihood Inference of Population Genetic Parameters. Bioinformatics 2006, 22, 341–345. [Google Scholar] [CrossRef]
- Tavaré, S. Some Probabilistic and Statistical Problems in the Analysis of DNA Sequences. In Some Mathematical Questions in Biology: DNA Sequence Analysis—Lectures on Mathematics in the Life Sciences, Vol. 17; Miura, R.M., Ed.; American Mathematical Society: Providence, RI, USA, 1986; pp. 57–86. [Google Scholar]
- Beerli, P.; Palczewski, M. Unified Framework to Evaluate Panmixia and Migration Direction among Multiple Sampling Locations. Genetics 2010, 185, 313–326. [Google Scholar] [CrossRef]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for Inference of Large Phylogenetic Trees. In Proceedings of the 2010 Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; Institute of Electrical and Electronics Engineers (IEEE): Piscataway, NJ, USA, 2010; pp. 1–8. [Google Scholar] [CrossRef]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.-H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.R.; Drummond, A.J. bModelTest: Bayesian Phylogenetic Site Model Averaging and Model Comparison. BMC Evol. Biol. 2017, 17, 42. [Google Scholar] [CrossRef] [PubMed]
- Gibb, G.C.; Condamine, F.L.; Kuch, M.; Enk, J.; Moraes-Barros, N.; Superina, M.; Poinar, H.N.; Delsuc, F. Shotgun Mitogenomics Provides a Reference Phylogenetic Framework and Timescale for Living Xenarthrans. Mol. Biol. Evol. 2016, 33, 621–642. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A. FigTree, v1.4.4. Available online: https://github.com/rambaut/figtree/releases/tag/v1.4.4 (accessed on 26 November 2018).
- Heled, J.; Drummond, A.J. Bayesian Inference of Population Size History from Multiple Loci. BMC Evol. Biol. 2008, 8, 289. [Google Scholar] [CrossRef]
- R Core Team R: A Language and Environment for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 21 April 2017).
- Hijmans, R.J.; Cameron, S.E.; Parra, J.L.; Jones, P.G.; Jarvis, A. Very High Resolution Interpolated Climate Surfaces for Global Land Areas. Int. J. Climatol. 2005, 25, 1965–1978. [Google Scholar] [CrossRef]
- Intergovernmental Panel on Climate Change. Summary for Policymakers. In Climate change 2013—The Physical Science Basis: Working Group I Contribution to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Stocker, T.F., Qin, D., Plattner, G.-K., Tignor, M., Allen, S.K., Boschung, J., Nauels, A., Xia, Y., Bex, V., Midgley, P.M., Eds.; Cambridge University Press: Cambridge, UK, 2014; pp. 1–30. ISBN 9781107057999. [Google Scholar]
- Thomson, A.M.; Calvin, K.V.; Smith, S.J.; Kyle, G.P.; Volke, A.; Patel, P.; Delgado-Arias, S.; Bond-Lamberty, B.; Wise, M.A.; Clarke, L.E.; et al. RCP4.5: A Pathway for Stabilization of Radiative Forcing by 2100. Clim. Chang. 2011, 109, 77–94. [Google Scholar] [CrossRef]
- Araújo, M.B.; New, M. Ensemble Forecasting of Species Distributions. Trends Ecol. Evol. 2007, 22, 42–47. [Google Scholar] [CrossRef]
- Hijmans, R.J.; Phillips, S.; Leathwick, J.; Elith, J. Dismo: Species Distribution Modeling, R Package Version 1.1-4. Available online: https://cran.r-project.org/package=dismo (accessed on 9 January 2017).
