
Geographic and Viral Etiology Patterns of TERT Promoter and CTNNB1 Exon 3 Mutations in Hepatocellular Carcinoma: A Comprehensive Review

 , , and
, , and
Abstract
1. Introduction
2. Structure and Function of Telomeres and Telomerase
3. The Role of TERT in HCC
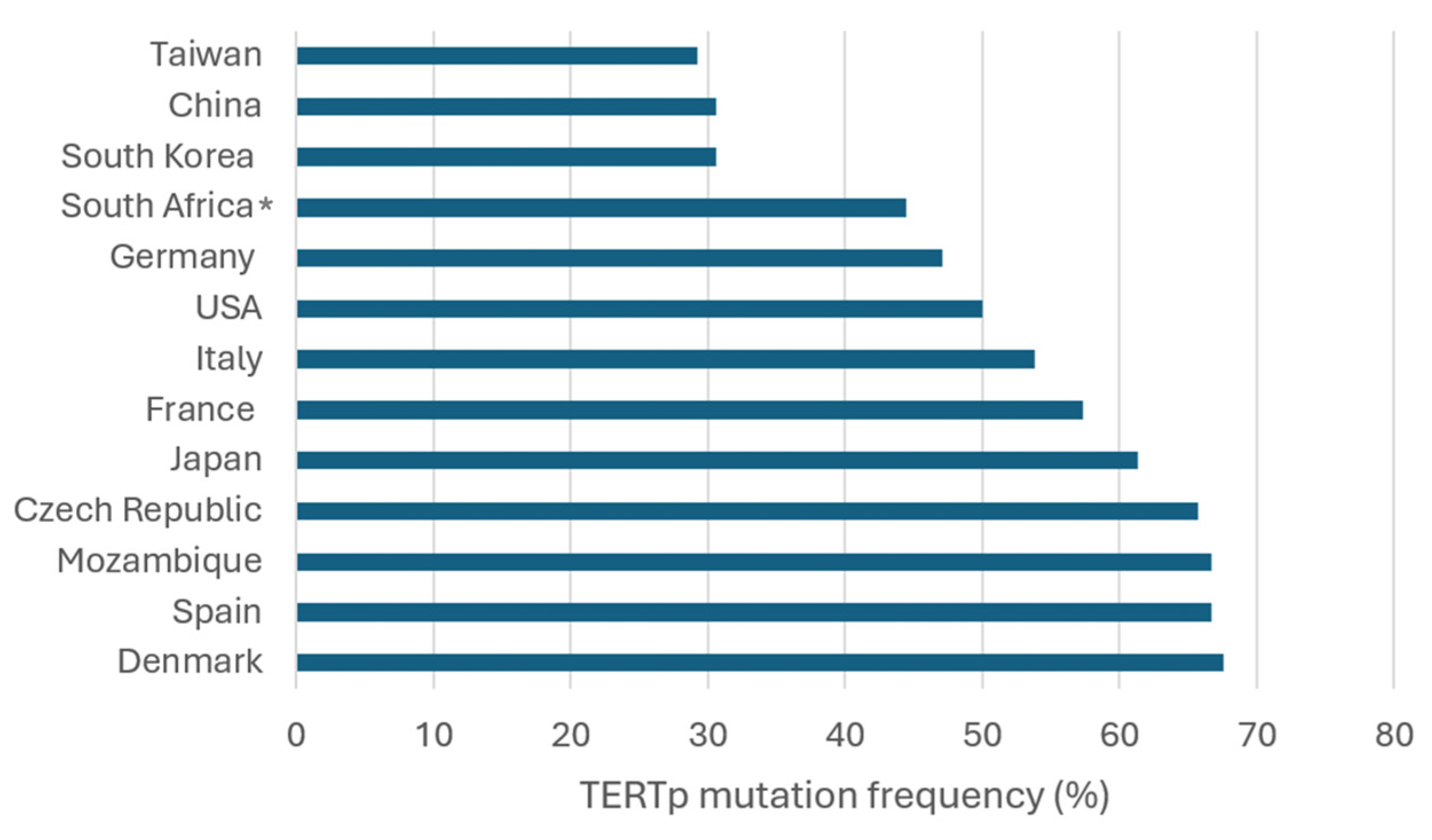
4. Frequency and Geographic Distribution of TERTp Mutations in HCC

{kind=link}
{kind=link}
{kind=link}
| Refs. | Country/Region | TERTp Mut (n, %) a | C228T (n, %) b | C250T (n, %) c | HBV + Mut (n, %) d | HCV + Mut (n, %) e |
|---|---|---|---|---|---|---|
| [66] | China (n = 190) | 57/190 (30) | 50/57 (87.7) | 7/57 (12.3) | 50/153 (32.7) | NA |
| [67] | China (n = 275) | 85/275 (30.9) | 84/85 (98.8) | 1/85 (1.2) | 78/259 (30.1) | NA |
| [75] | China (n = 35) | 11/35 (31.4) | 9/11 (81.8) | 2/11 (18.2) | NA | NA |
| [56] | Czech Republic (n = 67) | 44/67 (65.7) | 41/44 (93.2) | 3/44 (6.8) | NA | NA |
| [55] | Denmark (n = 34) | 23/34 (67.6) | 23/23 (100) | NA | NA | 4/5 (80) |
| [15] | France (n = 193) | 120/193 (62.1) f | 106/120 (88.3) | 5/120 (4.2) | 10/24 (41.7) | 27/36 (75) |
| [13] | France (n = 305) | 179/305 (58.7) | 168/179 (93.9) | 11/179 (6.1) | 26/67 (38.8) | 49/68 (72.1) |
| [17] | France (n = 75) | 23/75 (30.7) | 19/23 (82.6) | 4/23 (17.4) | NA | NA |
| [51] | France (n = 759) | 441/759 (58.1) f | 404/441 (91.6) | 19/441 (4.3) | NA | NA |
| [54] | Germany (n = 7) | 3/7 (42.9) | 2/3 (66.7) | 1/3 (33.3) | 1/3 (33.3) | NA |
| [53] | Germany (n = 78) | 37/78 (47.4) | 37/37 (100) | 0/37 (0) | NA | NA |
| [52] | Italy (n = 114) | 79/114 (69.3) | 79/79 (100) | 0/79 (0) | 6/10 (60) | 72/99 (72.7) |
| [70] | Italy (n = 127) | 64/127 (50.4) | 62/64 (96.9) | 2/64 (3.1) | 5/12 (41.7) | 59/110 (53.6) |
| [17] | Italy (n = 21) | 6/21 (28.6) | 6/6 (100) | 0/6 (0) | NA | NA |
| [15] | Italy (n = 41) | 21/41 (51.2) f | 20/21 (95.2) | 0/21 (0) | 7/10 (70) | 8/20 (40) |
| [71] | Italy (n = 67) | 29/67 (43.3) f | 28/29 (96.6) | 0/29 (0) | 2/3 (66.7) | 18/39 (46.2) |
| [59] | Japan (n = 104) | 68/104 (65.4) | 66/68 (97.1) | 2/68 (2.9) | 9/28 (32.1) | 40/50 (80) |
| [60] | Japan (n = 11) | 9/11 (81.8) | 9/9 (100) | NA | NA | NA |
| [54] | Japan (n = 11) | 4/11 (36.4) | 3/4 (75) | 1/4 (25) | 0/1 (0) | NA |
| [62] | Japan (n = 125) | 85/125 (68) | 83/85 (97.6) | 2/85 (2.4) | 14/27 (51.9) | 59/75 (78.7) |
| [57] | Japan (n = 36) | 21/36 (58.3) | 21/21 (100) | 0/21 (0) | NA | NA |
| [58] | Japan (n = 36) | 23/36 (63.9) | 23/23 (100) | NA | NA | NA |
| [19] | Japan (n = 374) | 224/374 (59.9) f | 208/224 (92.9) | 9/224 (4) | 40/107 (37.4) | 104/139 (74.8) |
| [61] | Japan (n = 97) | 53/97 (54.6) | 52/53 (98.1) | 1/53 (1.9) | 8/21 (38.1) | 21/30 (70) |
| [54] | Lesotho (n = 2) | 1/2 (50) | 0/1 (0) | 1/1 (100) | 1/2 (50) | NA |
| [54] | Mozambique (n = 6) | 4/6 (66.7) | 2/4 (50) | 2/4 (50) | 3/5 (60) | NA |
