

A Suite of Pea (Pisum sativum L.) Near-Isolines: Genetic Resources and Molecular Tools to Breed for Seed Carbohydrate and Protein Quality in Legumes

 , and
, and
Abstract

1. Introduction
2. Results
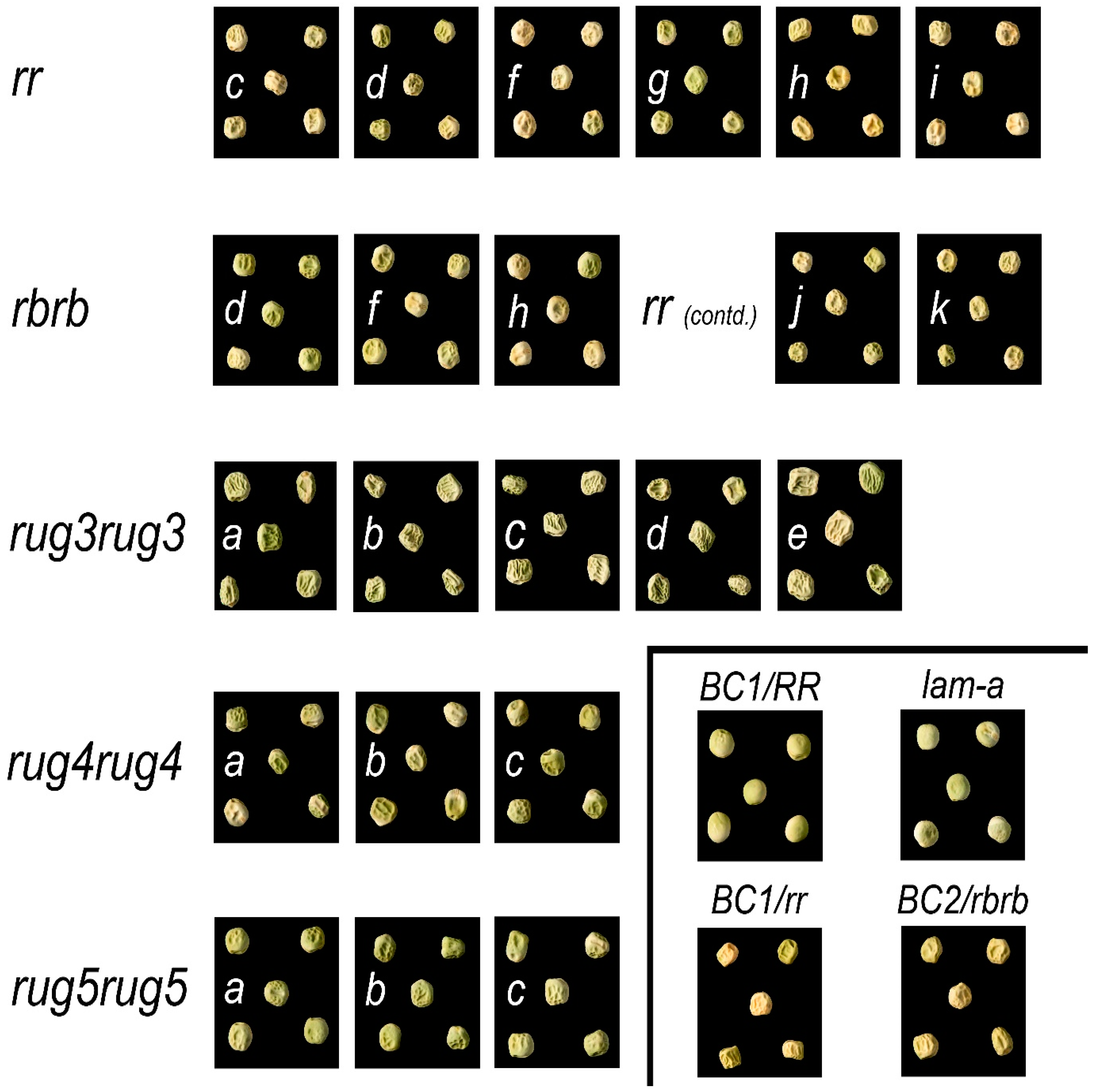
2.1. Seed Phenotypes
2.2. Unique Alleles
2.3. Protein Structures
2.3.1. Mutants at the rb Locus (ADP-Glucose Pyrophosphorylase Large Subunit 1; Figure 3 and Figure 4)
