1. Introduction
Polytrichum commune Hedw., the common hair-cap moss, is a generitype of the genus
Polytrichum, the type genus of Polytrichaceae (Polytrichales, Polytrichopsida). Polytrichopsida includes ca. 23 genera and ca. 200 species [
1,
2,
3]. The species classified in the genus
Polytrichum are common in the boreal zone and Arctic, where they occupy different ecotopes with disturbed vegetation. For instance,
P. commune is especially abundant in irrigated sphagnous bogs and forests, where it plays an important role replacing peat moss cover.
Polytrichum commune is a widely accepted model object for ecological, environmental, physiological, and genetic studies. Thus, it got ca. 12,300 results, obtained from scholar.google.com.
Due to their considerable variety of forms but relatively few species, Polytrichopsida has been considered as an apparently ancient group with a position strikingly analogous to that of the conifers within the seed plant clade [
2]. In terms of the peristome structure, which is traditionally used for establishing the high-level systematics of mosses, Polytrichopsida belongs to nematodontous mosses, which differ from arthrodontous mosses (Bryopsida). In nematodontous peristome, the teeth of peristome are solid and composed of bundles of dead cells. Each tooth of the arthrodontous peristome type is composed of periclinal (tangential) cell-wall remnants between two of the three concentric peristomial cell layers.
Representatives of Polytrichales (Polytrichopsida) and Tetraphidales (Tetraphidopsida) have a unique trait called nematodontous peristome, and therefore, they have recently been often united into a single class Polytrichopsida [
1,
3]. According to modern understanding, these mosses occupy a basal position in relation to the majority of mosses that bear arthrodontous peristome and are attributed to Bryopsida, while the earliest divergent lineages of mosses (Takakiopsida, Sphagnopsida, Andreaeopsida, Andreaeobryopsida and likely also Oedipodiopsida) are eperistomate. Earlier molecular research yielded conflicting results regarding relationships among the early-diverging peristomate moss lineages [
4]. Thus, monotypic Oedipodiales has been sometimes included in the class Polytrichopsida [
3], while Buxbaumiaceae, possessing peristome of a deviating type [
5] was considered closer to
Tetraphis [
6] or referred to as arthrodontous Bryopsida [
1,
3,
7,
8]. According to recent phylogenetic reconstructions, Oedipodiales occupies an intermediate position between Andreaeobryophytina and Tetraphidales, while Buxbaumiaceae are classified in arthrodontous Bryopsida [
7,
8].
Genomic data may be crucial for large-scale phylogenetic reconstructions. In this regard, at present, the greatest hopes are pinned on the analysis of the genomes of organelles—mitochondria and plastids. The size and structure of the mitochondrial genomes is extremely variable among the Viridiplantae. Nonetheless, mitogenomes of the liverworts and mosses are very stable and conservative. Only single rearrangements in their structure have been found so far [
9]. Sufficient steps toward understanding the nature of this phenomenon were performed recently [
10]. However, this fundamental issue is still far from complete, and additional efforts are required to shed light on the mechanisms behind the mitogenome stability in bryophytes. In this study, we contribute to understanding the nature of the above-mentioned phenomenon.
The variety of mitochondrial genomes of bryophytes, a highly diverse group of land plants, still remains insufficiently studied in comparison with angiosperms. Large-scale trends of mitogenome evolution are of special interest; however, to address these issues properly, a denser sampling of ancient, early-diverged lineages of bryophytes is crucial. The sequencing of the mitochondrial genome of the nematodontous moss Polytrichum commune extends the already-known diversity of moss mitogenomes.
2. Materials and Methods
2.1. Sample Collection and DNA Isolation
The P. commune samples were collected in the summer season of 2014 in Klin Distr. of Moscow Province, Middle European Russia; the reference specimen was deposited in MW (Herbarium of Moscow University).
A Nucleospin Plant DNA Kit (Macherey Nagel, Düren, Germany) was used for total DNA extraction from whole shoots of freshly collected plants according to the manufacturer’s protocol. A yield of about 2 μg DNA was obtained according to measurements determined with a Qubit fluorometer (Invitrogen, Carlsbad, CA, USA).