| [54] | South Africa (n = 2) | 1/2 (50) | 1/1 (100) | 0/1 (0) | 1/2 (50) | NA |
| [63] | South Korea (n = 105) | 41/105 (39) | 39/41 (95.1) | 2/41 (4.9) | 23/78 (29.5) | 5/6 (83.3) |
| [76] | South Korea (n = 160) | 46/160 (28.8) | 32/46 (69.6) | 14/46 (30.4) | 19/58 (32.8) | 3/5 (60) |
| [64] | South Korea (n = 205) | 57/205 (27.8) | 54/57 (94.7) | 3/57 (5.3) | 32/138 (23.2) | 7/16 (43.8) |
| [15] | Spain (n = 9) | 6/9 (66.7) | 6/6 (100) | 0/6 (0) | 1/1 (100) | 4/5 (80) |
| [54] | Swaziland (n = 1) | 0/1 (0) | 0/1 (0) | 0/1 (0) | 0/1 (0) | NA |
| [65] | Taiwan (n = 195) | 57/195 (29.2) | 54/57 (94.7) | 3/57 (5.3) | 27/121 (22.3) | 24/50 (48) |
| [54] | Transkei (n = 4) | 2/4 (50) | 2/2 (100) | 0/2 | 0/1 (0) | NA |
| [69] | USA (n = 61) | 27/61 (44.3) | 26/27 (96.3) | 1/27 (3.7) | 4/15 (26.7) | 10/16 (62.5) |
| [68] | USA (n = 70) | 50/70 (71.4) | 49/50 (98) | 1/50 (2) | 0/7 (0) | 36/40 (90) |
| [19] | USA (n = 89) | 33/89 (37.1) | 31/33 (93.9) | 2/33 (6.2) | 2/13 (15.4) | 20/51 (39.2) |
| Total (n = 4133) | 2034/4133 (49.2) | 1899/2035 (93.3) | 99/2035 (4.9) | 369/1167 (31.6) | 570/861 (66.2) |
5. Wnt/β-Catenin Signaling
6. Wnt/β-Catenin in HCC
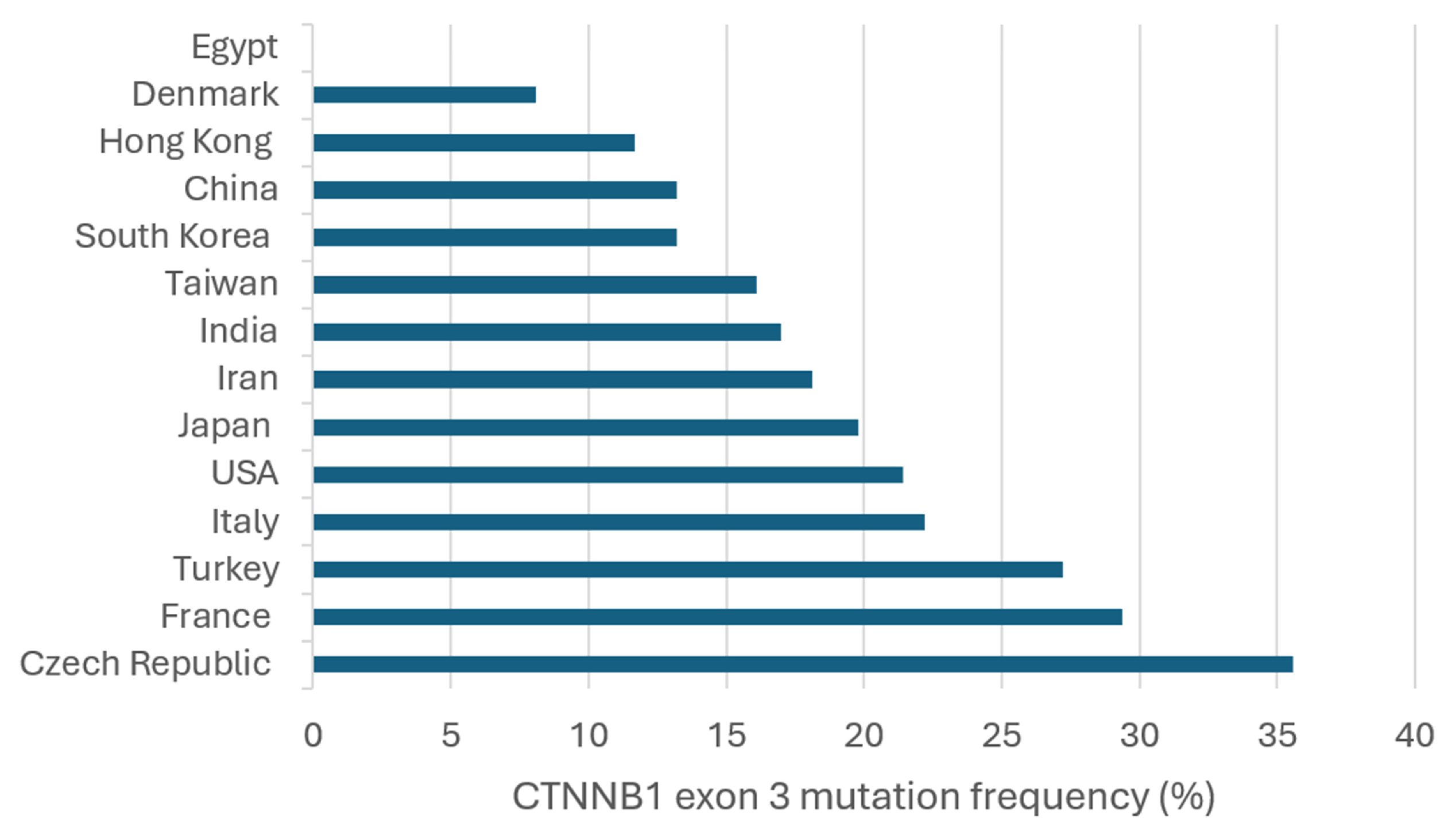
7. Frequency and Geographic Distribution of CTNNB1 Exon 3 Mutations in HCC
| Refs. | Country/Region | CTNNB1 Exon 3 (n, %) a | Mutation Sites | HBV+ Mut (n, %) b | HCV+ Mut (n, %) c |
|---|---|---|---|---|---|
| [120] | China (n = 156) | 15/156 (9.6) | D32G/Y G34E/V S37C T41A S45P | 12/109 (11) | NA |
| [112] | China (n = 39) | 2/39 (5.1) | D32N S37F | 2/29 (6.9) | NA |
| [66] | China (n = 70) | 17/70 (24.3) | NA | NA | NA |
| [56] | Czech Republic (n = 59) | 21/59 (35.6) | S29S D32V S33C/F/Y G34V S37C/Y T41A S45F/Y G48G E53E V57M | NA | NA |
| [109] | Denmark (n = 37) | 3/37 (8.1) | T41A | NA | 1/4 (25) |
| [102] | Egypt (n = 20) | 0/20 (0) | None | NA | NA |
| [121] | France (n = 137) | 26/137 (19) | S23R D32A/G S33C/F/L/S G34R/V I35S H36P S37A/Y T41A/I S45A/F/P | 1/42 (2.4) | 12/40 (30) |
| [13] | France (n = 304) | 101/304 (33.2) | H24P D32A/G/H/N/V/Y S33A/C/F/P/T/Y G34R/V I35S H36P S37C/F/P T41A/I S45A/F/P/Y | NA | NA |
| [104] | France (n = 42) | 21/42 (50) | D32G/N/Y S33A/C/F/P G34E/R S37A T41A/I S45F/P S45P | 3/7 (42.9) | 6/9 (66.7) |
| [106] | France (n = 45) | 20/45 (44.4) | D32G S33C/P/Y G34V S37Y T41A S45A/F/P/Y | 0/6 (0) | 5/8 (62.5) |
| [51] | France (n = 746) | 207/746 (27.7) | NA | NA | NA |
| [15] | France, Italy, and Spain (n = 235) | 82/235 (34.9) | NA | NA | NA |
| [114] | Hong Kong (n = 60) | 7/60 (11.7) | G34V I35S H36P T41A S45F/T | 5/48 (10.4) | 0/2 (0) |
| [117] | India (n = 15) | 2/15 (13.3) | G32C/S | 2/15 (13.3) | NA |
| [116] | India (n = 32) | 6/32 (18.8) | S33T T40P P52H E54A E55Q V57M | 6/32 (18.8) | NA |