- Nix, H.A. A Biogeographic Analysis of Australian Elapid Snakes. In Atlas of Elapid Snakes of Australia—Australian Flora and Fauna Series, No. 7; Longmore, R., Ed.; Bureau of Flora and Fauna: Canberra, Australia, 1986; pp. 4–15. [Google Scholar]
- Carpenter, G.; Gillison, A.N.; Winter, J. DOMAIN: A Flexible Modelling Procedure for Mapping Potential Distributions of Plants and Animals. Biodivers. Conserv. 1993, 2, 667–680. [Google Scholar] [CrossRef]
- Tax, D.M.J.; Duin, R.P.W. Support Vector Data Description. Mach. Learn. 2004, 54, 45–66. [Google Scholar] [CrossRef]
- Phillips, S.J.; Dudík, M. Modeling of Species Distributions with Maxent: New Extensions and a Comprehensive Evaluation. Ecography 2008, 31, 161–175. [Google Scholar] [CrossRef]
- Fielding, A.H.; Bell, J.F. A Review of Methods for the Assessment of Prediction Errors in Conservation Presence/Absence Models. Environ. Conserv. 1997, 24, 38–49. [Google Scholar] [CrossRef]
- Pearson, R.G.; Raxworthy, C.J.; Nakamura, M.; Townsend Peterson, A. Predicting Species Distributions from Small Numbers of Occurrence Records: A Test Case Using Cryptic Geckos in Madagascar. J. Biogeogr. 2007, 34, 102–117. [Google Scholar] [CrossRef]
- Bean, W.T.; Stafford, R.; Brashares, J.S. The Effects of Small Sample Size and Sample Bias on Threshold Selection and Accuracy Assessment of Species Distribution Models. Ecography 2012, 35, 250–258. [Google Scholar] [CrossRef]
- Shcheglovitova, M.; Anderson, R.P. Estimating Optimal Complexity for Ecological Niche Models: A Jackknife Approach for Species with Small Sample Sizes. Ecol. Model. 2013, 269, 9–17. [Google Scholar] [CrossRef]
- Meirmans, P.G. Seven Common Mistakes in Population Genetics and How to Avoid Them. Mol. Ecol. 2015, 24, 3223–3231. [Google Scholar] [CrossRef]
- Chikhi, L.; Sousa, V.C.; Luisi, P.; Goossens, B.; Beaumont, M.A. The Confounding Effects of Population Structure, Genetic Diversity and the Sampling Scheme on the Detection and Quantification of Population Size Changes. Genetics 2010, 186, 983–995. [Google Scholar] [CrossRef]
- Heller, R.; Chikhi, L.; Siegismund, H.R. The Confounding Effect of Population Structure on Bayesian Skyline Plot Inferences of Demographic History. PLoS ONE 2013, 8, e62992. [Google Scholar] [CrossRef]
- Franklin, J.; Wejnert, K.E.; Hathaway, S.A.; Rochester, C.J.; Fisher, R.N. Effect of Species Rarity on the Accuracy of Species Distribution Models for Reptiles and Amphibians in Southern California. Divers. Distrib. 2009, 15, 167–177. [Google Scholar] [CrossRef]
- Ministério do Meio Ambiente. Unidades de Conservação—2018. Available online: https://dados.gov.br/dataset/unidadesdeconservacao/resource/3562d093-14ed-434b-9fa3-411b664d6836 (accessed on 10 May 2022).
- Nardelli, M.; Ibáñez, E.A.; Dobler, D.; Justy, F.; Delsuc, F.; Abba, A.M.; Cassini, M.H.; Túnez, J.I. Genetic Structuring in a Relictual Population of Screaming Hairy Armadillo (Chaetophractus vellerosus) in Argentina Revealed by a Set of Novel Microsatellite Loci. Genetica 2016, 144, 469–476. [Google Scholar] [CrossRef]
- Lara-Ruiz, P.; Chiarello, A.G.; Santos, F.R. Extreme Population Divergence and Conservation Implications for the Rare Endangered Atlantic Forest Sloth, Bradypus torquatus (Pilosa: Bradypodidae). Biol. Conserv. 2008, 141, 1332–1342. [Google Scholar] [CrossRef]
- Schetino, M.A.A.; Coimbra, R.T.F.; Santos, F.R. Time Scaled Phylogeography and Demography of Bradypus torquatus (Pilosa: Bradypodidae). Glob. Ecol. Conserv. 2017, 11, 224–235. [Google Scholar] [CrossRef]
- Coimbra, R.T.F.; Miranda, F.R.; Lara, C.C.; Schetino, M.A.A.; Santos, F.R. Phylogeographic History of South American Populations of the Silky Anteater Cyclopes didactylus (Pilosa: Cyclopedidae). Genet. Mol. Biol. 2017, 40, 40–49. [Google Scholar] [CrossRef]
