
Combined Prokaryotic Transcriptomics and Proteomics Analysis of Clinical Trueperella pyogenes Isolates with Distinctive Cytotoxicity

Abstract
1. Introduction
2. Results
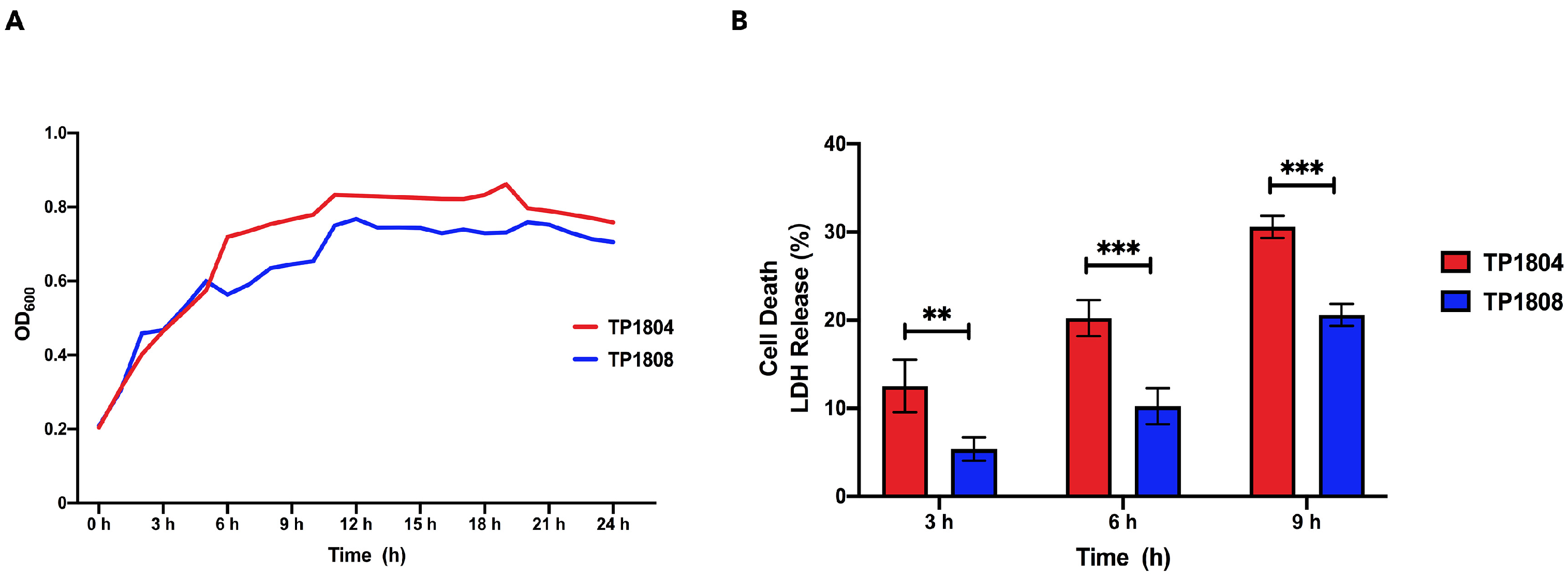
2.1. Bacterial Growth Characteristics and Toxicity
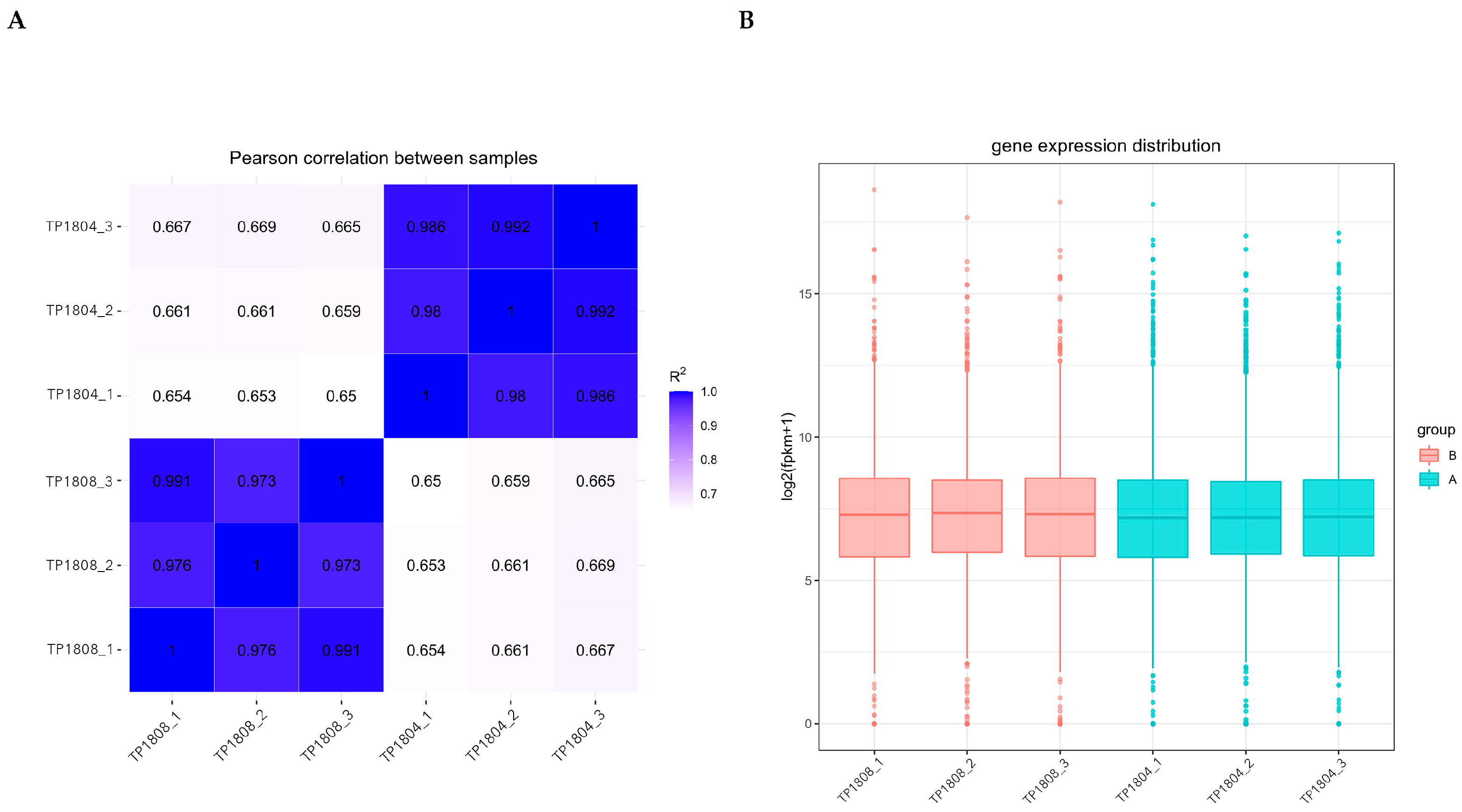
2.2. Sample Correlation and Gene Expression Distribution
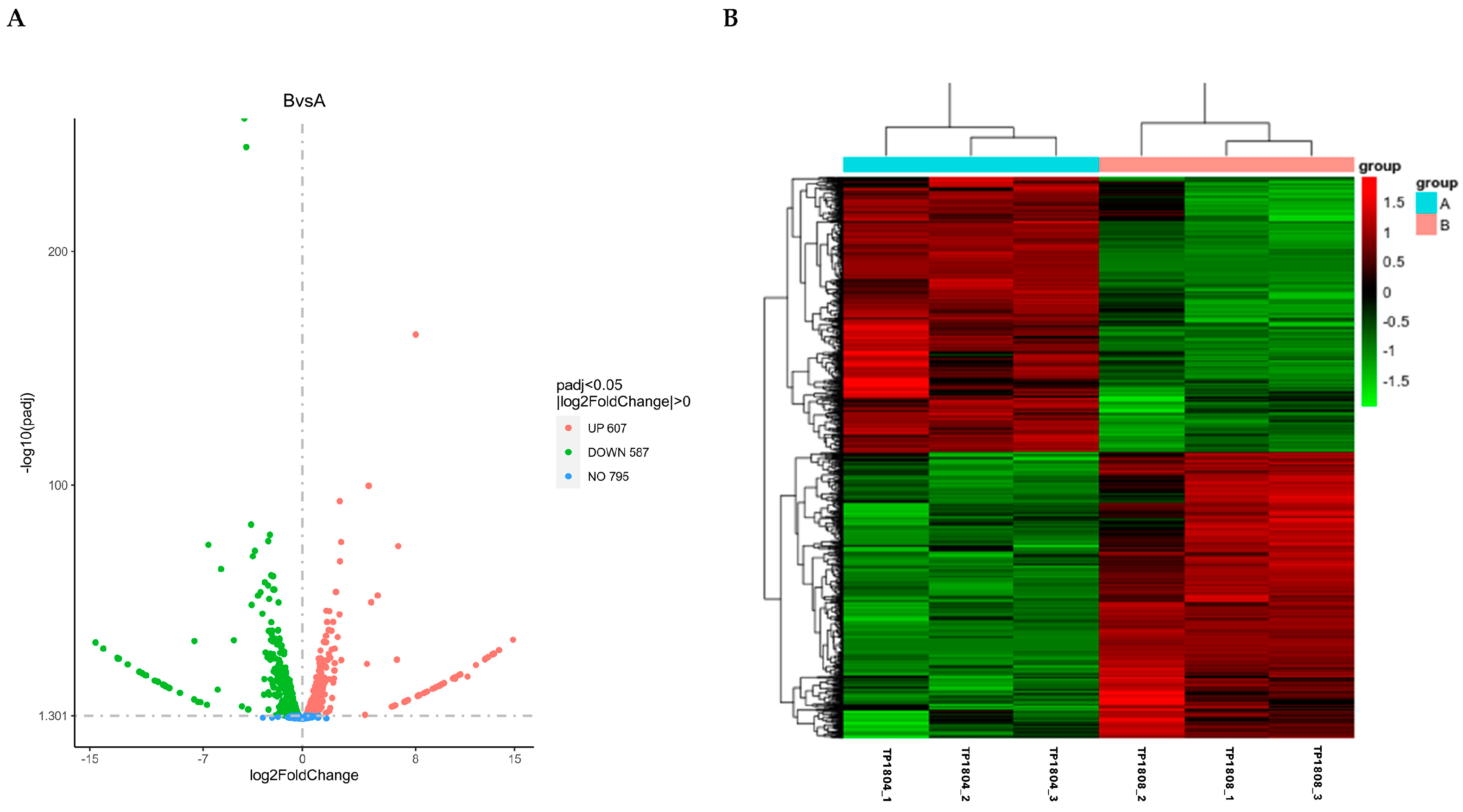
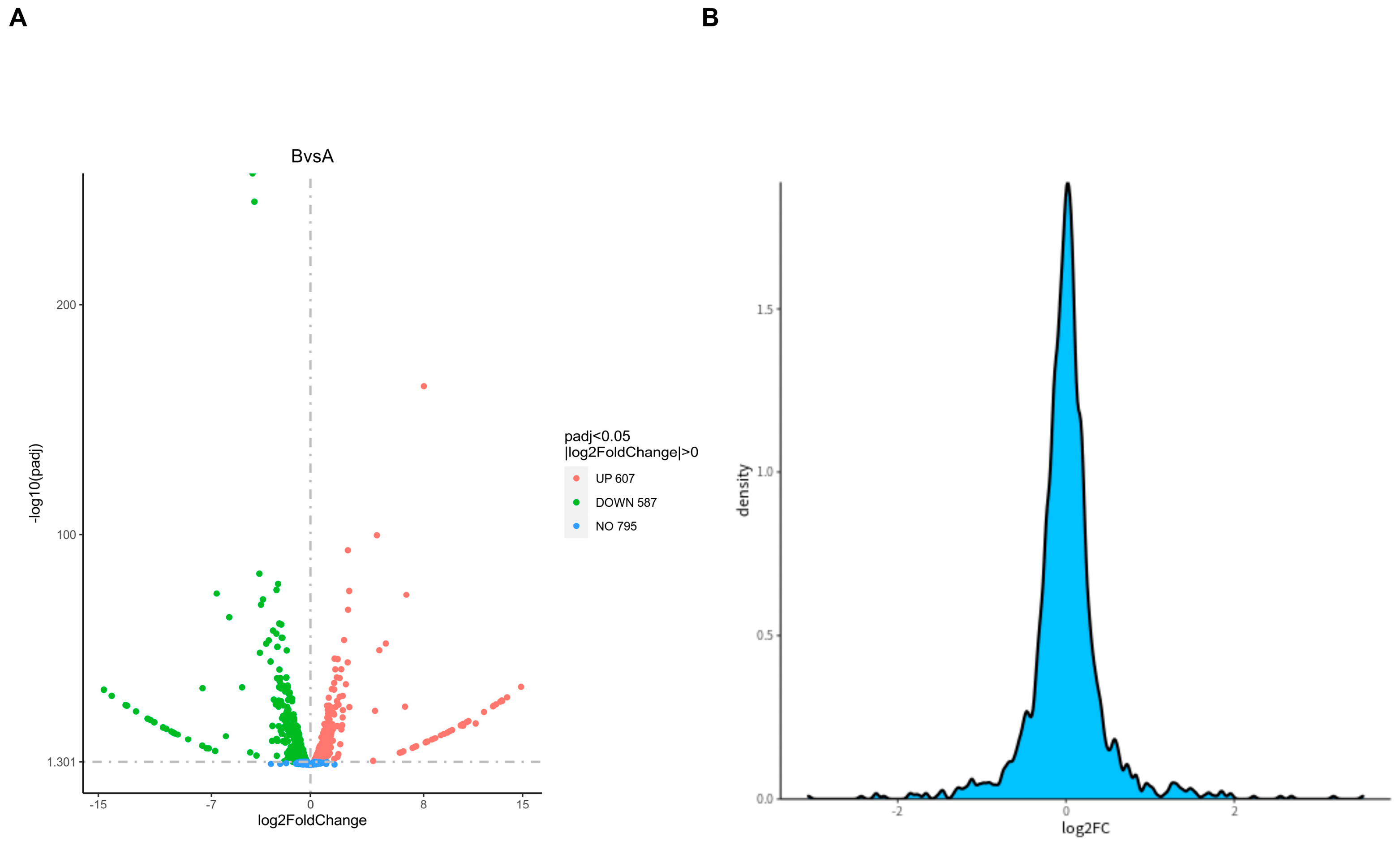
2.3. Differential Gene Analysis
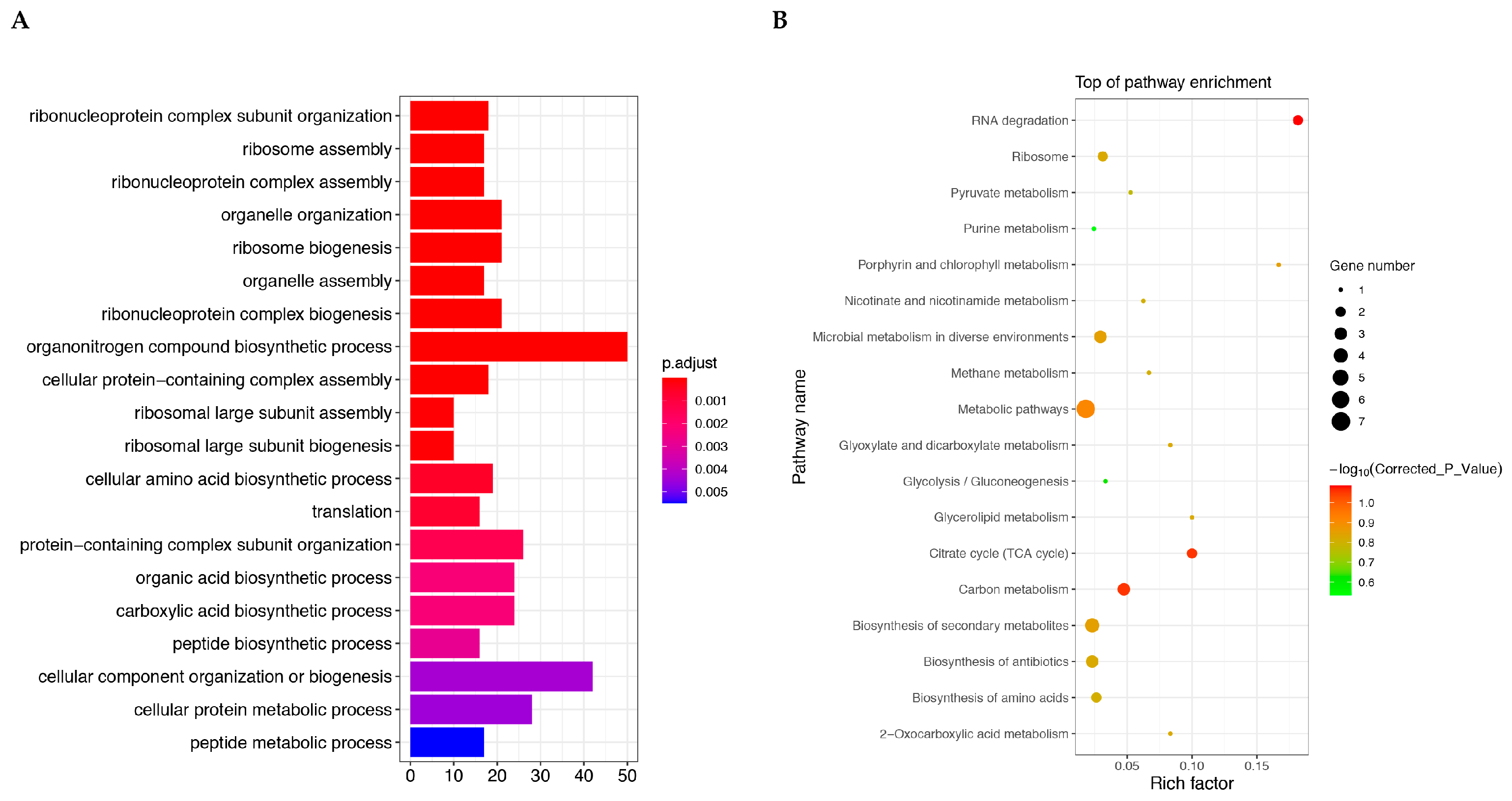
2.4. Enrichment Analysis
2.5. Principal Component Analysis (PCA)
2.6. Differential Expression Protein Analysis
2.7. Differential Protein Functional Enrichment Analysis
3. Discussion
4. Materials and Methods
4.1. Strain and Culture Medium
4.2. DNA Extraction and Sequencing
4.3. Bacterial Growth Curves
4.4. Cell Culture
4.5. Lactate Dehydrogenase (LDH) Assay
4.6. RNA Extraction and Transcriptomic Analysis
4.7. Proteomic Analysis
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Z.; Liang, Y.; Yu, L.; Chen, M.; Guo, Y.; Kang, Z.; Qu, C.; Tian, C.; Zhang, D.; Liu, M. TatD DNases Contribute to Biofilm Formation and Virulence in Trueperella pyogenes. Front. Microbiol. 2021, 12, 758465. [Google Scholar] [CrossRef]
- Qin, L.; Meng, F.; He, H.; Yang, Y.B.; Wang, G.; Tang, Y.D.; Sun, M.; Zhang, W.; Cai, X.; Wang, S. A Virulent Trueperella pyogenes Isolate, Which Causes Severe Bronchoconstriction in Porcine Precision-Cut Lung Slices. Front. Vet. Sci. 2021, 8, 824349. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Shan, Q.; Wu, X.; Xu, L.; Li, Y.; Wang, J.; Wang, X.; Zhu, Y. Phenotypic Characteristics, Antimicrobial Susceptibility and Virulence Genotype Features of Trueperella pyogenes Associated with Endometritis of Dairy Cows. Int. J. Mol. Sci. 2024, 25, 3974. [Google Scholar] [CrossRef]
