
Targeting Eukaryotic Elongation Factor 1A: How Small-Molecule Inhibitors Suppress Tumor Growth via Diverse Pathways


Abstract
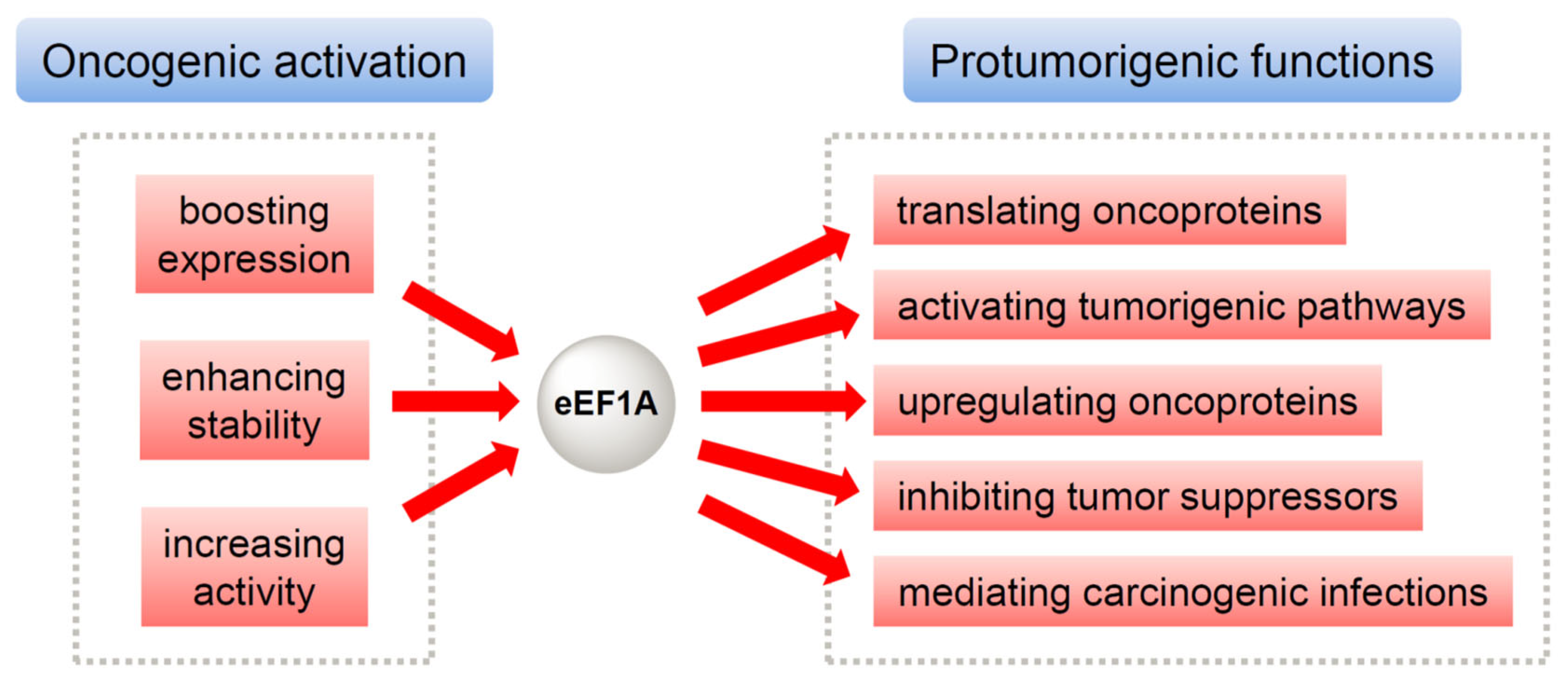
1. Introduction
2. The Mechanisms of eEF1A-Targeting Anticancer Inhibitors
2.1. Didemnins and Tamandarins
2.2. Cytotrienin A and Ansatrienin B
2.3. Ternatin
2.4. Nannocystins
2.5. Metarrestin
2.6. BE-43547A2
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar] [CrossRef]
- Lui, K.; Cheung, K.-K.; Ng, W.W.-M.; Wang, Y.; Au, D.W.H.; Cho, W.C. The Impact of Genetic Mutations on the Efficacy of Immunotherapies in Lung Cancer. Int. J. Mol. Sci. 2024, 25, 11954. [Google Scholar] [CrossRef]
- Kitano, H. Cancer as a robust system: Implications for anticancer therapy. Nat. Rev. Cancer 2004, 4, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Krol, K.; Mazur, A.; Stachyra-Strawa, P.; Grzybowska-Szatkowska, L. Non-Small Cell Lung Cancer Treatment with Molecularly Targeted Therapy and Concurrent Radiotherapy-A Review. Int. J. Mol. Sci. 2023, 24, 5858. [Google Scholar] [CrossRef]
- Wang, C.; Yao, S.; Zhang, T.; Sun, X.; Bai, C.; Zhou, P. RNA N6-Methyladenosine Modification in DNA Damage Response and Cancer Radiotherapy. Int. J. Mol. Sci. 2024, 25, 2597. [Google Scholar] [CrossRef] [PubMed]
- Mark, C.; Lee, J.S.; Cui, X.; Yuan, Y. Antibody-drug conjugates in breast cancer. Int. J. Mol. Sci. 2023, 24, 13726. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.; Suh, D.Y.; Frankel, A.E. Toxin conjugate therapy of cancer. Semin. Oncol. 2005, 32, 591–595. [Google Scholar] [CrossRef]
- Chiec, L.; Bruno, D.S. Immunotherapy for Treatment of Pleural Mesothelioma: Current and Emerging Therapeutic Strategies. Int. J. Mol. Sci. 2024, 25, 10861. [Google Scholar] [CrossRef]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduct. Target Ther. 2021, 6, 201. [Google Scholar] [CrossRef]
- Ohishi, T.; Kaneko, M.K.; Yoshida, Y.; Takashima, A.; Kato, Y.; Kawada, M. Current Targeted Therapy for Metastatic Colorectal Cancer. Int. J. Mol. Sci. 2023, 24, 1702. [Google Scholar] [CrossRef]
- Bhat, M.; Robichaud, N.; Hulea, L.; Sonenberg, N.; Pelletier, J.; Topisirovic, I. Targeting the translation machinery in cancer. Nat. Rev. Drug Discovery 2015, 14, 261–278. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Pelletier, J. Therapeutic opportunities in eukaryotic translation. Cold Spring Harbor Perspect. Biol. 2018, 10, a032995. [Google Scholar] [CrossRef] [PubMed]