2.2. Library Preparation and Sequencing
A 500 ng sample of genomic DNA was fragmented using a Covaris S220 sonicator (Covaris, Woburn, MA, USA), and a library was prepared using a TruSeq DNA sample preparation kit (Illumina, Mountain View, CA, USA). The concentration of the prepared library was measured with the Qubit fluorometer (Invitrogen, Carlsbad, CA, USA), and the qPCR and fragment length distribution were determined with Bioanalyzer 2100 (Agilent, Santa Clara, CA, USA). The library was diluted to 10 pM and used for cluster generation on a cBot instrument with TruSeq PE Cluster Kit v3 reagents (Illumina, Mountain View, CA, USA). Sequencing was performed on a HiSeq2000 machine (Illumina, San Diego, CA, USA) with a read length of 101 from both ends of the fragments. About 17.2 million paired-end reads were obtained.
2.3. Mitogenome Assembly and Annotation
Raw sequencing reads were preprocessed with Trimmomatic software [
11]. The whole genome assembly was then accomplished using the Spades assembler [
12]. A Blast database was generated from the assembled contigs, and a Blast search was performed against the
Physcomitrella patens (Hedw.) Bruch & Schimp. mitochondrial genome sequence [
13] using the standalone NCBI BLAST-2.2.29+ [
14]. Among the longest hits was the
P. commune complete mitogenome. Iterative mapping was carried out using Geneious R10 software (
https://www.geneious.com) [
15] to verify correctness of the assembled genome. The resulting sequence had almost 86× coverage depth. The genome boundaries were verified by PCR amplification followed by Sanger sequencing.
Genome annotation based on sequence similarity was performed using Geneious software. The mitogenome sequence of
Atrichum angustatum (Brid.) Bruch & Schimp. (GenBank accession number KC784956) was applied as a reference as it gave a maximal percent identity and total score in a blast search on the NCBI website (
https://www.ncbi.nlm.nih.gov) against a
P. commune complete mitogenome. The annotated
P. commune mitogenome sequence was submitted to the GenBank (accession number NC039775). A circular genome map was drawn using the CGView Server (
http://stothard.afns.ualberta.ca/cgview_server) [
16].
2.4. SSR Analysis
Simple sequence repeats (SSRs) were detected and located in the mitogenome of
P. commune using GMATo v1.2 software [
17].
2.5. Analysis of Repetive Elements
The search for repeating sequences was performed using the Repeat Finder plugin in the Geneious program with the minimum repeat length of 51 bp and maximum mismatches at 15%. Each identified repeat was observed at least twice in the analyzed genome. In case all the subunits of a particular repeat were nested in those of another longer repeat, the former was excluded from the consideration. If all the repeat subunits overlapped each other or were located in the same intergenic spacer, the repeat was also filtered out.
2.6. Phylogenomic and Genome-Wide Comparison Analyses
For phylogenetic reconstruction, only functional protein-coding sequences (CDS) of the whole bryophytes mitogenomes deposited in the GenBank (
http://www.ncbi.nlm.nih.gov) were selected. A total of 33 CDS of genes including
cox1,
atp4,
rps7,
atp1,
atp8,
atp6,
atp9,
cob,
cox2,
cox3,
nad1,
nad2,
nad3,
nad4,
nad4L,
nad5,
nad6,
nad9,
rpl2,
rpl5,
rpl6,
rpl16,
rps1,
rps2,
rps4,
rps12,
rps11,
rps13,
rps14,
rps19,
sdh3,
sdh4, and
tatC were used for the phylogeny inference. The CDS of 44 mosses were aligned with the default options of MAFFT plugin of Geneious Prime 2020.2 software, and the obtained alignment was adjusted manually in BioEdit 7.2.5 [
18]. The individual gene alignments were concatenated into a 24,667 bp matrix.
Phylogenetic reconstruction was performed by the maximum likelihood (ML) analyses with RAxML version 8.2.10 [
19], using the GTRGAMMA model. The number of bootstrap replications equal to 500 was set by the MRE (extended majority rules) criterion Mauve plugin with the default settings implemented in Geneious software was applied for genome-wide comparison of bryophyte mitogenomes.