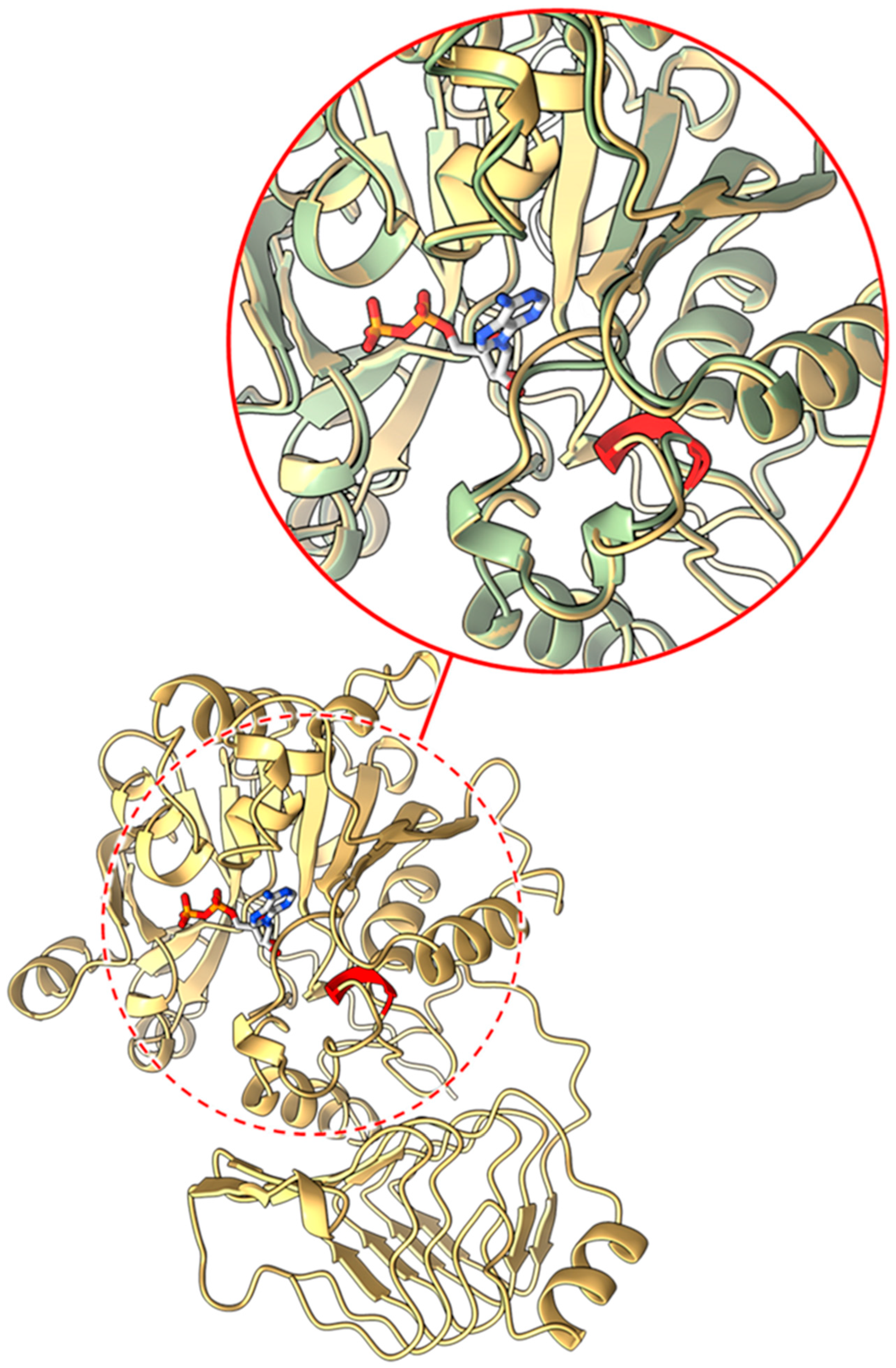
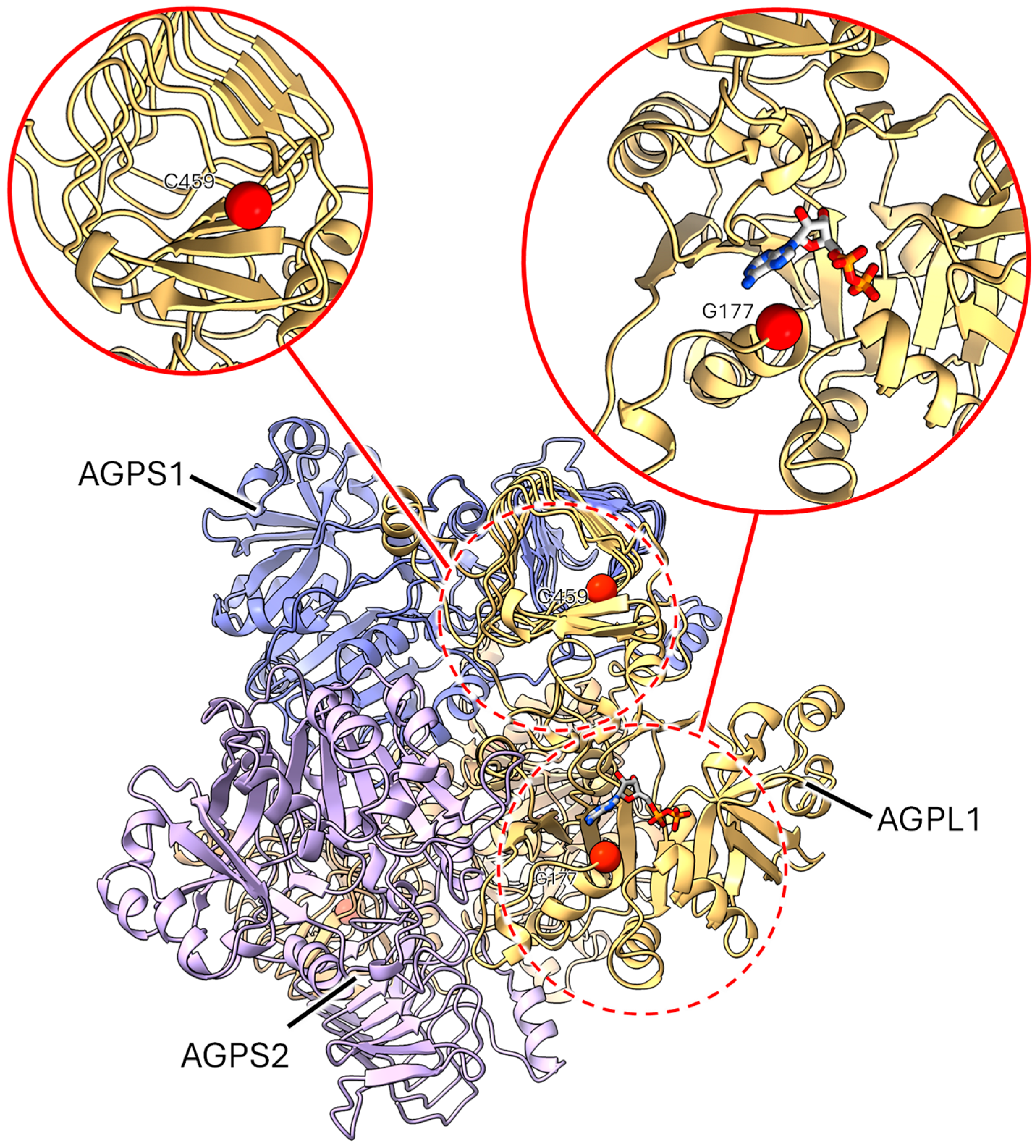
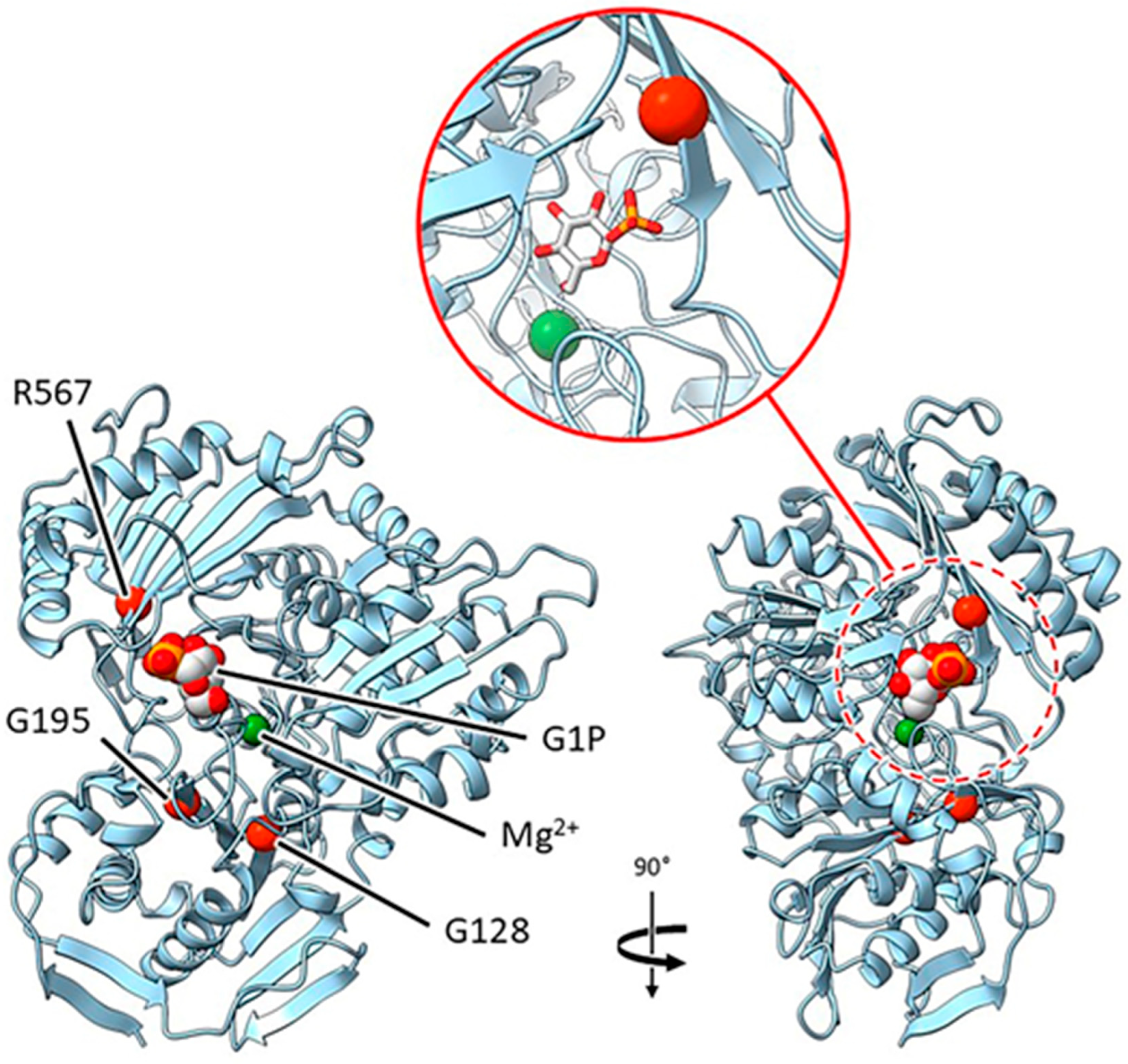
2.3.2. Mutants at the rug3 Locus (Plastidial Phosphoglucomutase; Figure 5)
2.3.3. Mutants at the rug4 Locus (Sucrose Synthase 1; Figure 6)

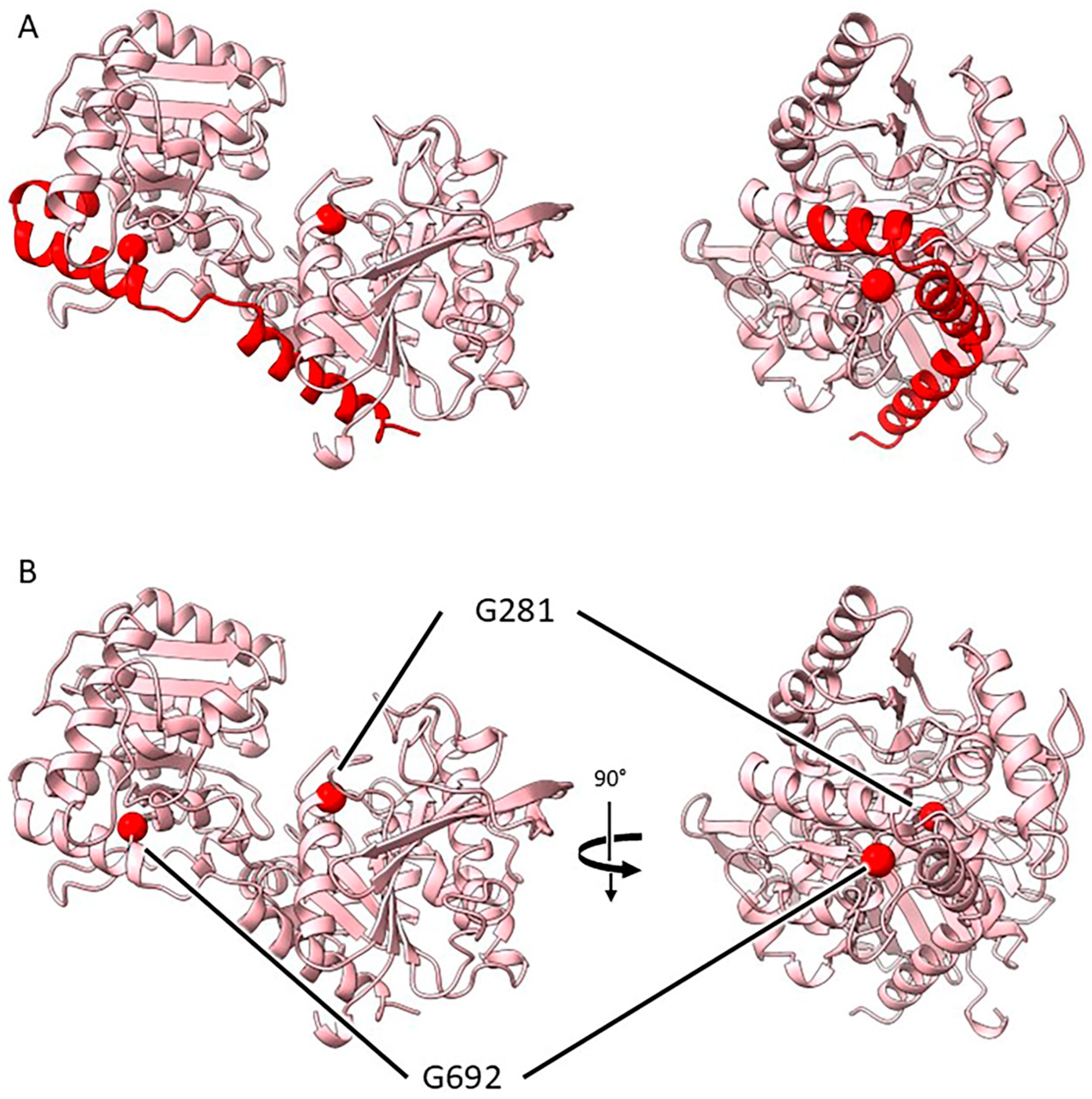
2.3.4. Mutants at the rug5 Locus (Starch Synthase 2; Figure 7)