- Fan, A.; Sharp, P.P. Inhibitors of Eukaryotic Translational Machinery as Therapeutic Agents. J. Med. Chem. 2021, 64, 2436–2465. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Cai, J.; Yu, S.; Sun, B.; Zhang, W. Anticancer Small-Molecule Agents Targeting Eukaryotic Elongation Factor 1A: State of the Art. Int. J. Mol. Sci. 2023, 24, 5184. [Google Scholar] [CrossRef]
- Negrutskii, B.S.; El’skaya, A.V. Eukaryotic translation elongation factor 1α: Structure, expression, functions, and possible role in aminoacyl-tRNA channeling. Prog. Nucleic Acid Res. Mol. Biol. 1998, 60, 47–78. [Google Scholar]
- Soares, D.C.; Barlow, P.N.; Newbery, H.J.; Porteous, D.J.; Abbott, C.M. Structural models of human eEF1A1 and eEF1A2 reveal two distinct surface clusters of sequence variation and potential differences in phosphorylation. PLoS ONE 2009, 4, e6315. [Google Scholar] [CrossRef]
- Mills, A.; Gago, F. On the Need to Tell Apart Fraternal Twins eEF1A1 and eEF1A2, and Their Respective Outfits. Int. J. Mol. Sci. 2021, 22, 6973. [Google Scholar] [CrossRef]
- Kahns, S.; Lund, A.; Kristensen, P.; Knudsen, C.R.; Clark, B.F.C.; Cavallius, J.; Merrick, W.C. The elongation factor 1 A-2 isoform from rabbit: Cloning of the cDNA and characterization of the protein. Nucleic Acids Res. 1998, 26, 1884–1890. [Google Scholar] [CrossRef]
- Carriles, A.A.; Mills, A.; Muñoz-Alonso, M.-J.; Gutiérrez, D.; Domínguez, J.M.; Hermoso, J.A.; Gago, F. Structural Cues for Understanding eEF1A2 Moonlighting. ChemBioChem 2021, 22, 374–391. [Google Scholar] [CrossRef]
- Mateyak, M.K.; Kinzy, T.G. eEF1A: Thinking outside the ribosome. J. Biol. Chem. 2010, 285, 21209–21213. [Google Scholar] [CrossRef]
- Sasikumar, A.N.; Perez, W.B.; Kinzy, T.G. The many roles of the eukaryotic elongation factor 1 complex. WIREs RNA 2012, 3, 543–555. [Google Scholar] [CrossRef]
- Thornton, S.; Anand, N.; Purcell, D.; Lee, J. Not just for housekeeping: Protein initiation and elongation factors in cell growth and tumorigenesis. J. Mol. Med. 2003, 81, 536–548. [Google Scholar] [CrossRef] [PubMed]