- Risseti, R.M.; Zastempowska, E.; Twaruzek, M.; Lassa, H.; Pantoja, J.C.F.; de Vargas, A.P.C.; Guerra, S.T.; Bolanos, C.A.D.; de Paula, C.L.; Alves, A.C.; et al. Virulence markers associated with Trueperella pyogenes infections in livestock and companion animals. Lett. Appl. Microbiol. 2017, 65, 125–132. [Google Scholar] [CrossRef]
- Ashrafi Tamai, I.; Mohammadzadeh, A.; Zahraei Salehi, T.; Mahmoodi, P. Genomic characterisation, detection of genes encoding virulence factors and evaluation of antibiotic resistance of Trueperella pyogenes isolated from cattle with clinical metritis. Antonie Van Leeuwenhoek 2018, 111, 2441–2453. [Google Scholar] [CrossRef] [PubMed]
- Shan, Q.; Ma, W.; Li, B.; Li, Q.; Wang, X.; Li, Y.; Wang, J.; Zhu, Y.; Liu, N. Revealing the Mechanism of NLRP3 Inflammatory Pathway Activation through K(+) Efflux Induced by PLO via Signal Point Mutations. Int. J. Mol. Sci. 2024, 25, 6703. [Google Scholar] [CrossRef]
- Tamai, I.A.; Mohammadzadeh, A.; Mahmoodi, P.; Pakbin, B.; Salehi, T.Z. Antimicrobial susceptibility, virulence genes and genomic characterization of Trueperella pyogenes isolated from abscesses in dairy cattle. Res. Vet. Sci. 2023, 154, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Rzewuska, M.; Kwiecien, E.; Chrobak-Chmiel, D.; Kizerwetter-Swida, M.; Stefanska, I.; Gierynska, M. Pathogenicity and Virulence of Trueperella pyogenes: A Review. Int. J. Mol. Sci. 2019, 20, 2737. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Liu, M.; Zhang, X.; Wang, H.; Yue, B. In vitro and in vivo expression of virulence genes in Trueperella pyogenes based on a mouse model. Vet. Microbiol. 