| [118] | Iran (n = 105) | 19/105 (18.1) | D32G/V S33C H36Q S37C G38R/S/V A39V T41P T42A P44R S45P | 9/71 (12.7) | NA |
| [70] | Italy (n = 127) | 33/127 (26) | S33 S37 S45 | 2/12 (16.7) | 29/110 (26.4) |
| [107] | Italy (n = 67) | 10/67 (14.9) | D32H S33A/C G34E/V I35S S37F/Y S45P | 0/10 (0) | 10/57 (17.5) |
| [17] | Italy and France (n = 7) | 1/7 (14.3) | NA | NA | NA |
| [59] | Japan (n = 104) | 31/104 (29.8) | NA | 3/31 (9.7) | 19/31 (61.3) |
| [60] | Japan (n = 11) | 5/11 (45.5) | NA | NA | NA |
| [62] | Japan (n = 125) | 31/125 (24.8) | D32N/Y S33C/F/P G34R/V H36P/R S37C/F T41A P44A S45P/F | 7/27 (25.9) | 21/75 (28) |
| [111] | Japan (n = 42) | 18/42 (42.9) | NA | NA | NA |
| [110] | Japan (n = 434) | 57/434 (13.1) | D32G/N/V/Y S33A/C/P G34E/R/V H36P S37C/F/Y T41A/I S45A/F/P | 30/323 (9.3) | 23/92 (25) |
| [122] | Japan and Switzerland (n = 22) | 9/22 (40.9) | D32A/G/N/Y S33Y S37P/Y T41A S45F/P | 0/22 (0) | 9/22 (40.9) |
| [63] | South Korea (n = 103) | 15/103 (14.6) | S29F D32A/G/N/V S33C S37A T41A S45A/F/P | 10/76 (13.2) | 1/7 (14.3) |
| [103] | South Korea (n = 36) | 1/36 (2.8) | T41A | 0/21 (0) | 0/4 (0) |
| [115] | South Korea (n = 81) | 13/81 (16) | D32G S33F/P G34R/V H36P S37Y T41A S45F | 13/78 (16.7) | 0/6 (0) |
| [113] | Taiwan (n = 115) | 21/115 (18.3) | S23N L31P D32G/N/V S33C/P G34E/R/V S37A T41A/I T42I S45P P52L G69E | 13/78 (16.7) | 5/24 (20.8) |
| [123] | Taiwan (n = 150) | 22/150 (14.7) | NA | NA | NA |
| [65] | Taiwan (n = 188) | 31/188 (16.5) | NA | 15/121 (12.4) | 14/50 (28) |
| [124] | Taiwan (n = 214) | 32/214 (15) | NA | NA | NA |
| [90] | Taiwan (n = 262) | 37/262 (14.1) | D32 S33 G34 H36 S37 T41 S45 | NA | NA |
| [125] | Taiwan (n = 73) | 18/73 (24.7) | NA | NA | NA |
| [108] | Turkey (n = 360) | 98/360 (27.2) | S45P/F/Y D32G/V/N | NA | NA |
| [119] | USA (n = 32) | 9/32 (28.1) | L30Q D32G/V S33C S37F T41F T42P S45P | NA | 3/6 (50) |
| [105] | USA (n = 7) The Netherlands (n = 1) China (n = 1) | 5/7 (71.4) 0/1 (0) 1/1 (100) | D32G/H G34V H36P | 0 0 1/1 (100) | 4/5 (80) 0/1 (0) 0/1 (0) |
| [68] | USA (n = 70) | 11/70 (15.7) | I35S S37C S45F/P/Y | 0/7 (0) | 9/40 (22.5) |
| [126] | USA (n = 73) | 14/73 (19.2) | D32G S33Y G34E/V T41A S45C/P/Y | NA | NA |
| [19] | USA and Japan (n = 469) | 146/469 (31.1) | Q28R D32G/H/N/V/Y S33A/C/F/P/Y G34E/R/V I35S H36P S37A/C/F/P/Y S45A/C/F/P/T/Y T41A/I T42K | 29/108 (26.9) | 69/188 (36.7) |
| Total (n = 5276) | 1218/5276 (23.1) | 163/1274 (12.8) | 240/782 (30.7) |
8. Interactions Between TERT and β-Catenin in HCC
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.E.M.; Lam, F.; Laversanne, M.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Today. Lyon, France: International Agency for Research on Cancer. Available online: https://gco.iarc.who.int/today (accessed on 18 January 2025).