- Lamberti, A.; Caraglia, M.; Longo, O.; Marra, M.; Abbruzzese, A.; Arcari, P. The translation elongation factor 1A in tumorigenesis, signal transduction and apoptosis: Review article. Amino Acids 2004, 26, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Abbas, W.; Kumar, A.; Herbein, G. The eEF1A Proteins: At the Crossroads of Oncogenesis, Apoptosis, and Viral Infections. Front. Oncol. 2015, 5, 75. [Google Scholar] [CrossRef] [PubMed]
- Anand, N.; Murthy, S.; Amann, G.; Wernick, M.; Porter, L.A.; Cukier, I.H.; Collins, C.; Gray, J.W.; Diebold, J.; Demetrick, D.J.; et al. Protein elongation factor EEF1A2 is a putative oncogene in ovarian cancer. Nat. Genet. 2002, 31, 301–305. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, J.; Shan, C. The eEF1A protein in cancer: Clinical significance, oncogenic mechanisms, and targeted therapeutic strategies. Pharmacol. Res. 2024, 204, 107195. [Google Scholar] [CrossRef]
- Yamato, M.; Dai, T.; Murata, Y.; Nakagawa, T.; Kikuchi, S.; Matsubara, D.; Noguchi, M. High expression of eukaryotic elongation factor 1-alpha-2 in lung adenocarcinoma is associated with poor prognosis. Pathol. Int. 2024, 74, 454–463. [Google Scholar] [CrossRef]
- Shi, Z.; Guo, X.; Hu, X.; Li, R.; Li, X.; Lu, J.; Jin, M.; Jiang, X. DNA methylation profiling identifies epigenetic signatures of early gastric cancer. Virchows Arch. 2024, 484, 687–695. [Google Scholar] [CrossRef]
- Chun, H.G.; Davies, B.; Hoth, D.; Suffness, M.; Plowman, J.; Flora, K.; Grieshaber, C.; Leyland-Jones, B.; Didemnin, B. The first marine compound entering clinical trials as an antineoplastic agent. Investig. New Drugs 1986, 4, 279–284. [Google Scholar] [CrossRef]
- Leisch, M.; Egle, A.; Greil, R. Plitidepsin: A potential new treatment for relapsed/refractory multiple myeloma. Future Oncol. 2019, 15, 109–120. [Google Scholar] [CrossRef]
- Jimenez, P.C.; Wilke, D.V.; Branco, P.C.; Bauermeister, A.; Rezende-Teixeira, P.; Gaudencio, S.P.; Costa-Lotufo, L.V. Enriching cancer pharmacology with drugs of marine origin. Brit. J. Pharmacol. 2020, 177, 3–27. [Google Scholar] [CrossRef]
- Metarrestin (ML-246) in Subjects with Metastatic Solid Tumors (Clinical trial ID: NCT04222413). Available online: www.clinicaltrials.gov (accessed on 1 July 2025).