2013, 163, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Jost, B.H.; Billington, S.J. Arcanobacterium pyogenes: Molecular pathogenesis of an animal opportunist. Antonie Van Leeuwenhoek 2005, 88, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi Tamai, I.; Mohammadzadeh, A.; Zahraei Salehi, T.; Mahmoodi, P.; Pakbin, B. Investigation of antimicrobial susceptibility and virulence factor genes in Trueperella pyogenes isolated from clinical mastitis cases of dairy cows. Food Sci. Nutr. 2021, 9, 4529–4538. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.; Peter, S.; Wagener, K.; Drillich, M.; Ehling-Schulz, M.; Einspanier, R.; Gabler, C. Bovine Endometrial Epithelial Cells Scale Their Pro-inflammatory Response In vitro to Pathogenic Trueperella pyogenes Isolated from the Bovine Uterus in a Strain-Specific Manner. Front. Cell Infect. Microbiol. 2017, 7, 264. [Google Scholar] [CrossRef] [PubMed]
- Rudnick, S.T.; Jost, B.H.; Billington, S.J. Transcriptional regulation of pyolysin production in the animal pathogen, Arcanobacterium pyogenes. Vet. Microbiol. 2008, 132, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Wang, M.; Lin, S.; Jian, R.; Li, X.; Chan, J.; Dong, G.; Fang, H.; Robinson, A.E.; Consortium, G.T.; et al. A Quantitative Proteome Map of the Human Body. Cell 2020, 183, 269–283.E19. [Google Scholar] [CrossRef]
- Liu, M.; Wang, B.; Liang, H.; Ma, B.; Wang, J.; Zhang, W. Determination of the expression of three fimbrial subunit proteins in cultured Trueperella pyogenes. Acta Vet. Scand. 2018, 60, 53. [Google Scholar] [CrossRef] [PubMed]
- Pietrocola, G.; Valtulina, V.; Rindi, S.; Jost, B.H.; Speziale, P. Functional and structural properties of CbpA, a collagen-binding protein from Arcanobacterium pyogenes. Microbiology 2007, 153, 3380–3389. [Google Scholar] [CrossRef] [PubMed]
- Alkasir, R.; Wang, J.; Gao, J.; Ali, T.; Zhang, L.; Szenci, O.; Bajcsy, A.C.; Han, B. Properties and antimicrobial susceptibility of Trueperella pyogenes isolated from bovine mastitis in China. Acta Vet. Hung. 2016, 64, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Graifer, D.; Karpova, G. Interaction of tRNA with eukaryotic ribosome. Int. J. Mol. Sci. 2015, 16, 7173–7194. [Google Scholar] [CrossRef] [PubMed]