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Ringelhan, M.; McKeating, J.A.; Protzer, U. Viral hepatitis and liver cancer. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160274. [Google Scholar] [CrossRef]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef]
- Maucort-Boulch, D.; de Martel, C.; Franceschi, S.; Plummer, M. Fraction and incidence of liver cancer attributable to hepatitis B and C viruses worldwide. Int. J. Cancer 2018, 142, 2471–2477. [Google Scholar] [CrossRef]
- Kulik, L.; El-Serag, H.B. Epidemiology and Management of Hepatocellular Carcinoma. Gastroenterology 2019, 156, 477–491.e1. [Google Scholar] [CrossRef]
- Calderaro, J.; Ziol, M.; Paradis, V.; Zucman-Rossi, J. Molecular and histological correlations in liver cancer. J. Hepatol. 2019, 71, 616–630. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23. [Google Scholar] [CrossRef]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef]
- Kotiyal, S.; Evason, K.J. Exploring the Interplay of Telomerase Reverse Transcriptase and beta-Catenin in Hepatocellular Carcinoma. Cancers 2021, 13, 4202. [Google Scholar] [CrossRef]
- Schulze, K.; Nault, J.C.; Villanueva, A. Genetic profiling of hepatocellular carcinoma using next-generation sequencing. J. Hepatol. 2016, 65, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.C.; Mallet, M.; Pilati, C.; Calderaro, J.; Bioulac-Sage, P.; Laurent, C.; Laurent, A.; Cherqui, D.; Balabaud, C.; Zucman-Rossi, J. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat. Commun. 2013, 4, 2218. [Google Scholar] [CrossRef]
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K.; et al. TERT promoter mutations in familial and sporadic melanoma. Science 2013, 339, 959–961. [Google Scholar] [CrossRef] [PubMed]
- Schulze, K.; Imbeaud, S.; Letouze, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Khemlina, G.; Ikeda, S.; Kurzrock, R. The biology of Hepatocellular carcinoma: Implications for genomic and immune therapies. Mol. Cancer 2017, 16, 149. [Google Scholar] [CrossRef]
- Nault, J.C.; Calderaro, J.; Di Tommaso, L.; Balabaud, C.; Zafrani, E.S.; Bioulac-Sage, P.; Roncalli, M.; Zucman-Rossi, J. Telomerase reverse transcriptase promoter mutation is an early somatic genetic alteration in the transformation of premalignant nodules in hepatocellular carcinoma on cirrhosis. Hepatology 2014, 60, 1983–1992. [Google Scholar] [CrossRef]
- Pezzuto, F.; Buonaguro, L.; Buonaguro, F.M.; Tornesello, M.L. Frequency and geographic distribution of TERT promoter mutations in primary hepatocellular carcinoma. Infect. Agent. Cancer 2017, 12, 27. [Google Scholar] [CrossRef]
- Totoki, Y.; Tatsuno, K.; Covington, K.R.; Ueda, H.; Creighton, C.J.; Kato, M.; Tsuji, S.; Donehower, L.A.; Slagle, B.L.; Nakamura, H.; et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat. Genet. 2014, 46, 1267–1273. [Google Scholar] [CrossRef]
- Blackburn, E.H. Switching and signaling at the telomere. Cell 2001, 106, 661–673. [Google Scholar] [CrossRef]
- Palm, W.; de Lange, T. How shelterin protects mammalian telomeres. Annu. Rev. Genet. 2008, 42, 301–334. [Google Scholar] [CrossRef]
- Griffith, J.D.; Comeau, L.; Rosenfield, S.; Stansel, R.M.; Bianchi, A.; Moss, H.; de Lange, T. Mammalian telomeres end in a large duplex loop. Cell 1999, 97, 503–514. [Google Scholar] [CrossRef]
- Greider, C.W.; Blackburn, E.H. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell 1985, 43, 405–413. [Google Scholar] [CrossRef]
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.P.; Morin, G.B.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of life-span by introduction of telomerase into normal human cells. Science 1998, 279, 349–352. [Google Scholar] [CrossRef]
- Armanios, M.; Blackburn, E.H. The telomere syndromes. Nat. Rev. Genet. 2012, 13, 693–704. [Google Scholar] [CrossRef]
- Oh, H.; Taffet, G.E.; Youker, K.A.; Entman, M.L.; Overbeek, P.A.; Michael, L.H.; Schneider, M.D. Telomerase reverse transcriptase promotes cardiac muscle cell proliferation, hypertrophy, and survival. Proc. Natl. Acad. Sci. USA 2001, 98, 10308–10313. [Google Scholar] [CrossRef]
- Wu, K.J.; Grandori, C.; Amacker, M.; Simon-Vermot, N.; Polack, A.; Lingner, J.; Dalla-Favera, R. Direct activation of TERT transcription by c-MYC. Nat. Genet. 1999, 21, 220–224. [Google Scholar] [CrossRef]
- Ulaner, G.A.; Hu, J.F.; Vu, T.H.; Giudice, L.C.; Hoffman, A.R. Tissue-specific alternate splicing of human telomerase reverse transcriptase (hTERT) influences telomere lengths during human development. Int. J. Cancer 2001, 91, 644–649. [Google Scholar] [CrossRef]
- Haendeler, J.; Hoffmann, J.; Rahman, S.; Zeiher, A.M.; Dimmeler, S. Regulation of telomerase activity and anti-apoptotic function by protein-protein interaction and phosphorylation. FEBS Lett. 2003, 536, 180–186. [Google Scholar] [CrossRef]
- Singhapol, C.; Pal, D.; Czapiewski, R.; Porika, M.; Nelson, G.; Saretzki, G.C. Mitochondrial telomerase protects cancer cells from nuclear DNA damage and apoptosis. PLoS ONE 2013, 8, e52989. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Saginc, G.; Leow, S.C.; Khattar, E.; Shin, E.M.; Yan, T.D.; Wong, M.; Zhang, Z.; Li, G.; Sung, W.K.; et al. Telomerase directly regulates NF-kappaB-dependent transcription. Nat. Cell Biol. 2012, 14, 1270–1281. [Google Scholar] [CrossRef]
- Nault, J.C.; Zucman-Rossi, J. TERT promoter mutations in primary liver tumors. Clin. Res. Hepatol. Gastroenterol. 2016, 40, 9–14. [Google Scholar] [CrossRef]
- Shay, J.W.; Wright, W.E. Telomeres and telomerase: Three decades of progress. Nat. Rev. Genet. 2019, 20, 299–309. [Google Scholar] [CrossRef]
- Fujimoto, A.; Furuta, M.; Shiraishi, Y.; Gotoh, K.; Kawakami, Y.; Arihiro, K.; Nakamura, T.; Ueno, M.; Ariizumi, S.; Nguyen, H.H.; et al. Whole-genome mutational landscape of liver cancers displaying biliary phenotype reveals hepatitis impact and molecular diversity. Nat. Commun. 2015, 6, 6120. [Google Scholar] [CrossRef]
- Zhu, R.X.; Seto, W.K.; Lai, C.L.; Yuen, M.F. Epidemiology of Hepatocellular Carcinoma in the Asia-Pacific Region. Gut Liver 2016, 10, 332–339. [Google Scholar] [CrossRef]
- Hoffmeyer, K.; Raggioli, A.; Rudloff, S.; Anton, R.; Hierholzer, A.; Del Valle, I.; Hein, K.; Vogt, R.; Kemler, R. Wnt/beta-catenin signaling regulates telomerase in stem cells and cancer cells. Science 2012, 336, 1549–1554. [Google Scholar] [CrossRef]
- Park, J.I.; Venteicher, A.S.; Hong, J.Y.; Choi, J.; Jun, S.; Shkreli, M.; Chang, W.; Meng, Z.; Cheung, P.; Ji, H.; et al. Telomerase modulates Wnt signalling by association with target gene chromatin. Nature 2009, 460, 66–72. [Google Scholar] [CrossRef]
- Relitti, N.; Saraswati, A.P.; Federico, S.; Khan, T.; Brindisi, M.; Zisterer, D.; Brogi, S.; Gemma, S.; Butini, S.; Campiani, G. Telomerase-based Cancer Therapeutics: A Review on their Clinical Trials. Curr. Top. Med. Chem. 2020, 20, 433–457. [Google Scholar] [CrossRef]
- Asai, A.; Oshima, Y.; Yamamoto, Y.; Uochi, T.A.; Kusaka, H.; Akinaga, S.; Yamashita, Y.; Pongracz, K.; Pruzan, R.; Wunder, E.; et al. A novel telomerase template antagonist (GRN163) as a potential anticancer agent. Cancer Res. 2003, 63, 3931–3939. [Google Scholar]
- Djojosubroto, M.W.; Chin, A.C.; Go, N.; Schaetzlein, S.; Manns, M.P.; Gryaznov, S.; Harley, C.B.; Rudolph, K.L. Telomerase antagonists GRN163 and GRN163L inhibit tumor growth and increase chemosensitivity of human hepatoma. Hepatology 2005, 42, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- The American Association for Cancer Research (AACR). Imetelstat Approved for Certain Myelodysplastic Syndromes. Available online: https://www.aacr.org/patients-caregivers/progress-against-cancer/imetelstat-approved-for-certain-myelodysplastic-syndromes/ (accessed on 18 January 2024).