- Miranda, F.R.; Casali, D.M.; Perini, F.A.; Machado, F.A.; Santos, F.R. Taxonomic Review of the Genus Cyclopes Gray, 1821 (Xenarthra: Pilosa), with the Revalidation and Description of New Species. Zool. J. Linn. Soc. 2018, 183, 687–721. [Google Scholar] [CrossRef]
- Eizirik, E.; Kim, J.-H.; Menotti-Raymond, M.; Crawshaw, P.G., Jr.; O’Brien, S.J.; Johnson, W.E. Phylogeography, Population History and Conservation Genetics of Jaguars (Panthera onca, Mammalia, Felidae). Mol. Ecol. 2001, 10, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Medri, Í.M.; Mourão, G. Home Range of Giant Anteaters (Myrmecophaga tridactyla) in the Pantanal Wetland, Brazil. J. Zool. 2005, 266, 365–375. [Google Scholar] [CrossRef]
- Huchon, D.; Delsuc, F.; Catzeflis, F.M.; Douzery, E.J.P. Armadillos Exhibit Less Genetic Polymorphism in North America than in South America: Nuclear and Mitochondrial Data Confirm a Founder Effect in Dasypus novemcinctus (Xenarthra). Mol. Ecol. 1999, 8, 1743–1748. [Google Scholar] [CrossRef] [PubMed]
- Redford, K.H. The Edentates of the Cerrado. Edentata 1994, 1, 4–10. [Google Scholar]
- Desbiez, A.L.J.; Medri, Í.M. Density and Habitat Use by Giant Anteaters (Myrmecophaga tridactyla) and Southern Tamanduas (Tamandua tetradactyla) in the Pantanal Wetland, Brazil. Edentata 2010, 11, 4–10. [Google Scholar] [CrossRef]
- Haffer, J. Speciation in Amazonian Forest Birds. Science 1969, 165, 131–137. [Google Scholar] [CrossRef]
- Werneck, F.P.; Nogueira, C.; Colli, G.R.; Sites, J.W.; Costa, G.C. Climatic Stability in the Brazilian Cerrado: Implications for Biogeographical Connections of South American Savannas, Species Richness and Conservation in a Biodiversity Hotspot. J. Biogeogr. 2012, 39, 1695–1706. [Google Scholar] [CrossRef]
- Bueno, M.L.; Pennington, R.T.; Dexter, K.G.; Kamino, L.H.Y.; Pontara, V.; Neves, D.M.; Ratter, J.A.; de Oliveira-Filho, A.T. Effects of Quaternary Climatic Fluctuations on the Distribution of Neotropical Savanna Tree Species. Ecography 2017, 40, 403–414. [Google Scholar] [CrossRef]
- Hyseni, C.; Garrick, R.C. The Role of Glacial-interglacial Climate Change in Shaping the Genetic Structure of Eastern Subterranean Termites in the Southern Appalachian Mountains, USA. Ecol. Evol. 2019, 9, 4621–4636. [Google Scholar] [CrossRef]
- Veera Singham, G.; Othman, A.S.; Lee, C.-Y. Phylogeography of the Termite Macrotermes gilvus and Insight into Ancient Dispersal Corridors in Pleistocene Southeast Asia. PLoS ONE 2017, 12, e0186690. [Google Scholar] [CrossRef]
- Leppänen, J.; Vepsäläinen, K.; Savolainen, R. Phylogeography of the Ant Myrmica rubra and Its Inquiline Social Parasite. Ecol. Evol. 2011, 1, 46–62. [Google Scholar] [CrossRef]
- Mendoza-Ramírez, M.; Gutiérrez-Rodríguez, J.; Poteaux, C.; Ornelas-García, P.; Zaldívar-Riverón, A. Late Pleistocene Genetic Diversification and Demographic Expansion in the Widely Distributed Neotropical Ant Neoponera villosa (Ponerinae). Mitochondrial DNA Part A 2019, 30, 296–306. [Google Scholar] [CrossRef]
- Barnosky, A.D. Megafauna Biomass Tradeoff as a Driver of Quaternary and Future Extinctions. Proc. Natl. Acad. Sci. USA 2008, 105, 11543–11548. [Google Scholar] [CrossRef]
- Barnosky, A.D.; Lindsey, E.L. Timing of Quaternary Megafaunal Extinction in South America in Relation to Human Arrival and Climate Change. Quat. Int. 2010, 217, 10–29. [Google Scholar] [CrossRef]
- Prado, J.L.; Martinez-Maza, C.; Alberdi, M.T. Megafauna Extinction in South America: A New Chronology for the Argentine Pampas. Palaeogeogr. Palaeoclimatol. Palaeoecol. 2015, 425, 41–49. [Google Scholar] [CrossRef]