- Hinnebusch, A.G. Molecular mechanism of scanning and start codon selection in eukaryotes. Microbiol. Mol. Biol. Rev. 2011, 75, 434–467. [Google Scholar] [CrossRef]
- Filbeck, S.; Cerullo, F.; Pfeffer, S.; Joazeiro, C.A.P. Ribosome-associated quality-control mechanisms from bacteria to humans. Mol. Cell 2022, 82, 1451–1466. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Foo, M.; Eren, A.M.; Pan, T. tRNA modification dynamics from individual organisms to metaepitranscriptomics of microbiomes. Mol. Cell 2022, 82, 891–906. [Google Scholar] [CrossRef] [PubMed]
- Mandler, M.D.; Maligireddy, S.S.; Guiblet, W.M.; Fitzsimmons, C.M.; McDonald, K.S.; Warrell, D.L.; Batista, P.J. The modification landscape of Pseudomonas aeruginosa tRNAs. RNA 2024, 30, 1025–1040. [Google Scholar] [CrossRef]
- Srinivasan, R.; Ramadoss, R.; Kandasamy, V.; Ranganadin, P.; Green, S.R.; Kasirajan, A.; Pillai, A.B. Exploring the regulatory role of small RNAs in modulating host-pathogen interactions: Implications for bacterial and viral infections. Mol. Biol. Rep. 2025, 52, 115. [Google Scholar] [CrossRef] [PubMed]
- Bjork, G.R.; Hagervall, T.G. Transfer RNA Modification: Presence, Synthesis, and Function. EcoSal Plus 2014, 6, 1–68. [Google Scholar] [CrossRef]
- de Crécy-Lagard, V.; Ross, R.L.; Jaroch, M.; Marchand, V.; Eisenhart, C.; Brégeon, D.; Motorin, Y.; Limbach, P.A. Survey and Validation of tRNA Modifications and Their Corresponding Genes in sp Subtilis Strain 168. Biomolecules 2020, 10, 977. [Google Scholar] [CrossRef]
- Lin, Q.; Huang, J.; Liu, Z.; Chen, Q.; Wang, X.; Yu, G.; Cheng, P.; Zhang, L.H.; Xu, Z. tRNA modification enzyme MiaB connects environmental cues to activation of Pseudomonas aeruginosa type III secretion system. PLoS Pathog. 2022, 18, e1011027. [Google Scholar] [CrossRef] [PubMed]