- Vonderheide, R.H. Telomerase as a universal tumor-associated antigen for cancer immunotherapy. Oncogene 2002, 21, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Vonderheide, R.H. Prospects and challenges of building a cancer vaccine targeting telomerase. Biochimie 2008, 90, 173–180. [Google Scholar] [CrossRef]
- Vahidi, S.; Zabeti Touchaei, A. Telomerase-based vaccines: A promising frontier in cancer immunotherapy. Cancer Cell Int. 2024, 24, 421. [Google Scholar] [CrossRef]
- Chehelgerdi, M.; Chehelgerdi, M.; Khorramian-Ghahfarokhi, M.; Shafieizadeh, M.; Mahmoudi, E.; Eskandari, F.; Rashidi, M.; Arshi, A.; Mokhtari-Farsani, A. Comprehensive review of CRISPR-based gene editing: Mechanisms, challenges, and applications in cancer therapy. Mol. Cancer 2024, 23, 9. [Google Scholar] [CrossRef]
- Yu, S.; Zhao, R.; Zhang, B.; Lai, C.; Li, L.; Shen, J.; Tan, X.; Shao, J. Research progress and application of the CRISPR/Cas9 gene-editing technology based on hepatocellular carcinoma. Asian J. Pharm. Sci. 2023, 18, 100828. [Google Scholar] [CrossRef]
- Jaskelioff, M.; Muller, F.L.; Paik, J.H.; Thomas, E.; Jiang, S.; Adams, A.C.; Sahin, E.; Kost-Alimova, M.; Protopopov, A.; Cadinanos, J.; et al. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature 2011, 469, 102–106. [Google Scholar] [CrossRef]
- Tang, Q.; Hu, G.; Sang, Y.; Chen, Y.; Wei, G.; Zhu, M.; Chen, M.; Li, S.; Liu, R.; Peng, Z. Therapeutic targeting of PLK1 in TERT promoter-mutant hepatocellular carcinoma. Clin. Transl. Med. 2024, 14, e1703. [Google Scholar] [CrossRef]
- Rashid, S.; Sun, Y.; Ali Khan Saddozai, U.; Hayyat, S.; Munir, M.U.; Akbar, M.U.; Khawar, M.B.; Ren, Z.; Ji, X.; Ihsan Ullah Khan, M. Circulating tumor DNA and its role in detection, prognosis and therapeutics of hepatocellular carcinoma. Chin. J. Cancer Res. 2024, 36, 195–214. [Google Scholar] [CrossRef]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef]
- Nault, J.C.; Martin, Y.; Caruso, S.; Hirsch, T.Z.; Bayard, Q.; Calderaro, J.; Charpy, C.; Copie-Bergman, C.; Ziol, M.; Bioulac-Sage, P.; et al. Clinical Impact of Genomic Diversity From Early to Advanced Hepatocellular Carcinoma. Hepatology 2020, 71, 164–182. [Google Scholar] [CrossRef] [PubMed]
- Pezzuto, F.; Izzo, F.; De Luca, P.; Biffali, E.; Buonaguro, L.; Tatangelo, F.; Buonaguro, F.M.; Tornesello, M.L. Clinical Significance of Telomerase Reverse-Transcriptase Promoter Mutations in Hepatocellular Carcinoma. Cancers 2021, 13, 3771. [Google Scholar] [CrossRef] [PubMed]
- Quaas, A.; Oldopp, T.; Tharun, L.; Klingenfeld, C.; Krech, T.; Sauter, G.; Grob, T.J. Frequency of TERT promoter mutations in primary tumors of the liver. Virchows Arch. 2014, 465, 673–677. [Google Scholar] [CrossRef]
- Cevik, D.; Yildiz, G.; Ozturk, M. Common telomerase reverse transcriptase promoter mutations in hepatocellular carcinomas from different geographical locations. World J. Gastroenterol. 2015, 21, 311–317. [Google Scholar] [CrossRef]
- Oversoe, S.K.; Clement, M.S.; Pedersen, M.H.; Weber, B.; Aagaard, N.K.; Villadsen, G.E.; Gronbaek, H.; Hamilton-Dutoit, S.J.; Sorensen, B.S.; Kelsen, J. TERT promoter mutated circulating tumor DNA as a biomarker for prognosis in hepatocellular carcinoma. Scand. J. Gastroenterol. 2020, 55, 1433–1440. [Google Scholar] [CrossRef]
- Ambrozkiewicz, F.; Trailin, A.; Cervenkova, L.; Vaclavikova, R.; Hanicinec, V.; Allah, M.A.O.; Palek, R.; Treska, V.; Daum, O.; Tonar, Z.; et al. CTNNB1 mutations, TERT polymorphism and CD8+ cell densities in resected hepatocellular carcinoma are associated with longer time to recurrence. BMC Cancer 2022, 22, 884. [Google Scholar] [CrossRef]
- Ako, S.; Nouso, K.; Kinugasa, H.; Matsushita, H.; Terasawa, H.; Adachi, T.; Wada, N.; Takeuchi, Y.; Mandai, M.; Onishi, H.; et al. Human Telomerase Reverse Transcriptase Gene Promoter Mutation in Serum of Patients with Hepatocellular Carcinoma. Oncology 2020, 98, 311–317. [Google Scholar] [CrossRef]
- Akuta, N.; Kawamura, Y.; Kobayashi, M.; Arase, Y.; Saitoh, S.; Fujiyama, S.; Sezaki, H.; Hosaka, T.; Kobayashi, M.; Suzuki, Y.; et al. TERT Promoter Mutation in Serum Cell-Free DNA Is a Diagnostic Marker of Primary Hepatocellular Carcinoma in Patients with Nonalcoholic Fatty Liver Disease. Oncology 2021, 99, 114–123. [Google Scholar] [CrossRef]
- Kawai-Kitahata, F.; Asahina, Y.; Tanaka, S.; Kakinuma, S.; Murakawa, M.; Nitta, S.; Watanabe, T.; Otani, S.; Taniguchi, M.; Goto, F.; et al. Comprehensive analyses of mutations and hepatitis B virus integration in hepatocellular carcinoma with clinicopathological features. J. Gastroenterol. 2016, 51, 473–486. [Google Scholar] [CrossRef]
- Ki Kim, S.; Ueda, Y.; Hatano, E.; Kakiuchi, N.; Takeda, H.; Goto, T.; Shimizu, T.; Yoshida, K.; Ikura, Y.; Shiraishi, Y.; et al. TERT promoter mutations and chromosome 8p loss are characteristic of nonalcoholic fatty liver disease-related hepatocellular carcinoma. Int. J. Cancer 2016, 139, 2512–2518. [Google Scholar] [CrossRef]
- Kwa, W.T.; Effendi, K.; Yamazaki, K.; Kubota, N.; Hatano, M.; Ueno, A.; Masugi, Y.; Sakamoto, M. Telomerase reverse transcriptase (TERT) promoter mutation correlated with intratumoral heterogeneity in hepatocellular carcinoma. Pathol. Int. 2020, 70, 624–632. [Google Scholar] [CrossRef]
- Nishida, N.; Nishimura, T.; Kaido, T.; Minaga, K.; Yamao, K.; Kamata, K.; Takenaka, M.; Ida, H.; Hagiwara, S.; Minami, Y.; et al. Molecular Scoring of Hepatocellular Carcinoma for Predicting Metastatic Recurrence and Requirements of Systemic Chemotherapy. Cancers 2018, 10, 367. [Google Scholar] [CrossRef]
- Lee, S.E.; Chang, S.H.; Kim, W.Y.; Lim, S.D.; Kim, W.S.; Hwang, T.S.; Han, H.S. Frequent somatic TERT promoter mutations and CTNNB1 mutations in hepatocellular carcinoma. Oncotarget 2016, 7, 69267–69275. [Google Scholar] [CrossRef]
- Jang, J.W.; Kim, J.S.; Kim, H.S.; Tak, K.Y.; Lee, S.K.; Nam, H.C.; Sung, P.S.; Kim, C.M.; Park, J.Y.; Bae, S.H.; et al. Significance of TERT Genetic Alterations and Telomere Length in Hepatocellular Carcinoma. Cancers 2021, 13, 2160. [Google Scholar] [CrossRef]