3. Results
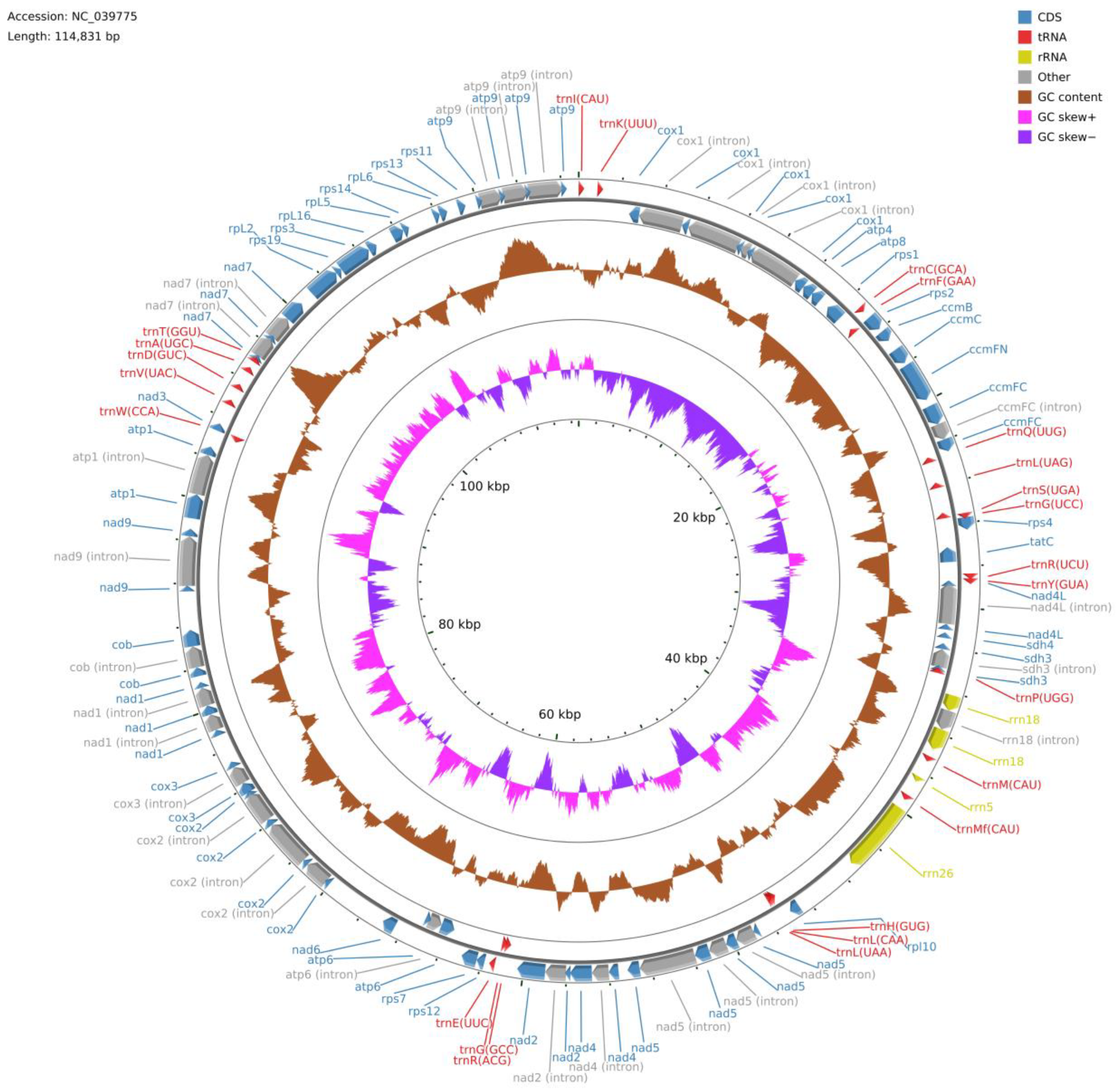
3.1. The General Mitogenome Structure of P. commune
Polytrichum commune mitogenome (
Figure 1) consists of a single circular molecule; its length is 114,831 bp, which exceeds the length of most published bryophyte mitochondrial genomes in the GenBank database, with the exception of
Sphagnum palustre L. NC024521 (141,276 bp),
Andreaea wangiana P.C. Chen NC046759, (117,857 bp), and
Atrichum angustatum NC024520 (115,146 bp). The size of mitogenome of
Pogonatum inflexum (Lindb.) Sande Lac., which is one of three Polytrichopsida representatives involved in the analysis, is slightly smaller and equal to 113,866 bp, while the mitogenome length of all the other mosses is less than 110 Kb and varies from 109,586 bp (
Funaria hygrometrica Hedw.) to 100,342 bp (
Mielichhoferia elongata (Hoppe & Hornsch.) Nees & Hornsch.).