- Romsang, A.; Duang-Nkern, J.; Khemsom, K.; Wongsaroj, L.; Saninjuk, K.; Fuangthong, M.; Vattanaviboon, P.; Mongkolsuk, S. Pseudomonas aeruginosa ttcA encoding tRNA-thiolating protein requires an iron-sulfur cluster to participate in hydrogen peroxide-mediated stress protection and pathogenicity. Sci. Rep. 2018, 8, 11882. [Google Scholar] [CrossRef]
- Thongdee, N.; Jaroensuk, J.; Atichartpongkul, S.; Chittrakanwong, J.; Chooyoung, K.; Srimahaeak, T.; Chaiyen, P.; Vattanaviboon, P.; Mongkolsuk, S.; Fuangthong, M. TrmB, a tRNA m7G46 methyltransferase, plays a role in hydrogen peroxide resistance and positively modulates the translation of katA and katB mRNAs in Pseudomonas aeruginosa. Nucleic Acids Res. 2019, 47, 9271–9281. [Google Scholar] [CrossRef]
- Liu, N.; Wang, X.; Shan, Q.; Xu, L.; Li, Y.; Chu, B.; Yang, L.; Wang, J.; Zhu, Y. Lactobacillus rhamnosus Ameliorates Multi-Drug-Resistant Bacillus cereus-Induced Cell Damage through Inhibition of NLRP3 Inflammasomes and Apoptosis in Bovine Endometritis. Microorganisms 2022, 10, 137. [Google Scholar] [CrossRef] [PubMed]
- Shan, Q.; Liu, N.; Wang, X.; Zhu, Y.; Yin, J.; Wang, J. Lactobacillus rhamnosus GR-1 attenuates foodborne Bacillus cereus-induced NLRP3 inflammasome activity in bovine mammary epithelial cells by protecting intercellular tight junctions. J. Anim. Sci. Biotechnol. 2022, 13, 101. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Min, H.; Luo, K.; Zhang, R.; Chen, Q.; Chen, Z. Transcriptomics and proteomics revealed the psychrotolerant and antibiotic-resistant mechanisms of strain Pseudomonas psychrophila RNC-1 capable of assimilatory nitrate reduction and aerobic denitrification. Sci. Total Environ. 2022, 820, 153169. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, Y.; Zhang, J. Proteomic mechanisms for the regulation of growth, photosynthetic activity and nitrogen fixation in Nostoc sp. PCC 7120 exposed to three antibiotic contaminants. Ecotoxicol. Environ. Saf. 2021, 225, 112753. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID/Name | Description | Log2 FC | p-Value |
|---|---|---|---|
| CQ11_RS06575 | hypothetical protein | 8.01881517709806 | 6.24680407290023 × 10−168 |
| CQ11_RS07205 | choice-of-anchor M domain-containing protein | 4.69405827857662 | 3.98052216466355 × 10−103 |
| CQ11_RS00725 | hypothetical protein | 2.6437161209102 | 1.7332343504935 × 10−96 |
| CQ11_RS07305 | HD domain-containing protein | 2.73722865310865 | 1.07719646567085 × 10−78 |
| CQ11_RS11205 | DUF1349 domain-containing protein | 6.77907877522527 | 7.36392725346365 × 10−77 |
| CQ11_RS00105 | rhodanese-like domain-containing protein | 2.65861336514393 | 3.45483260566808 × 10−70 |
| CQ11_RS08465 | dimethyl sulfoxide reductase anchor subunit | 2.37509451739112 | 6.22678372311554 × 10−57 |
| CQ11_RS05835 | hypothetical protein | 5.32834123917815 | 2.18027298280556 × 10−55 |
| CQ11_RS10335 | tRNA-Met | 4.86792626174594 | 2.23813376131865 × 10−52 |
| CQ11_RS02580/pheT | Phenylalanine tRNA ligase subunit beta/Ferredoxin-fold anticodon binding domain | 1.70567200296519 | 1.02370047692426 × 10−48 |
| ID/Name | Description | Log2 FC | p-Value |
|---|---|---|---|
| CQ11_RS04815 | peptidoglycan bridge formation glycyltransferase FemA/FemB family protein | −4.09598434309614 | 0 |
| CQ11_RS04810 | UDP-N-acetylmuramate dehydrogenase/UDP-N-acetylenolpyruvoylglucosamine reductase | −3.96207095264488 | 2.17463477730769 × 10 −248 |
| CQ11_RS04820/rpmG | 50S ribosomal protein L33 | −3.61149921577885 | 2.49897333558599 × 10 −86 |
| CQ11_RS06175 | universal stress protein | −2.28635433173304 | 7.53326393741449 × 10 −82 |