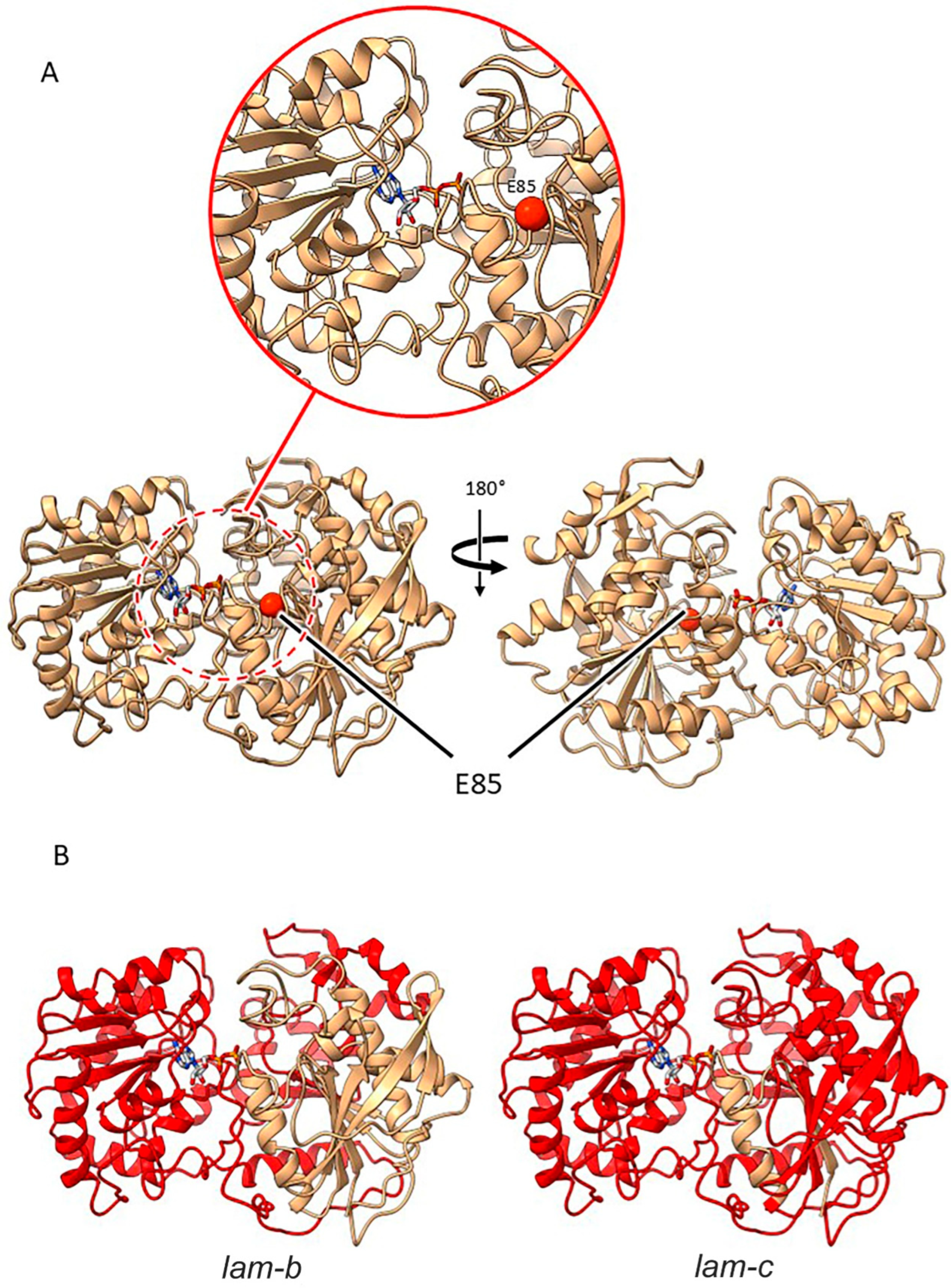
2.3.5. Mutants at the lam Locus (Granule-Bound Starch Synthase 1; Figure 8)
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Detection of Mutations
4.3. Protein Structure Modelling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Plant-Based Protein Market by Source (Soy, Wheat, Pea, Canola, Oats, Rice, Potato, Beans and Seeds, Fermented Protein), Type (Concentrates, Isolates, Textured, by Application (Food and Feed)—Global Forecast to 2029. MarketsandMarkets. Available online: https://www.marketsandmarkets.com/Market-Reports/plant-based-protein-market-14715651.html#:~:text=The%20global%20plant%2Dbased%20protein,2027%20in%20terms%20of%20value (accessed on 16 December 2024).