The mitogenome of P. commune contains 67 genes including genes for three rRNAs (rrn18, rrn26, and rrn5), 24 tRNAs and 40 conserved mitochondrial proteins (15 ribosomal proteins, four ccm proteins, nine nicotinamide adenine dinucleotide dehydrogenase subunits, five ATPase subunits, two succinate dehydrogenase subunits, one apocytochrome b, three cytochrome oxidase subunits, and one twin-arginine translocation complex subunit). However, the gene set of sequenced mitogenomes differs among mosses. For instance, the rpl16 gene is absent in the GenBank annotations of Racomitrium elongatum Ehrh. ex Frisvoll, Codriophorus varius (Mitt.) Bednarek-Ochyra & Ochyra, Racomitrium lanuginosum (Hedw.) Brid., Racomitrium emersum (Müll. Hal.) A. Jaeger, Codriophorus acicularis (Hedw.) P. Beauv., Codriophorus laevigatus (A. Jaeger) Bednarek-Ochyra & Ochyra, Bucklandiella orthotrichacea (Müll. Hal.) Bednarek-Ochyra & Ochyra, and Racomitrium ericoides (Brid.) Brid., but in fact, is present in their genomes. This situation is similar to the rps4 gene in the Oxystegus tenuirostris (Hook. & Taylor) A.J.E. Sm. mitogenome and the rpl5 gene in the Tetraphis pellucida Hedw. mitogenome.
The length of genes, exones, and introns in the mitochondrial genome of
P. commune is presented in
Figure 2. The shortest genes are
trnG (UCC),
trnC (GCA), and
trnQ (UUG), with a length of (71 bp). The first two genes
trnC (GCA) and
trnG (UCC) have the same length in all other 43 mitogenomes under investigation. At the same time, due to a discrepancy in annotation of the
trnQ (UUG) gene its length (and orientation in
Oxystegus tenuirostris) differs among Bryophyta mitogenomes: 68 bp in
Pogonatum inflexum, 71 bp in
Andreaea wangiana,
Atrichum angustatum,
Bartramia pomiformis Hedw.
Buxbaumia aphylla Hedw.,
Funaria hygrometrica,
Mielichhoferia elongata, and
Sphagnum palustre, and 72 bp in all other analyzed mosses (
Table S1). In all mitogenomes under consideration, the
cox1 gene is the largest; the maximum length among them is 14,934 bp in
Sphagnum palustre, and 9450 bp in
P. commune. In other mosses, its length varies from 7081 bp in
Tetraplodon fuegianus Besch. to 9443 bp in
Pogonatum inflexum. Among exons, exon 3 of the
atp9 gene (8 bp) and exon 2 of the
nad2 gene (1314 bp) have boundary values in length distribution. Intron length varies from 480 bp (intron 3 of the
cox1 gene) up to 2649 bp (intron 3 of the
nad5 gene).
Most of the compared mitochondrial genomes do not differ from the one presented herein in the number of exons. Among the analyzed genomes, the cox1 gene contains the largest number of exons: 7 exons in Sphagnum palustre and 5 in all others. Fifty genes of P. commune consist of a single exon.