- Chen, Y.L.; Jeng, Y.M.; Chang, C.N.; Lee, H.J.; Hsu, H.C.; Lai, P.L.; Yuan, R.H. TERT promoter mutation in resectable hepatocellular carcinomas: A strong association with hepatitis C infection and absence of hepatitis B infection. Int. J. Surg. 2014, 12, 659–665. [Google Scholar] [CrossRef]
- Yuan, X.; Cheng, G.; Yu, J.; Zheng, S.; Sun, C.; Sun, Q.; Li, K.; Lin, Z.; Liu, T.; Li, P.; et al. The TERT promoter mutation incidence is modified by germline TERT rs2736098 and rs2736100 polymorphisms in hepatocellular carcinoma. Oncotarget 2017, 8, 23120–23129. [Google Scholar] [CrossRef]
- Yang, X.; Guo, X.; Chen, Y.; Chen, G.; Ma, Y.; Huang, K.; Zhang, Y.; Zhao, Q.; Winkler, C.A.; An, P.; et al. Telomerase reverse transcriptase promoter mutations in hepatitis B virus-associated hepatocellular carcinoma. Oncotarget 2016, 7, 27838–27847. [Google Scholar] [CrossRef]
- Chianchiano, P.; Pezhouh, M.K.; Kim, A.; Luchini, C.; Cameron, A.; Weiss, M.J.; He, J.; Voltaggio, L.; Oshima, K.; Anders, R.A.; et al. Distinction of intrahepatic metastasis from multicentric carcinogenesis in multifocal hepatocellular carcinoma using molecular alterations. Hum. Pathol. 2018, 72, 127–134. [Google Scholar] [CrossRef]
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A., Jr.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026. [Google Scholar] [CrossRef]
- Pezzuto, F.; Izzo, F.; Buonaguro, L.; Annunziata, C.; Tatangelo, F.; Botti, G.; Buonaguro, F.M.; Tornesello, M.L. Tumor specific mutations in TERT promoter and CTNNB1 gene in hepatitis B and hepatitis C related hepatocellular carcinoma. Oncotarget 2016, 7, 54253–54262. [Google Scholar] [CrossRef]
- Lombardo, D.; Saitta, C.; Giosa, D.; Di Tocco, F.C.; Musolino, C.; Caminiti, G.; Chines, V.; Franze, M.S.; Alibrandi, A.; Navarra, G.; et al. Frequency of somatic mutations in TERT promoter, TP53 and CTNNB1 genes in patients with hepatocellular carcinoma from Southern Italy. Oncol. Lett. 2020, 19, 2368–2374. [Google Scholar] [CrossRef]
- Machida, K. HCV and tumor-initiating stem-like cells. Front. Physiol. 2022, 13, 903302. [Google Scholar] [CrossRef]
- An, P.; Xu, J.; Yu, Y.; Winkler, C.A. Host and Viral Genetic Variation in HBV-Related Hepatocellular Carcinoma. Front. Genet. 2018, 9, 261. [Google Scholar] [CrossRef]
- Tu, T.; Budzinska, M.A.; Shackel, N.A.; Urban, S. HBV DNA Integration: Molecular Mechanisms and Clinical Implications. Viruses 2017, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.S.; Wang, Z.; He, X.J.; Diplas, B.H.; Yang, R.; Killela, P.J.; Meng, Q.; Ye, Z.Y.; Wang, W.; Jiang, X.T.; et al. Recurrent TERT promoter mutations identified in a large-scale study of multiple tumour types are associated with increased TERT expression and telomerase activation. Eur. J. Cancer 2015, 51, 969–976. [Google Scholar] [CrossRef]
- Lee, H.W.; Park, T.I.; Jang, S.Y.; Park, S.Y.; Park, W.J.; Jung, S.J.; Lee, J.H. Clinicopathological characteristics of TERT promoter mutation and telomere length in hepatocellular carcinoma. Medicine 2017, 96, e5766. [Google Scholar] [CrossRef]
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef]
- Clevers, H. Wnt/beta-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef]
- Kimelman, D.; Xu, W. beta-catenin destruction complex: Insights and questions from a structural perspective. Oncogene 2006, 25, 7482–7491. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Clevers, H. Wnt/beta-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Valenta, T.; Hausmann, G.; Basler, K. The many faces and functions of beta-catenin. EMBO J. 2012, 31, 2714–2736. [Google Scholar] [CrossRef]
- Shah, K.; Kazi, J.U. Phosphorylation-Dependent Regulation of WNT/Beta-Catenin Signaling. Front. Oncol. 2022, 12, 858782. [Google Scholar] [CrossRef]
- Clevers, H.; Nusse, R. Wnt/beta-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.E.; Huang, H.; Zhao, M.; Zhang, X.; Zhang, A.; Semonov, M.V.; MacDonald, B.T.; Zhang, X.; Garcia Abreu, J.; Peng, L.; et al. Wnt stabilization of beta-catenin reveals principles for morphogen receptor-scaffold assemblies. Science 2013, 340, 867–870. [Google Scholar] [CrossRef]
- Liu, J.; Xiao, Q.; Xiao, J.; Niu, C.; Li, Y.; Zhang, X.; Zhou, Z.; Shu, G.; Yin, G. Wnt/beta-catenin signalling: Function, biological mechanisms, and therapeutic opportunities. Signal Transduct. Target. Ther. 2022, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Trejo-Solis, C.; Escamilla-Ramirez, A.; Jimenez-Farfan, D.; Castillo-Rodriguez, R.A.; Flores-Najera, A.; Cruz-Salgado, A. Crosstalk of the Wnt/beta-Catenin Signaling Pathway in the Induction of Apoptosis on Cancer Cells. Pharmaceuticals 2021, 14, 871. [Google Scholar] [CrossRef]
- Jia, D.; Dong, R.; Jing, Y.; Xu, D.; Wang, Q.; Chen, L.; Li, Q.; Huang, Y.; Zhang, Y.; Zhang, Z.; et al. Exome sequencing of hepatoblastoma reveals novel mutations and cancer genes in the Wnt pathway and ubiquitin ligase complex. Hepatology 2014, 60, 1686–1696. [Google Scholar] [CrossRef]
- Gao, C.; Wang, Y.; Broaddus, R.; Sun, L.; Xue, F.; Zhang, W. Exon 3 mutations of CTNNB1 drive tumorigenesis: A review. Oncotarget 2018, 9, 5492–5508. [Google Scholar] [CrossRef]
- Lu, G.; Lin, J.; Song, G.; Chen, M. Prognostic significance of CTNNB1 mutation in hepatocellular carcinoma: A systematic review and meta-analysis. Aging 2023, 15, 9759–9778. [Google Scholar] [CrossRef]
- Mao, T.L.; Chu, J.S.; Jeng, Y.M.; Lai, P.L.; Hsu, H.C. Expression of mutant nuclear beta-catenin correlates with non-invasive hepatocellular carcinoma, absence of portal vein spread, and good prognosis. J. Pathol. 2001, 193, 95–101. [Google Scholar] [CrossRef]
- Wang, Z.; Sheng, Y.Y.; Gao, X.M.; Wang, C.Q.; Wang, X.Y.; Lu, X.U.; Wei, J.W.; Zhang, K.L.; Dong, Q.Z.; Qin, L.X. beta-catenin mutation is correlated with a favorable prognosis in patients with hepatocellular carcinoma. Mol. Clin. Oncol. 2015, 3, 936–940. [Google Scholar] [CrossRef]
- Khalaf, A.M.; Fuentes, D.; Morshid, A.I.; Burke, M.R.; Kaseb, A.O.; Hassan, M.; Hazle, J.D.; Elsayes, K.M. Role of Wnt/beta-catenin signaling in hepatocellular carcinoma, pathogenesis, and clinical significance. J. Hepatocell. Carcinoma 2018, 5, 61–73. [Google Scholar] [CrossRef]