- Aimutis, W.R. Plant-Based Proteins: The Good, Bad, and Ugly. Annu. Rev. Food Sci. Technol. 2022, 13, 1–17. [Google Scholar] [CrossRef]
- Schneider, A.V.C. Overview of the market and consumption of pulses in Europe. Br. J. Nutrit. 2002, 88, S243–S250. [Google Scholar] [CrossRef] [PubMed]
- McClements, D.J.; Grossmann, L. A brief review of the science behind the design of healthy and sustainable plant-based foods. npj Sci. Food 2021, 5, 17. [Google Scholar] [CrossRef]
- Wang, T.L.; Bogracheva, T.Y.; Hedley, C.L. Starch: As simple as A, B, C? J. Exp. Bot. 1998, 49, 481–502. [Google Scholar] [CrossRef]
- Raghunathan, R.; Hoover, R.; Waduge, R.; Liu, Q.; Warkentin, T.D. Impact of molecular structure on the physicochemical properties of starches isolated from different field pea (Pisum sativum L.) cultivars grown in Saskatchewan, Canada. Food Chem. 2017, 221, 1514–1521. [Google Scholar] [CrossRef] [PubMed]
- Ratnayake, W.; Hoover, R.; Warkentin, T. Pea starch: Composition, structure and properties—A review. Starch–Stärke 2002, 54, 217–234. [Google Scholar] [CrossRef]
- Morales-Hernández, N.; Mondragón-Cortéz, P.; Prieto-Vázquez del Mercado, P.A. Pea Starch: Functionality and Potential Applications. Non-Conventional Starch Sources: Properties, Functionality, and Applications; Lorenzo, J.M., Bangar, S.P., Eds.; Academic Press: London, UK, 2024; pp. 495–521. ISBN 9780443189821. [Google Scholar]
- Fenn, D.; Wang, N.; Maximiuk, L. Physicochemical, anti-nutritional, and functional properties of air-classified protein concentrates from commercially grown Canadian yellow pea (Pisum sativum) varieties with variable protein levels. Cereal Chem. 2022, 99, 157–168. [Google Scholar] [CrossRef]
- Farshi, P.; Mirmohammadali, S.N.; Rajpurohit, B.; Smith, J.S.; Li, Y. Pea protein and starch: Functional properties and applications in edible films. J. Agri. Food Res. 2024, 15, 100927. [Google Scholar] [CrossRef]
- Petropoulou, K.; Salt, L.J.; Edwards, C.H.; Warren, F.J.; Garcia-Perez, I.; Chambers, E.S.; Alshaalan, R.; Khatib, M.; Perez-Moral, N.; Cross, K.L.; et al. A natural mutation in Pisum sativum L. (pea) alters starch assembly and improves glucose homeostasis in humans. Nat. Food 2020, 1, 693–704. [Google Scholar] [CrossRef]
- Wu, D.-T.; Li, W.-X.; Wan, J.-J.; Hu, Y.-C.; Gan, R.-Y.; Zou, L. A comprehensive review of pea (Pisum sativum L.): Chemical composition, processing, health benefits, and food applications. Foods 2023, 12, 2527. [Google Scholar] [CrossRef]
- Olías, R.; Rayner, T.; Clemente, A.; Domoney, D. Combination of three null mutations affecting seed protein accumulation in pea (Pisum sativum L.) impacts positively on digestibility. Food Res. Int. 2023, 169, 112825. [Google Scholar] [CrossRef]
- Hughes, R.K.; Desforges, N.; Selwood, C.; Smith, R.; Speirs, C.I.; Sinnaeve, G.; Gorton, P.G.; Wiseman, J.; Jumel, K.; Harding, S.E.; et al. Genes affecting starch biosynthesis exert pleiotropic effects on the protein content and composition of pea seeds. J. Sci. Food Agric. 2001, 81, 877–882. [Google Scholar] [CrossRef]
- Yu, B.; Xiang, D.; Mahfuz, H.; Patterson, N.; Bing, D. Understanding starch metabolism in pea seeds towards tailoring functionality for value-added utilization. Int. J. Mol. Sci. 2021, 22, 8972. [Google Scholar] [CrossRef] [PubMed]
- Mendel, G. Versuche über pflanzen-hybriden. In Verhandlungen des Naturforschenden Vereines in Brünn; Verlage des Vereines: Brünn, Czech Republic, 1866; Volume 4, pp. 3–47. [Google Scholar]
- Bhattacharyya, M.K.; Smith, A.M.; Ellis, T.H.N.; Hedley, C.; Martin, C. The wrinkled-seed character of pea described by Mendel is caused by a transposon-like insertion in a gene encoding starch-branching enzyme. Cell 1990, 60, 115–122. [Google Scholar] [CrossRef]
- Kooistra, E. On the differences between smooth and three types of wrinkled peas. Euphytica 1962, 11, 357–373. [Google Scholar] [CrossRef]