- Lee, J.; Currano, J.N.; Carroll, P.J.; Joullie, M.M. Didemnins, tamandarins and related natural products. Nat. Prod. Rep. 2012, 29, 404–424. [Google Scholar] [CrossRef]
- Zhang, H.-p.; Kakeya, H.; Osada, H. Novel triene-ansamycins, cytotrienins A and B, inducing apoptosis on human leukemia HL-60 cells. Tetrahedron Lett. 1997, 38, 1789–1792. [Google Scholar] [CrossRef]
- Yamada, Y.; Taketani, S.; Osada, H.; Kataoka, T. Cytotrienin A, a translation inhibitor that induces ectodomain shedding of TNF receptor 1 via activation of ERK and p38 MAP kinase. Eur. J. Pharmacol. 2011, 667, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chen, S.; Wang, J.; Zhang, M.; Wu, W.; Liu, W.; Sun, P. Ansafurantrienins, Unprecedented Ansatrienin Derivatives Formed via Photocatalytic Intramolecular [3+ 2] Oxidative Cycloaddition. Org. Lett. 2022, 24, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Van Goietsenoven, G.; Hutton, J.; Becker, J.P.; Lallemand, B.; Robert, F.; Lefranc, F.; Pirker, C.; Vandenbussche, G.; Van Antwerpen, P.; Evidente, A.; et al. Targeting of eEF1A with Amaryllidaceae isocarbostyrils as a strategy to combat melanomas. FASEB J. 2010, 24, 4575–4584. [Google Scholar] [CrossRef]
- Yao, N.; Chen, C.Y.; Wu, C.Y.; Motonishi, K.; Kung, H.J.; Lam, K.S. Novel flavonoids with antiproliferative activities against breast cancer cells. J. Med. Chem. 2011, 54, 4339–4349. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Yang, H.; Holm, M.; Tom, H.; Oltion, K.; Al-Khdhairawi, A.A.Q.; Weber, J.-F.F.; Blanchard, S.C.; Ruggero, D.; Taunton, J. Synthesis and single-molecule imaging reveal stereospecific enhancement of binding kinetics by the antitumour eEF1A antagonist SR-A3. Nat. Chem. 2022, 14, 1443–1450. [Google Scholar] [CrossRef]
- Hoffmann, H.; Kogler, H.; Heyse, W.; Matter, H.; Caspers, M.; Schummer, D.; Klemke-Jahn, C.; Bauer, A.; Penarier, G.; Debussche, L.; et al. Discovery, Structure Elucidation, and Biological Characterization of Nannocystin A, a Macrocyclic Myxobacterial Metabolite with Potent Antiproliferative Properties. Angew. Chem. Int. Ed. 2015, 54, 10145–10148. [Google Scholar] [CrossRef]
- Krastel, P.; Roggo, S.; Schirle, M.; Ross, N.T.; Perruccio, F.; Aspesi, P., Jr.; Aust, T.; Buntin, K.; Estoppey, D.; Liechty, B.; et al. Nannocystin A: An Elongation Factor 1 Inhibitor from Myxobacteria with Differential Anti-Cancer Properties. Angew. Chem. Int. Ed. 2015, 54, 10149–10154. [Google Scholar] [CrossRef]
- Hou, Y.; Liu, R.; Xia, M.; Sun, C.; Zhong, B.; Yu, J.; Ai, N.; Lu, J.-J.; Ge, W.; Liu, B.; et al. Nannocystin ax, an eEF1A inhibitor, induces G1 cell cycle arrest and caspase-independent apoptosis through cyclin D1 downregulation in colon cancer in vivo. Pharmacol. Res. 2021, 173, 105870. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xie, F.; Yuan, X.-Y.; Dai, X.-T.; Tian, Y.-F.; Sun, M.-M.; Yu, S.-Q.; Cai, J.-Y.; Sun, B.; Zhang, W.-C.; et al. Discovery of a nitroaromatic nannocystin with potent in vivo anticancer activity against colorectal cancer by targeting AKT1. Acta Pharmacol. Sin. 2024, 45, 1044–1059. [Google Scholar] [CrossRef]