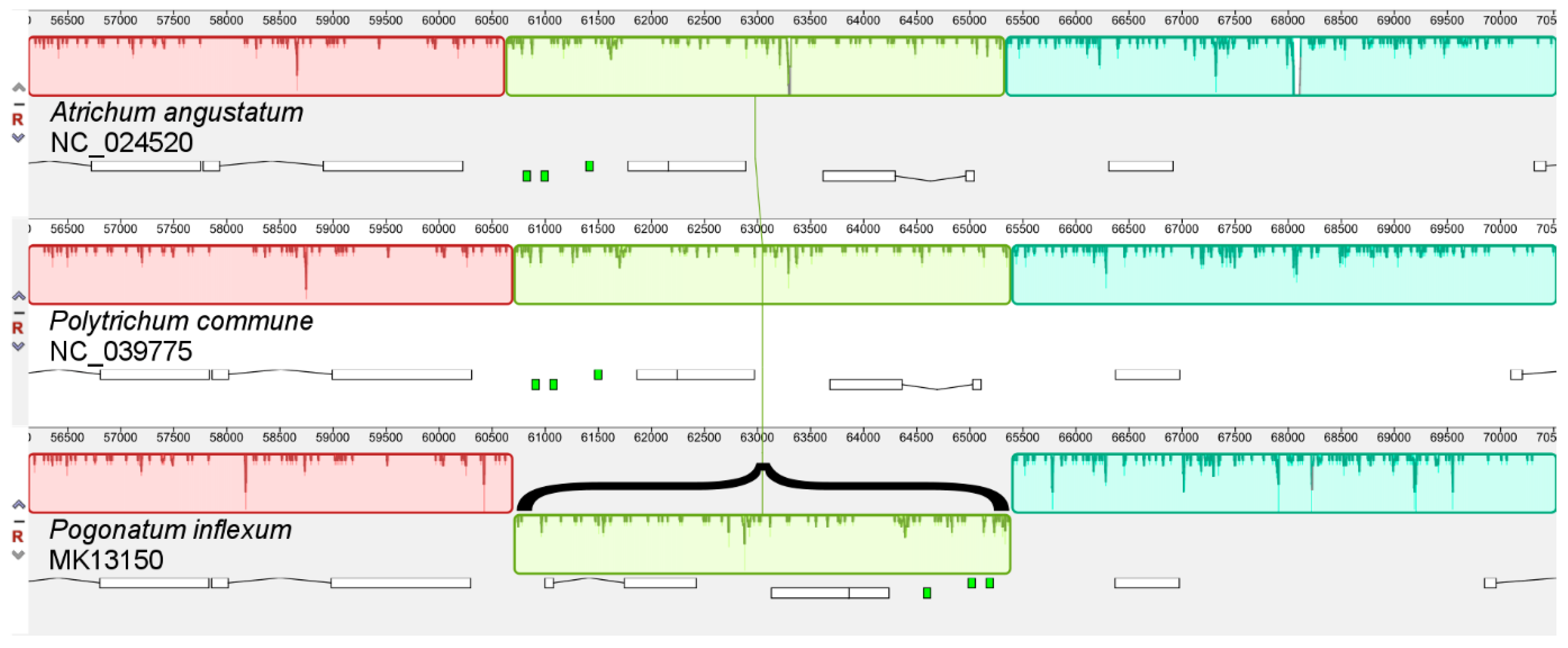
When comparing the structures of the mitogenomes of
P. commune and other Polytrichopsida species, an inversion of about 4700 bp was revealed in the
Pogonatum inflexum (
Figure 3). The
atp6,
rps7, and
rps12 genes, as well as the
trnE,
trnR, and
trnG tRNA genes, are located within the inverted genome fragment of
P. inflexum. It is interesting to note that two forward repeat subunits A
7(TA)T
8 were identified in the regions flanking the inversion. When comparing 44 bryophyte mitogenomes in the Mauve program, this rearrangement was not discovered in other mosses.
3.2. Microsatellite Content
In total, 89 microsatellite loci (SSR) were found in the mitogenome of
P. commune (
Table S2) under the following identification criteria: minimal number of repeating units ≥10 for mononucleotides, ≥5 for dinucleotides, ≥4 for trinucleotides, and ≥3 for tetra-, penta-, and hexa-nucleotides. Of these, 13 are located in coding sequences, and 76 in noncoding sequences. The SSR length distribution is shown in
Figure 4.
Most microsatellites refer to mono- and di-nucleotides classes (33 and 35 loci, respectively). Penta- and hexa-nucleotides are the least frequent SSR groups in the genome (one locus each). Among all the SSRs, 84.44% are composed only of A/T bases. The total length of the SSR loci is 1021 bp, which comprises approximately 0.89% of the genome length.