- Hylton, C.; Smith, A.M. The rb mutation of peas causes structural and regulatory changes in ADP glucose pyrophosphorylase from developing embryos. Plant Physiol. 1992, 99, 1626–1634. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.J.; Mould, R.M.; Leech, M.J.; Johnson, S.; Turner, L.; Schreck, S.L.; Baird, K.; Jack, P.; Rawsthorne, S.; Hedley, C.L.; et al. The rug3 locus of pea (Pisum sativum L.) encodes plastidial phosphoglucomutase. Plant Physiol. 2000, 122, 1187–1192. [Google Scholar] [CrossRef]
- Harrison, C.J.; Hedley, C.L.; Wang, T.L. Evidence that the rug3 locus of pea (Pisum sativum L.) encodes plastidial phosphoglucomutase confirms that the imported substrate for starch synthesis in pea amyloplasts is glucose-6-phosphate. Plant J. 1998, 13, 753–762. [Google Scholar] [CrossRef]
- Craig, J.; Barratt, P.; Tatge, H.; Déjardin, A.; Handley, L.; Gardner, C.D.; Barber, L.; Wang, T.L.; Hedley, C.L.; Martin, C.; et al. Mutations at the rug4 locus alter the carbon and nitrogen metabolism of pea plants through an effect on sucrose synthase. Plant J. 1999, 17, 353–362. [Google Scholar] [CrossRef]
- Barratt, D.H.; Barber, L.; Kruger, N.J.; Smith, A.M.; Wang, T.L.; Martin, C. Multiple, distinct isoforms of sucrose synthase in pea. Plant Physiol. 2001, 127, 655–664. [Google Scholar] [CrossRef]
- Craig, J.; Lloyd, J.R.; Tomlinson, K.; Barber, L.; Edwards, A.; Wang, T.L.; Martin, C.; Hedley, C.L.; Smith, A.M. Mutations in the gene encoding starch synthase II profoundly alter amylopectin structure in pea embryos. Plant Cell 1998, 10, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Denyer, K.; Barber, L.R.; Burton, R.; Hedley, C.L.; Hylton, C.M.; Johnson, S.; Jones, D.A.; Marshall, J.; Smith, A.M.; Tatge, H.; et al. The isolation and characterisation of novel low-amylose mutants of Pisum sativum L. Plant Cell Environ. 1995, 18, 1019–1026. [Google Scholar]
- Xing, Q.; Utami, D.P.; Demattey, M.B.; Kyriakopoulou, K.; de Wit, M.; Boom, R.M.; Schutyser, M.A.I. A two-step air classification and electrostatic separation process for protein enrichment of starch-containing legumes. Inn. Food Sci. Emerg. Tech. 2020, 66, 102480. [Google Scholar] [CrossRef]
- Reichert, R. Air Classification of peas (Pisum sativum) varying widely in protein content. J. Food Sci. 2006, 47, 1263–1267. [Google Scholar] [CrossRef]
- Burstin, J.; Marget, P.; Huart, M.; Moessner, A.; Mangin, B.; Duchene, C.; Desprez, B.; Munier-Jolain, N.; Duc, G. Developmental genes have pleiotropic effects on plant morphology and source capacity, eventually impacting on seed protein content and productivity in pea. Plant Physiol. 2007, 144, 768–781. [Google Scholar] [CrossRef] [PubMed]
- Warsame, A.O.; Balk, J.; Domoney, C. Identification of significant genome-wide associations and QTL underlying variation in seed protein composition in pea (Pisum sativum L.). bioRxiv 2024. [Google Scholar] [CrossRef]
- Moreau, C.; Warren, F.J.; Rayner, T.; Perez-Moral, N.; Lawson, D.M.; Wang, T.L.; Domoney, C. An allelic series of starch-branching enzyme mutants in pea (Pisum sativum L.) reveals complex relationships with seed starch phenotypes. Carb. Polym. 2022, 288, 119386. [Google Scholar] [CrossRef]
- Rayner, T.; Saalbach, G.; Vickers, M.; Paajanen, P.; Martins, C.; Wouters, R.H.M.; Chinoy, C.; Mulholland, F.; Bal, M.; Isaac, P.; et al. Rebalancing the seed proteome following deletion of vicilin-related genes in pea (Pisum sativum L.). J. Exp. Bot. 2024; in press. [Google Scholar] [CrossRef]
- Wang, T.L.; Hedley, C.L. Seed development in peas: Knowing your three “r’s” (or four, or five). Seed Sci. Res. 1991, 1, 3–14. [Google Scholar] [CrossRef]
- Wang, T.L.; Hedley, C.L. Seed mutants in Pisum. Pisum Genet. 1993, 25, 64–70. [Google Scholar]
- Wang, T.L.; Hadavizadeh, A.; Harwood, A.; Welham, T.J.; Harwood, W.A.; Faulks, R.; Hedley, C.L. An analysis of seed development in Pisum sativum. XIII. The chemical induction of storage product mutants. Plant Breed. 1990, 105, 311–320. [Google Scholar] [CrossRef]
- Zheng, Y.; Anderson, S.; Zhang, Y.; Garavito, R.M. The structure of sucrose synthase-1 from Arabidopsis thaliana and its functional implications. J. Biol. Chem. 2011, 286, 36108–36118. [Google Scholar] [CrossRef] [PubMed]