| CQ11_RS04790 | phosphotransferase | −2.39848722472287 | 3.50225282301118 × 10 −79 |
| CQ11_RS05950 | hypothetical protein | −6.63226646505396 | 1.89968005619395 × 10 −77 |
| CQ11_RS04825 | tRNA-Met | −3.35664880655572 | 9.57703127155594 × 10 −75 |
| CQ11_RS07665 | type IV toxin-antitoxin system AbiEi family antitoxin domain-containing protein | −3.49604862585357 | 2.04467148122348 × 10 −72 |
| CQ11_RS03615 | tRNA-Glu | −5.74227957327654 | 6.64897669722015 × 10 −67 |
| CQ11_RS01540 | RNA-binding S4 domain-containing protein | −2.20257447774072 | 3.71201280550628 × 10 −64 |
| ID/Name | Description | Log2 FC | p-Value |
|---|---|---|---|
| WP_024963008.1 | sulfatase-like hydrolase/transferase | 0.976030237642828 | 1.02255789360905 × 10−53 |
| WP_024963088.1 | hypothetical protein | 0.48038724806113 | 9.16585334898564 × 10−21 |
| WP_024963127.1/pknB | Stk1 family PASTA domain-containing Ser/Thr kinase | 0.0682783903146863 | 0.016432574900883 |
| WP_024963128.1 | penicillin-binding protein | 0.069473085083476 | 0.0497705393613076 |
| WP_024963167.1 | threonylcarbamoyl-AMP synthase | 0.183202749033643 | 0.0000800968930751222 |
| WP_024963168.1 | hypothetical protein | 0.19200766313386 | 0.000144392977367676 |
| WP_024963245.1 | dCTP deaminase | 0.128853301697393 | 0.0114288489838083 |
| WP_024963273.1/sufB | Fe-S cluster assembly protein SufB | 0.0871993586367944 | 0.000183042840201825 |
| WP_024963278.1 | SUF system NifU family Fe-S cluster assembly protein | 0.797769603520733 | 4.35880831787613 × 10−15 |
| WP_024963289.1 | NfeD family protein | 0.441682051391175 | 1.42689523273307 × 10−8 |
| ID/Name | Description | Log2 FC | p-Value |
|---|---|---|---|
| WP_024963019.1 | DUF3039 domain-containing protein | −0.296962054944321 | 0.0000746811041672553 |
| WP_024963030.1 | peroxiredoxin | −0.319549484183203 | 7.9950611280789 × 10−27 |