3.3. Phylogenetic Analysis
The identity between all the analyzed complete genome sequences was calculated (
Figure 5). The mitogenome of
P. commune is most similar to those of other Polytrichopsida representatives (
Atrichum angustatum (97.9%) and
Pogonatum inflexum (93.2%)), and it was the least similar to
Sphagnum palustre (65.3%) and
Buxbaumia aphylla (67.8%). Overall, the least identity was between
Buxbaumia aphylla and
Sphagnum palustre (54.3%).
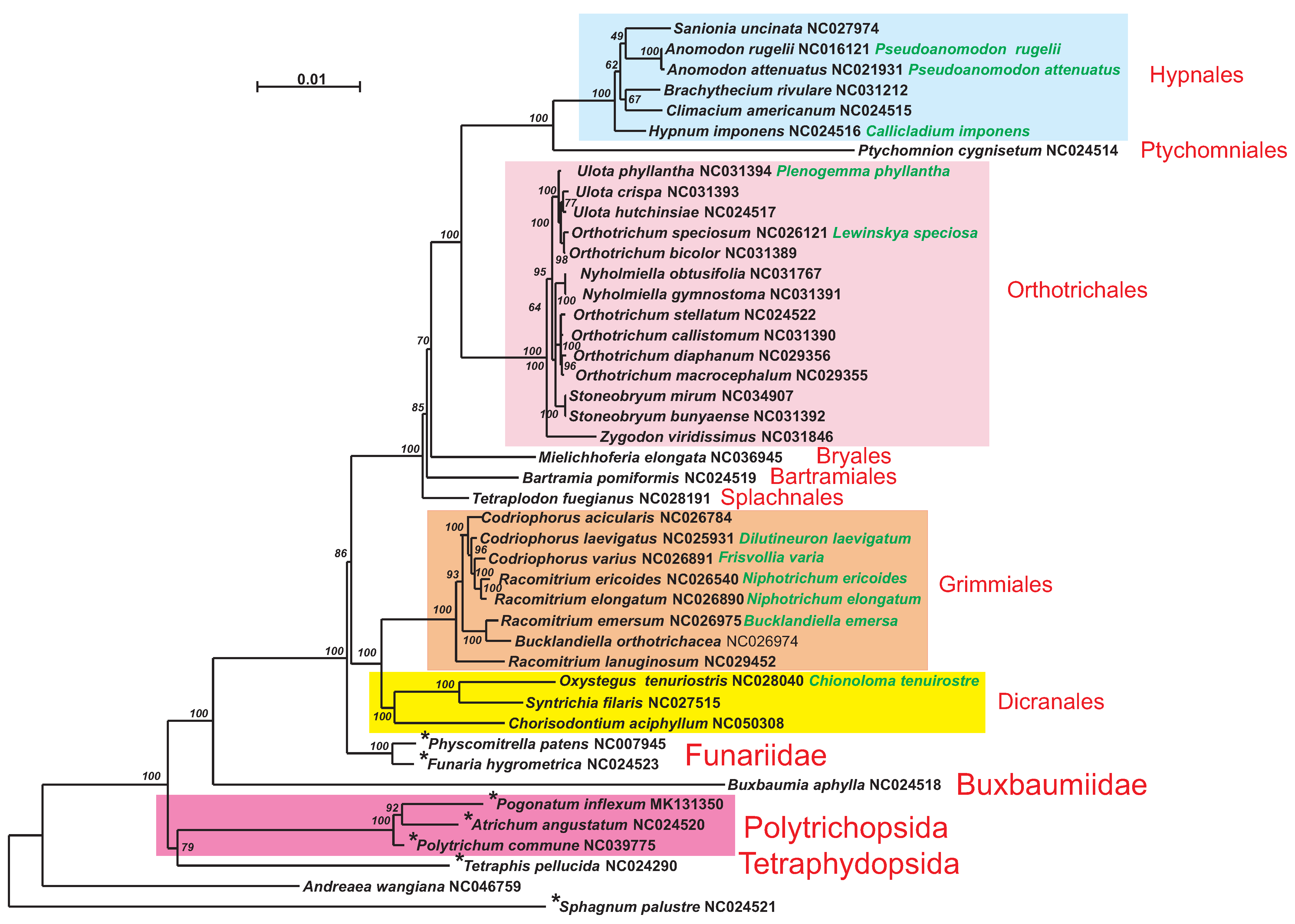
A phylogenetic tree constructed from 33 mitogenome CDS is presented in
Figure 6.