- Rebouissou, S.; Franconi, A.; Calderaro, J.; Letouze, E.; Imbeaud, S.; Pilati, C.; Nault, J.C.; Couchy, G.; Laurent, A.; Balabaud, C.; et al. Genotype-phenotype correlation of CTNNB1 mutations reveals different beta-catenin activity associated with liver tumor progression. Hepatology 2016, 64, 2047–2061. [Google Scholar] [CrossRef]
- Tran, F.H.; Zheng, J.J. Modulating the wnt signaling pathway with small molecules. Protein Sci. 2017, 26, 650–661. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Pan, S.; Hsieh, M.H.; Ng, N.; Sun, F.; Wang, T.; Kasibhatla, S.; Schuller, A.G.; Li, A.G.; Cheng, D.; et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc. Natl. Acad. Sci. USA 2013, 110, 20224–20229. [Google Scholar] [CrossRef]
- Huang, S.M.; Mishina, Y.M.; Liu, S.; Cheung, A.; Stegmeier, F.; Michaud, G.A.; Charlat, O.; Wiellette, E.; Zhang, Y.; Wiessner, S.; et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 2009, 461, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Font, E.; Perez-Capo, M.; Ramos, R.; Felipe, I.; Garcias, C.; Luna, P.; Terrasa, J.; Martin-Broto, J.; Vogler, O.; Alemany, R.; et al. Impact of Wnt/beta-Catenin Inhibition on Cell Proliferation through CDC25A Downregulation in Soft Tissue Sarcomas. Cancers 2020, 12, 2556. [Google Scholar] [CrossRef]
- Xiao, X.; Mo, H.; Tu, K. CTNNB1 mutation suppresses infiltration of immune cells in hepatocellular carcinoma through miRNA-mediated regulation of chemokine expression. Int. Immunopharmacol. 2020, 89, 107043. [Google Scholar] [CrossRef] [PubMed]
- Ruiz de Galarreta, M.; Bresnahan, E.; Molina-Sanchez, P.; Lindblad, K.E.; Maier, B.; Sia, D.; Puigvehi, M.; Miguela, V.; Casanova-Acebes, M.; Dhainaut, M.; et al. beta-Catenin Activation Promotes Immune Escape and Resistance to Anti-PD-1 Therapy in Hepatocellular Carcinoma. Cancer Discov. 2019, 9, 1124–1141. [Google Scholar] [CrossRef]
- National Cancer Center Japan. Cancer Gene Panel Test OncoGuideTM NCC Oncopanel System Added to Health Insurance Coverage List. Available online: https://www.ncc.go.jp/en/information/press_release/2019/0717/index.html (accessed on 15 March 2025).
- Terashima, T.; Yamashita, T.; Arai, K.; Takata, N.; Hayashi, T.; Seki, A.; Nakagawa, H.; Nio, K.; Iida, N.; Yamada, S.; et al. Comprehensive genomic profiling for advanced hepatocellular carcinoma in clinical practice. Hepatol. Int. 2025, 19, 212–221. [Google Scholar] [CrossRef]
- Hosny, G.; Farahat, N.; Tayel, H.; Hainaut, P. Ser-249 TP53 and CTNNB1 mutations in circulating free DNA of Egyptian patients with hepatocellular carcinoma versus chronic liver diseases. Cancer Lett. 2008, 264, 201–208. [Google Scholar] [CrossRef]
- Kim, Y.D.; Park, C.H.; Kim, H.S.; Choi, S.K.; Rew, J.S.; Kim, D.Y.; Koh, Y.S.; Jeung, K.W.; Lee, K.H.; Lee, J.S.; et al. Genetic alterations of Wnt signaling pathway-associated genes in hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2008, 23, 110–118. [Google Scholar] [CrossRef]
- Cavard, C.; Terris, B.; Grimber, G.; Christa, L.; Audard, V.; Radenen-Bussiere, B.; Simon, M.T.; Renard, C.A.; Buendia, M.A.; Perret, C. Overexpression of regenerating islet-derived 1 alpha and 3 alpha genes in human primary liver tumors with beta-catenin mutations. Oncogene 2006, 25, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhao, H.; Zhang, X.; Wood, L.D.; Anders, R.A.; Choti, M.A.; Pawlik, T.M.; Daniel, H.D.; Kannangai, R.; Offerhaus, G.J.; et al. Inactivating mutations of the chromatin remodeling gene ARID2 in hepatocellular carcinoma. Nat. Genet. 2011, 43, 828–829. [Google Scholar] [CrossRef]
- Zucman-Rossi, J.; Benhamouche, S.; Godard, C.; Boyault, S.; Grimber, G.; Balabaud, C.; Cunha, A.S.; Bioulac-Sage, P.; Perret, C. Differential effects of inactivated Axin1 and activated β-catenin mutations in human hepatocellular carcinomas. Oncogene 2007, 26, 774–780. [Google Scholar] [CrossRef]
- Tornesello, M.L.; Buonaguro, L.; Tatangelo, F.; Botti, G.; Izzo, F.; Buonaguro, F.M. Mutations in TP53, CTNNB1 and PIK3CA genes in hepatocellular carcinoma associated with hepatitis B and hepatitis C virus infections. Genomics 2013, 102, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Biterge Sut, B. Computational analysis of TP53 vs. CTNNB1 mutations in hepatocellular carcinoma suggests distinct cancer subtypes with differential gene expression profiles and chromatin states. Comput. Biol. Chem. 2020, 89, 107404. [Google Scholar] [CrossRef]
- Oversoe, S.K.; Clement, M.S.; Weber, B.; Gronbaek, H.; Hamilton-Dutoit, S.J.; Sorensen, B.S.; Kelsen, J. Combining tissue and circulating tumor DNA increases the detection rate of a CTNNB1 mutation in hepatocellular carcinoma. BMC Cancer 2021, 21, 376. [Google Scholar] [CrossRef]
- Hsu, H.C.; Jeng, Y.M.; Mao, T.L.; Chu, J.S.; Lai, P.L.; Peng, S.Y. Beta-catenin mutations are associated with a subset of low-stage hepatocellular carcinoma negative for hepatitis B virus and with favorable prognosis. Am. J. Pathol. 2000, 157, 763–770. [Google Scholar] [CrossRef]
- Sekine, S.; Ogawa, R.; Ojima, H.; Kanai, Y. Expression of SLCO1B3 is associated with intratumoral cholestasis and CTNNB1 mutations in hepatocellular carcinoma. Cancer Sci. 2011, 102, 1742–1747. [Google Scholar] [CrossRef]
- Ke, L.; Shen, J.; Feng, J.; Chen, J.; Shen, S.; Li, S.; Kuang, M.; Liang, L.; Lu, C.; Li, D.; et al. Somatic Mutation Profiles Revealed by Next Generation Sequencing (NGS) in 39 Chinese Hepatocellular Carcinoma Patients. Front. Mol. Biosci. 2021, 8, 800679. [Google Scholar] [CrossRef]
- Lu, L.C.; Shao, Y.Y.; Lee, Y.H.; Hsieh, M.S.; Hsiao, C.H.; Lin, H.H.; Kao, H.F.; Ma, Y.Y.; Yen, F.C.; Cheng, A.L.; et al. beta-catenin (CTNNB1) mutations are not associated with prognosis in advanced hepatocellular carcinoma. Oncology 2014, 87, 159–166. [Google Scholar] [CrossRef]