- Villavicencio, N.A.; Lindsey, E.L.; Martin, F.M.; Borrero, L.A.; Moreno, P.I.; Marshall, C.R.; Barnosky, A.D. Combination of Humans, Climate, and Vegetation Change Triggered Late Quaternary Megafauna Extinction in the Última Esperanza Region, Southern Patagonia, Chile. Ecography 2016, 39, 125–140. [Google Scholar] [CrossRef]
- Avilla, L.S.; Figueiredo, A.M.G.; Kinoshita, A.; Bertoni-Machado, C.; Mothé, D.; Asevedo, L.; Baffa, O.; Dominato, V.H. Extinction of a Gomphothere Population from Southeastern Brazil: Taphonomic, Paleoecological and Chronological Remarks. Quat. Int. 2013, 305, 85–90. [Google Scholar] [CrossRef]
- Arruda, D.M.; Schaefer, C.E.G.R.; Fonseca, R.S.; Solar, R.R.C.; Fernandes-Filho, E.I. Vegetation Cover of Brazil in the Last 21 Ka: New Insights into the Amazonian Refugia and Pleistocenic Arc Hypotheses. Glob. Ecol. Biogeogr. 2018, 27, 47–56. [Google Scholar] [CrossRef]
- Zimbres, B.Q.C.; Aquino, P.D.P.U.; Machado, R.B.; Silveira, L.; Jácomo, A.T.A.; Sollmann, R.; Tôrres, N.M.; Furtado, M.M.; Marinho-Filho, J. Range Shifts under Climate Change and the Role of Protected Areas for Armadillos and Anteaters. Biol. Conserv. 2012, 152, 53–61. [Google Scholar] [CrossRef]
- Frankham, R. Genetics and Extinction. Biol. Conserv. 2005, 126, 131–140. [Google Scholar] [CrossRef]
- Françoso, R.D.; Brandão, R.; Nogueira, C.C.; Salmona, Y.B.; Machado, R.B.; Colli, G.R. Habitat Loss and the Effectiveness of Protected Areas in the Cerrado Biodiversity Hotspot. Nat. Conserv. 2015, 13, 35–40. [Google Scholar] [CrossRef]
- Diniz, M.F.; Brito, D. Protected Areas Effectiveness in Maintaining Viable Giant Anteater (Myrmecophaga tridactyla) Populations in an Agricultural Frontier. Nat. Conserv. 2015, 13, 145–151. [Google Scholar] [CrossRef][Green Version]
- Beerli, P. How to Use MIGRATE or Why Are Markov Chain Monte Carlo Programs Difficult to Use? In Population Genetics for Animal Conservation; Bertorelle, G., Bruford, M.W., Hauffe, H.C., Rizzoli, A., Vernesi, C., Eds.; Conservation Biology; Cambridge University Press: Cambridge, UK, 2009; pp. 42–79. ISBN 9780521866309. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Description | Marginal Likelihood | BF | Model Probability |
|---|---|---|---|---|
| I | Panmixia | −1901.6788 | <1 × 10−5 | <1 × 10−5 |
| II | Bidirectional migration | −1892.3102 | 0.1051 | 0.0891 |
| III | Migration from AM to CEPTAF | −1890.0575 | 1 | 0.8476 |
| IV | Migration from CEPTAF to AM | −1892.6514 | 0.0747 | 0.0633 |
| V | Isolated populations | −1927.6109 | <1 × 10−16 | <1 × 10−16 |
| Cluster | n | H | h ± SD | π ± SD |
|---|---|---|---|---|
| AM | 9 | 6 | 0.8889 ± 0.0910 | 0.0027 ± 0.0018 |
| CEPTAF | 97 | 29 | 0.7204 ± 0.0512 | 0.0019 ± 0.0012 |
| Total | 106 | 33 | 0.7623 ± 0.0445 | 0.0021 ± 0.0013 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coimbra, R.T.F.; Magalhães, R.F.; Lemes, P.; Miranda, F.R.; Santos, F.R. Integrative Phylogeography Reveals Conservation Priorities for the Giant Anteater Myrmecophaga tridactyla in Brazil. Diversity 2022, 14, 542. https://doi.org/10.3390/d14070542
Coimbra RTF, Magalhães RF, Lemes P, Miranda FR, Santos FR. Integrative Phylogeography Reveals Conservation Priorities for the Giant Anteater Myrmecophaga tridactyla in Brazil. Diversity. 2022; 14(7):542. https://doi.org/10.3390/d14070542
Chicago/Turabian StyleCoimbra, Raphael T. F., Rafael F. Magalhães, Priscila Lemes, Flávia R. Miranda, and Fabrício R. Santos. 2022. "Integrative Phylogeography Reveals Conservation Priorities for the Giant Anteater Myrmecophaga tridactyla in Brazil" Diversity 14, no. 7: 542. https://doi.org/10.3390/d14070542
APA StyleCoimbra, R. T. F., Magalhães, R. F., Lemes, P., Miranda, F. R., & Santos, F. R. (2022). Integrative Phylogeography Reveals Conservation Priorities for the Giant Anteater Myrmecophaga tridactyla in Brazil. Diversity, 14(7), 542. https://doi.org/10.3390/d14070542