Polytrichum commune is found among other nematodontous mosses, occupying a basal position to two other Polytrichaceae species. The overall topology of the phylogenetic tree corresponds to the topology of a coalescent tree based on 272 protein-coding genes of the nucleus and organelles [
7] and reconstructions from 39 mitochondrial protein genes of 157 bryophytes [
8].
3.4. Analysis of Repeating Elements
It was suggested based on the analysis of 14 mitogenomes that the complete lack of intergenic repeat sequences, considered to be essential for intragenomic recombinations, likely accounts for the evolutionary stability of moss mitogenomes [
10]. Study of the presence of repeats, especially those localized in intergenic spacers, is extremely important for understanding the forces that maintain the highly stable mitogenome structure in mosses. We carried out a search for such repetitive elements in 44 moss mitogenomes using the same search criteria as Liu et al. [
10]: nonadjacent repeats longer than 50 bp and having at least 85% of pairwise identity between repeat subunits. It was believed that repeats with such parameters are most efficiently involved in the intragenomic recombination processes [
10].
As a result, the presence of intergenic repeats was revealed in 7 out of 44 analyzed species, which is about 16% (
Table 1). Intergenic repeats were found in
Sphagnum,
Tetraphis, in all analyzed representatives of Polytrichopsida (
P. inflexum,
A. augustatum, and
P. commune), and Funariidae (
Physcomitrella patens and
Funaria hygrometrica). All members of the Dicranales, Grimmiales, Hypnales, and Orthotrichales completely lack intergenic repeats. The largest number of repeats in intergenic spacers was found in
P. inflexum.Thus, our data on a wider sample confirm the conclusion about the small repeat numbers in the intergenic spacers of the mosses mitogenomes.
4. Discussion
Due to the high level of conservation of bryophyte mitogenomes, it is possible to construct a reliable multiple alignment not only for CDS but for complete genome sequences. When analyzing pairwise identity of whole mitogenome sequences on a heatmap, four groups are well distinguished (
Figure 5). The first one consists of
P. commune,
A. angustatum, and
P. inflexum from Polytrichopsida (intragroup identity varies from 92.85% to 97.90%). The second group is composed of Grimmiales species (intragroup identity varies from 97.62% to 99.56%). The third group is formed by members of the Orthotrichales order (intragroup identity ranges from 96.22% to 99.98%), and the fourth group includes representatives of Hypnales (intragroup identity ranges from 95.66% to 99.87%). All groups are presented as maximally supported clades on the phylogenetic tree (
Figure 6). All Bryopsida representatives are combined in a large group with a minimal pairwise identity of 79.19%. Thus, clustering of bryophytes taxa on pairwise identity of the whole genomes corresponds to phylogenetic reconstruction only from coding sequences but with the two exceptions explained in detail below.
It is worth noting that along with the basal eperistomate mosses
Sphagnum palustre and
Andreaea wangiana, there are two other taxa with surprisingly lower identity to other Bryophyta:
Tetraphis pellucida and
Buxbaumia aphylla. The pairwise identity between these and all the other mosses was not higher than 68.81% for
B. aphylla and 74.33% for
T. pellucida, respectively. Remarkably,
T. pellucida possesses the same nematodontous peristome type as other Polytrichopsida species but differs sufficiently from them in the value of the pairwise identity of entire genomes. This finding questions the treatment of these four-toothed mosses within Polytrichopsida [
1,
3]. Moreover, the joining of
Tetraphis pellucida with other Polytrichopsida species is not highly supported on the phylogenetic tree. The taxonomical placement of the
Buxbaumia genus, which has a peculiar peristome structure [
5], is not currently well defined and significantly differs according to various estimations [
1,
3,
6,
7,
8]. Unfortunately, it was not possible to conclude whether
Tetraphis or arthrodontous Bryopsida species are significantly closer to
Buxbaumia based on the performed whole mitogenomes identity analysis. However,
B. aphylla forms maximally supported clade together with another Bryopsida representatives on the phylogenetic tree.
The application of organellar CDS for phylogeny reconstruction is widely used for plants. However, the introduction in the analysis of complete genome sequences may contribute to the relatedness assessment. Moreover, it is important to involve the noncoding sequences for phylogeny reconstructions, as those are not constrained by selection.