- Edwards, A.; Marshall, J.; Denyer, K.; Sidebottom, C.; Visser, R.G.F.; Martin, C.; Smith, A.M. Evidence that a 77-kilodalton protein from the starch of pea embryos is an isoform of starch synthase that is both soluble and granule bound. Plant Physiol. 1996, 112, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Blixt, S. Mutation genetics in Pisum. Agri Hort. Genet. 1972, 30, 1–293. [Google Scholar]
- Rayner, T.; Moreau, C.; Ambrose, M.; Isaac, P.G.; Ellis, N.; Domoney, C. Genetic variation controlling wrinkled seed phenotypes in Pisum: How lucky was Mendel? Int. J. Mol. Sci. 2017, 18, 1205. [Google Scholar] [CrossRef]
- Lacey, C.N.D.; Hughes, S.G.; Harrison, C.J.; Wang, T.L.; Hedley, C.L. Method for Increasing Sucrose Content of Plants. WO1998001574A1, 15 January 1998. [Google Scholar]
- Coxon, D.T.; Wright, D.J. Analysis of pea lipid content by gas chromatographic and microgravimetric methods. genotype variation in lipid content and fatty acid composition. J. Sci. Food. Agri. 1985, 36, 847–856. [Google Scholar] [CrossRef]
- Jones, D.A.; Arthur, A.E.; Adams, H.M.; Coxon, D.T.; Wang, T.L.; Hedley, C.L. An analysis of seed development in Pisum sativum. IX. Genetic Analysis of Lipid Content. Plant Breed. 1990, 104, 144–151. [Google Scholar] [CrossRef]
- Shen, Y.; Hong, S.; Li, Y. Pea protein composition, functionality, modification, and food applications: A review. Adv. Food Nutr. Res. 2022, 101, 71–127. [Google Scholar] [PubMed]
- Van Oosten, J.-J.M.; Kaplan, C.P. Method for Increasing Vitamin C Content of Plants. WO-1999053041-A2, 20 October 1999. [Google Scholar]
- Perry, J.; Welham, T.; Brachmann, A.; Binder, A.; Charpentier, M.; Groth, M.; Haage, K.; Markmann, K.; Wang, T.L.; Parniske, M. TILLING in Lotus japonicus identified large allelic series for symbiosis genes and revealed a bias in functionally defective ethyl methane sulfonate alleles towards glycine replacements. Plant Physiol. 2009, 151, 1281–1291. [Google Scholar] [CrossRef]
- Wang, T.L.; Bogracheva, T.Y.; Hedley, C.L. Manipulation of starch quality in seeds: A genetic approach. In Seed Biology: Advances and Applications, Proceedings of the Sixth International Workshop on Seeds, Merida, Mexico, 24–28 January 1999; Black, M., Bradford, K.J., Vázquez Ramos, J., Eds.; CAB International: Wallingford, UK, 2000; pp. 439–448. ISBN 0851994040. [Google Scholar]
- Figueroa, C.M.; Asencion Diez, M.D.; Ballicora, M.A.; Iglesias, A.A. Structure, function, and evolution of plant ADP-glucose pyrophosphorylase. Plant Mol. Biol. 2022, 108, 307–323. [Google Scholar] [CrossRef]
- Weigelt, K.; Küster, H.; Rutten, T.; Fait, A.; Fernie, A.R.; Miersch, O.; Wasternack, C.; Emery, R.J.N.; Desel, C.; Hosein, F.; et al. ADP-glucose pyrophosphorylase-deficient pea embryos reveal specific transcriptional and metabolic changes of carbon-nitrogen metabolism and stress responses. Plant Physiol. 2009, 149, 395–411. [Google Scholar] [CrossRef]
- Trindler, C.; Kopf-Bolanz, K.A.; Denkel, C. Aroma of peas, its constituents and reduction strategies—Effects from breeding to processing. Food Chem. 2022, 376, 131892. [Google Scholar] [CrossRef]
- Bhowmik, P.; Konkin, D.; Polowick, P.; Hodgins, C.L.; Subedi, M.; Xiang, D.; Yu, B.; Patterson, N.; Rajagopalan, N.; Babic, V.; et al. CRISPR/Cas9 gene editing in legume crops: Opportunities and challenges. Legume Sci. 2021, 3, e96. [Google Scholar] [CrossRef]
- Rasheed, A.; Barqawi, A.A.; Mahmood, A.; Nawaz, M.; Shah, A.N.; Bay, D.H.; Alahdal, M.A.; Hassan, M.U.; Qari, S.H. CRISPR/Cas9 is a powerful tool for precise genome editing of legume crops: A review. Mol. Biol. Rep. 2022, 49, 5595–5609. [Google Scholar] [CrossRef]
- Hodgins, C.L.; Salama, E.M.; Kumar, R.; Zhao, Y.; Roth, S.A.; Cheung, I.Z.; Chen, J.; Arganosa, G.C.; Warkentin, T.D.; Bhowmik, P.; et al. Creating saponin-free yellow pea seeds by CRISPR/Cas9-enabled mutagenesis on β-amyrin synthase. Plant Direct 2024, 8, e563. [Google Scholar] [CrossRef] [PubMed]
- Hofer, J.; Turner, L.; Moreau, C.; Ambrose, M.; Isaac, P.; Butcher, S.; Weller, J.; Dupin, A.; Dalmais, M.; Le Signor, C.; et al. Tendril-less regulates tendril formation in pea leaves. Plant Cell 2009, 21, 420–428. [Google Scholar] [CrossRef]
- Available online: https://bio.tools/targetp (accessed on 26 March 2024).