- Wong, C.M.; Fan, S.T.; Ng, I.O. beta-Catenin mutation and overexpression in hepatocellular carcinoma: Clinicopathologic and prognostic significance. Cancer 2001, 92, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Park, W.S.; Nam, S.W.; Kim, S.Y.; Lee, S.H.; Yoo, N.J.; Lee, J.Y.; Park, C.K. Mutations of beta-catenin and AXIN I genes are a late event in human hepatocellular carcinogenesis. Liver Int. 2005, 25, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Nadda, N.; Quadri, A.; Kumar, R.; Paul, S.; Tanwar, P.; Gamanagatti, S.; Dash, N.R.; Saraya, A.; Nayak, B. Assessments of TP53 and CTNNB1 gene hotspot mutations in circulating tumour DNA of hepatitis B virus-induced hepatocellular carcinoma. Front. Genet. 2023, 14, 1235260. [Google Scholar] [CrossRef] [PubMed]
- Vivekanandan, P.; Torbenson, M.; Ramakrishna, B. Hepatitis B virus-associated hepatocellular carcinoma from India: Role of viral genotype and mutations in CTNNB1 (beta-catenin) and TP53 genes. J. Gastrointest. Cancer 2011, 42, 20–25. [Google Scholar] [CrossRef]
- Javanmard, D.; Najafi, M.; Babaei, M.R.; Karbalaie Niya, M.H.; Esghaei, M.; Panahi, M.; Safarnezhad Tameshkel, F.; Tavakoli, A.; Jazayeri, S.M.; Ghaffari, H.; et al. Investigation of CTNNB1 gene mutations and expression in hepatocellular carcinoma and cirrhosis in association with hepatitis B virus infection. Infect. Agent. Cancer 2020, 15, 37. [Google Scholar] [CrossRef]
- Cieply, B.; Zeng, G.; Proverbs-Singh, T.; Geller, D.A.; Monga, S.P. Unique phenotype of hepatocellular cancers with exon-3 mutations in beta-catenin gene. Hepatology 2009, 49, 821–831. [Google Scholar] [CrossRef]
- Ding, X.; Yang, Y.; Han, B.; Du, C.; Xu, N.; Huang, H.; Cai, T.; Zhang, A.; Han, Z.G.; Zhou, W.; et al. Transcriptomic characterization of hepatocellular carcinoma with CTNNB1 mutation. PLoS ONE 2014, 9, e95307. [Google Scholar] [CrossRef] [PubMed]
- Laurent-Puig, P.; Legoix, P.; Bluteau, O.; Belghiti, J.; Franco, D.; Binot, F.; Monges, G.; Thomas, G.; Bioulac-Sage, P.; Zucman-Rossi, J. Genetic alterations associated with hepatocellular carcinomas define distinct pathways of hepatocarcinogenesis. Gastroenterology 2001, 120, 1763–1773. [Google Scholar] [CrossRef]
- Huang, H.; Fujii, H.; Sankila, A.; Mahler-Araujo, B.M.; Matsuda, M.; Cathomas, G.; Ohgaki, H. Beta-catenin mutations are frequent in human hepatocellular carcinomas associated with hepatitis C virus infection. Am. J. Pathol. 1999, 155, 1795–1801. [Google Scholar] [CrossRef]
- Lin, Z.Z.; Jeng, Y.M.; Hu, F.C.; Pan, H.W.; Tsao, H.W.; Lai, P.L.; Lee, P.H.; Cheng, A.L.; Hsu, H.C. Significance of Aurora B overexpression in hepatocellular carcinoma. Aurora B Overexpression in HCC. BMC Cancer 2010, 10, 461. [Google Scholar] [CrossRef]
- Yuan, R.H.; Chang, K.T.; Chen, Y.L.; Hsu, H.C.; Lee, P.H.; Lai, P.L.; Jeng, Y.M. S100P expression is a novel prognostic factor in hepatocellular carcinoma and predicts survival in patients with high tumor stage or early recurrent tumors. PLoS ONE 2013, 8, e65501. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.Y.; Chang, T.T.; Steffen, J.D.; Chen, S.; Jain, S.; Song, W.; Lin, Y.J.; Su, Y.H. Detection of CTNNB1 Hotspot Mutations in Cell-Free DNA from the Urine of Hepatocellular Carcinoma Patients. Diagnostics 2021, 11, 1475. [Google Scholar] [CrossRef]
- Taniguchi, K.; Roberts, L.R.; Aderca, I.N.; Dong, X.; Qian, C.; Murphy, L.M.; Nagorney, D.M.; Burgart, L.J.; Roche, P.C.; Smith, D.I.; et al. Mutational spectrum of beta-catenin, AXIN1, and AXIN2 in hepatocellular carcinomas and hepatoblastomas. Oncogene 2002, 21, 4863–4871. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.C.; Villanueva, A. Biomarkers for Hepatobiliary Cancers. Hepatology 2021, 73 (Suppl. S1), 115–127. [Google Scholar] [CrossRef] [PubMed]
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.C.; Llovet, J.M. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology 2015, 149, 1226–1239.e4. [Google Scholar] [CrossRef]
- Zhang, Y.; Toh, L.; Lau, P.; Wang, X. Human telomerase reverse transcriptase (hTERT) is a novel target of the Wnt/beta-catenin pathway in human cancer. J. Biol. Chem. 2012, 287, 32494–32511. [Google Scholar] [CrossRef]

| Reference | Country | TERTp (n, %) | CTNNB1 (n, %) | Mutation Correlation |
|---|---|---|---|---|
| [15] | France, Italy, and Spain | 147/243 (60.5) | 82/235 (34.9) | Yes (p = 0.03) |
| [51] | France | 441/759 (58.1) | 207/746 (27.7) | Yes (p = 0.0000001) |
| [13] | France | 179/305 (58.7) | 101/304 (33.2) | Yes (p < 0.0001) |
| [19] | USA and Japan | 257/463 (55.5) | 146/469 (31.1) | Yes (p < 0.0001) |
| [66] | China | 57/190 (30) | 17/70 (24.3) | No (p = 0.535) |
| [63] | South Korea | 41/105 (39) | 15/103 (14.6) | No (p = 0.568) |
| [60] | Japan | 9/11 (81.8) | 5/11 (45.5) | No (p = 0.4545) |
| [70] | Italy | 64/127 (50.4) | 33/127 (26) | No (p = 0.4192) |
| [65] | Taiwan | 57/195 (29.2) | 31/188 (16.5) | No (p = 0.2055) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terra, M.L.; Sant’Anna, T.B.F.; de Barros, J.J.F.; de Araujo, N.M. Geographic and Viral Etiology Patterns of TERT Promoter and CTNNB1 Exon 3 Mutations in Hepatocellular Carcinoma: A Comprehensive Review. Int. J. Mol. Sci. 2025, 26, 2889. https://doi.org/10.3390/ijms26072889
Terra ML, Sant’Anna TBF, de Barros JJF, de Araujo NM. Geographic and Viral Etiology Patterns of TERT Promoter and CTNNB1 Exon 3 Mutations in Hepatocellular Carcinoma: A Comprehensive Review. International Journal of Molecular Sciences. 2025; 26(7):2889. https://doi.org/10.3390/ijms26072889
Chicago/Turabian StyleTerra, Mariana Leonardo, Thaís Barbosa Ferreira Sant’Anna, José Junior França de Barros, and Natalia Motta de Araujo. 2025. "Geographic and Viral Etiology Patterns of TERT Promoter and CTNNB1 Exon 3 Mutations in Hepatocellular Carcinoma: A Comprehensive Review" International Journal of Molecular Sciences 26, no. 7: 2889. https://doi.org/10.3390/ijms26072889
APA StyleTerra, M. L., Sant’Anna, T. B. F., de Barros, J. J. F., & de Araujo, N. M. (2025). Geographic and Viral Etiology Patterns of TERT Promoter and CTNNB1 Exon 3 Mutations in Hepatocellular Carcinoma: A Comprehensive Review. International Journal of Molecular Sciences, 26(7), 2889. https://doi.org/10.3390/ijms26072889