The low structural variability of bryophyte mitochondrial genomes was broadly noted recently. Despite the inversion in the
Pogonatum inflexum mitogenome detected in the present study, the performed analysis shows a high evolutionary conservatism of the mitogenome structure in mosses. By contrast, it is known that in flowering plants, mitochondrial genomes undergo extensive and high frequency recombination that results in rearrangements and rapid evolution of their general structure. In this case, recombinations occur most often between long (several kb in size) repeats [
20,
21]. Most angiosperms have several pairs of such repeats in their mitogenomes. For example, in
Silene conica L., a species with an extremely large mitogenome, the number of large (>1 kb) repeats is 1121, and in the
Silene species, the relative frequency of recombinant genomes increases with repeat size [
22]. At the same time, recombination between the intermediate size repeats (ISRs, usually ranging from 50 to 600 bp in size) is generally infrequent [
20].
In the analyzed Bryophyta mitochondrial genomes (
Table 1), the abundance of the intergenic repeats is wider than previously indicated [
10], but the length of the repeats does not exceed 257 bp. Mitochondrial genomes of liverworts are also characterized by evolutionary stability, yet contain a considerable number of repeats in intergenic spacers. In
Gymnomitrion concinnatum (Lightf.) Corda, the only liverwort species in which structural rearrangements of the mitogenome were revealed, ten pairs of intergenic repeats were identified, with sizes varying from 107 to 566 bp. The two longest repeats, 566 bp (97.7% sequence identity) and 435 bp length (95.9% sequence identity), were located on junctions between conservative blocks. In each of the six analyzed liverwort species except
Aneura pinguis (L.) Dumort., two pairs of repeated sequences exceeding 400 bp and 90% of identity were found [
9]. Thus, evolutionarily stable liverwort genomes contain only intermediate size repeats but lack long repeats. In chlorophytes and charophytes, which are the groups of algae closest to land plants, the length of repeats in the mitochondrial genome also does not exceed five hundred base pairs [
23].
Thus, long repeats exceeding 1 kb were observed only in vascular plants. Hence, it can be concluded that long repeats do have a key influence on mitogenome stability in Viridiplantae, and the greater stability of the mitochondrial genomes of mosses apparently is associated mainly with the absence of long repeats, although recombination events between shorter repetitive sequences can occur with a low frequency. Therefore, the inversion observed in the polytrichopsid moss Pogonatum inflexum that is flanked by a short 17 bp repeat demonstrates that even those shorter than 50 bp facilitate rearrangements in mitogenomes.
Thus, the extremely low structural variability and lack of the long repetitive elements in moss mitogenomes is an ancient trait. This statement is in good agreement with the conclusion of Emily Wynn and Alan Christensen, that the first vascular common ancestor of lycophytes, ferns, gymnosperms, and angiosperms acquired new mechanisms of mitochondrial genome replication and repair that led to an expansion of repeats and increasing repeat size and mitochondrial genome length [
23].
It is worth noting that distribution of intergenic repeats along the phylogenetic tree of mosses is not random (
Figure 6). The repeats are present in mitogenomes of most basal species but absent in the species of terminal clades. A mitogenome size reduction during the evolution of mosses has recently been noted in the analysis of the
Andreaea wangiana mitogenome [
24]. The above-mentioned trends to miniaturization of the mitochondrial genome and loss of intergenic repeats may indicate a strong selection toward further stabilization of the genome structure in mosses.
The revealed inversion within Pogonatum inflexum mitogenome is the only currently known rearrangement in bryophytes, and the sequencing of new moss mitogenomes is crucial for an adequate assessment of the level of conservatism in its structure as well as the deep insight into the evolutionary history of bryophytes.


 and
and 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}