- Emanuelsson, O.; Nielsen, H.; Brunak, S.; Von Heijne, G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol. 2000, 300, 1005–1016. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb (accessed on 26 March 2024).
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Meth. 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- EMBL-EBI Protein Data Bank in Europe. PDBeFold. Available online: https://www.ebi.ac.uk/msd-srv/ssm/ (accessed on 10 April 2024).
- AlphaFill AlphaFold Models Enriched with Ligands and Co-Factors. Available online: https://alphafill.eu/ (accessed on 10 April 2024).
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.L.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Allele | Isolation Number (Prefix − E = EMS; M = MNU) | Line Number † | Mutation | ||
|---|---|---|---|---|---|
| Position | Genomic | Protein | |||
r | N/A | BC1/rr | Exon 22 | A27379-insertion of ~1 kb ‘element’ | Predicted E 860 →, ≤30 aberrant amino acids * |
| r-c | M(850)7065 | SIM53 | Exon 15 | G22948A | R 571 H |
| r-d | M(718)7339 | SIM54 | Exon 13 | G20772A | G 495 D |
| r-f | E499(1677) | SIM56 | Exon 18 | G25551A | W 684 * |
| r-g | E(117)22 | SIM57 | Exon 18 | G25633A | R 712 C |
| r-h | E(349)7277 | SIM58 | Intron 15 acceptor splice site | G24153A | 10 aberrant amino acids: 586–595 1* |
| r-i | E(625)113 | SIM59 | Intron 8 acceptor splice site | G13242A (84 nucleotide deletion: 957–1040) | 28 amino acid deletion (319–346) 2 |
| r-j | E(122)1154 | SIM61 | Exon 16 | G24218A | E 607 K |
| r-k | M(32)7325 | SIM71 | Intron 16 donor splice site | G24299A | Variants include: loss of 13 amino acids (621–633) 3; four aberrant amino acids (634–637) * |
| rb | N/A | BC2/rbrb | Exon 2 | T470-G471 (9 bp deletion) | A98-(ATP or TPA missing)-A99 |
| rb-d | E(162)1232 | SIM15 | Exon 13 | G2802A | C 456 Y |
| rb-f | E(1663)5142 | SIM101 | Exon 8 | G1924A | W 304 * |
| rb-h | E(1258)3892 | SIM103 | Exon 4 | G1153A | G 174 R |
| rug3-a | E(543)1836 | SIM1 | Exon 4 | G703A | G 128 D |
| rug3-b | E(626)124 | SIM32 | Exon 20 | C4367T | R 567 C |
| rug3-c | E(476)1589 | SIM41 | Exon 7 | G1479A | G 195 S |
| rug3-d | E(549)548 | SIM42 | Exon 4 | C711T | R 131 * |
| rug3-e | E(1780)5702 | SIM43 | Exon 17 | CT deletion 3757/3758 | S 477 C, 479 * |
| rug4-a | E(620)121 | SIM11 | Exon 4 | C788T | L 164 F |
| rug4-b | E(77)16 | SIM91 | Exon 11 | G2668A | R 578 K |
| rug4-c | M(3)545 | SIM201A | Intron 8 donor splice site | G1966A | C 432 I, 37 aberrant amino acids, 468 * |
| rug5-a | E(640)2239 | SIM51 | Exon 8 | G3702A | W 708 * |
| rug5-b | E(167)1236 | SIM52 | Exon 3 | G1926A | G 281 R |
| rug5-c | E(509)1728 | SIM81 | Exon 8 | G3653A | G 692 D |
| lam-a | E(1470)4322 | SIM501 | Exon 1 | G253A | E 85 K |
| lam-b | E(0.3)151 4/ E(722)149 | SIM502/512 | Intron 6 acceptor splice site | G1654A | A 254 R, 12 aberrant amino acids, 266 * |
| lam-c | E(1599)4812 | SIM503/504 | Intron 1 acceptor splice site | G454A | G 108 V, 11 aberrant amino acids, 119 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rayner, T.; Mundy, J.E.A.; Bilham, L.J.; Moreau, C.; Lawson, D.M.; Domoney, C.; Wang, T.L. A Suite of Pea (Pisum sativum L.) Near-Isolines: Genetic Resources and Molecular Tools to Breed for Seed Carbohydrate and Protein Quality in Legumes. Int. J. Mol. Sci. 2025, 26, 2612. https://doi.org/10.3390/ijms26062612
Rayner T, Mundy JEA, Bilham LJ, Moreau C, Lawson DM, Domoney C, Wang TL. A Suite of Pea (Pisum sativum L.) Near-Isolines: Genetic Resources and Molecular Tools to Breed for Seed Carbohydrate and Protein Quality in Legumes. International Journal of Molecular Sciences. 2025; 26(6):2612. https://doi.org/10.3390/ijms26062612
Chicago/Turabian StyleRayner, Tracey, Julia E. A. Mundy, Lorelei J. Bilham, Carol Moreau, David M. Lawson, Claire Domoney, and Trevor L. Wang. 2025. "A Suite of Pea (Pisum sativum L.) Near-Isolines: Genetic Resources and Molecular Tools to Breed for Seed Carbohydrate and Protein Quality in Legumes" International Journal of Molecular Sciences 26, no. 6: 2612. https://doi.org/10.3390/ijms26062612
APA StyleRayner, T., Mundy, J. E. A., Bilham, L. J., Moreau, C., Lawson, D. M., Domoney, C., & Wang, T. L. (2025). A Suite of Pea (Pisum sativum L.) Near-Isolines: Genetic Resources and Molecular Tools to Breed for Seed Carbohydrate and Protein Quality in Legumes. International Journal of Molecular Sciences, 26(6), 2612. https://doi.org/10.3390/ijms26062612