| WP_024963103.1 | ferredoxin family protein | −0.179468051587051 | 0.00566840136579069 |
| WP_024963105.1 | citrate synthase | −0.415582660837812 | 5.97700695284514 × 10−67 |
| WP_024963116.1 | DNA topoisomerase (ATP-hydrolyzing) subunit B | −0.127299178762501 | 0.0000394313454208064 |
| WP_024963121.1 | hypothetical protein | −0.24636030814353 | 0.0398927209768755 |
| WP_024963122.1 | peptidylprolyl isomerase | −0.0830256030105712 | 0.000222111411178628 |
| WP_024963141.1 | ribonuclease HI | −0.13186639640718 | 0.0419950883999149 |
| WP_024963170.1 | DUF3145 domain-containing protein | −0.343841482144734 | 0.0231910525838472 |
| WP_024963187.1 | deoxyguanosinetriphosphate triphosphohydrolase | −0.0796591823712023 | 0.0354396623845714 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, N.; Li, Q.; Shan, Q. Combined Prokaryotic Transcriptomics and Proteomics Analysis of Clinical Trueperella pyogenes Isolates with Distinctive Cytotoxicity. Int. J. Mol. Sci. 2025, 26, 1490. https://doi.org/10.3390/ijms26041490
Liu N, Li Q, Shan Q. Combined Prokaryotic Transcriptomics and Proteomics Analysis of Clinical Trueperella pyogenes Isolates with Distinctive Cytotoxicity. International Journal of Molecular Sciences. 2025; 26(4):1490. https://doi.org/10.3390/ijms26041490
Chicago/Turabian StyleLiu, Ning, Qian Li, and Qiang Shan. 2025. "Combined Prokaryotic Transcriptomics and Proteomics Analysis of Clinical Trueperella pyogenes Isolates with Distinctive Cytotoxicity" International Journal of Molecular Sciences 26, no. 4: 1490. https://doi.org/10.3390/ijms26041490
APA StyleLiu, N., Li, Q., & Shan, Q. (2025). Combined Prokaryotic Transcriptomics and Proteomics Analysis of Clinical Trueperella pyogenes Isolates with Distinctive Cytotoxicity. International Journal of Molecular Sciences, 26(4), 1490. https://doi.org/10.3390/ijms26041490