- Frankowski, K.J.; Wang, C.; Patnaik, S.; Schoenen, F.J.; Southall, N.; Li, D.; Teper, Y.; Sun, W.; Kandela, I.; Hu, D.; et al. Metarrestin, a perinucleolar compartment inhibitor, effectively suppresses metastasis. Sci. Transl. Med. 2018, 10, eaap8307. [Google Scholar] [CrossRef] [PubMed]
- Frankowski, K.J.; Patnaik, S.; Wang, C.; Southall, N.; Dutta, D.; De, S.; Li, D.; Dextras, C.; Lin, Y.-H.; Bryant-Connah, M.; et al. Discovery and Optimization of Pyrrolopyrimidine Derivatives as Selective Disruptors of the Perinucleolar Compartment, a Marker of Tumor Progression toward Metastasis. J. Med. Chem. 2022, 65, 8303–8331. [Google Scholar] [CrossRef]
- Burglová, K.; Rylová, G.; Markos, A.; Prichystalova, H.; Soural, M.; Petracek, M.; Medvedikova, M.; Tejral, G.; Sopko, B.; Hradil, P.; et al. Identification of Eukaryotic Translation Elongation Factor 1-α 1 Gamendazole-Binding Site for Binding of 3-Hydroxy-4(1H)-quinolinones as Novel Ligands with Anticancer Activity. J. Med. Chem. 2018, 61, 3027–3036. [Google Scholar] [CrossRef]
- Klein, V.G.; Bray, W.M.; Wang, H.-Y.; Edmondson, Q.; Schwochert, J.; Ono, S.; Naylor, M.R.; Turmon, A.C.; Faris, J.H.; Okada, O.; et al. Identifying the Cellular Target of Cordyheptapeptide A and Synthetic Derivatives. ACS Chem. Biol. 2021, 16, 1354–1364. [Google Scholar] [CrossRef]
- Sun, Y.; Ding, Y.; Li, D.; Zhou, R.; Su, X.; Yang, J.; Guo, X.; Chong, C.; Wang, J.; Zhang, W.; et al. Cyclic Depsipeptide BE-43547A2: Synthesis and Activity against Pancreatic Cancer Stem Cells. Angew. Chem. Int. Ed. 2017, 56, 14627–14631. [Google Scholar] [CrossRef]
- Liu, C.; Wang, L.; Sun, Y.; Zhao, X.; Chen, T.; Su, X.; Guo, H.; Wang, Q.; Xi, X.; Ding, Y.; et al. Probe Synthesis Reveals Eukaryotic Translation Elongation Factor 1 Alpha 1 as the Anti-Pancreatic Cancer Target of BE-43547A2. Angew. Chem. Int. Ed. 2022, 61, e202206953. [Google Scholar] [CrossRef]
- Losada, A.; Munoz-Alonso, M.J.; Garcia, C.; Sanchez-Murcia, P.A.; Martinez-Leal, J.F.; Dominguez, J.M.; Lillo, M.P.; Gago, F.; Galmarini, C.M. Translation Elongation Factor eEF1A2 is a Novel Anticancer Target for the Marine Natural Product Plitidepsin. Sci. Rep. 2016, 6, 35100. [Google Scholar] [CrossRef]
- Crews, C.M.; Collins, J.L.; Lane, W.S.; Snapper, M.L.; Schreiber, S.L. GTP-dependent binding of the antiproliferative agent didemnin to elongation factor 1α. J. Biol. Chem. 1994, 269, 15411–15414. [Google Scholar] [CrossRef]
- García-Fernández, L.F.; Losada, A.; Alcaide, V.; Álvarez, A.M.; Cuadrado, A.; Gonzalez, L.; Nakayama, K.; Nakayama, K.I.; Fernández-Sousa, J.M.; Munoz, A. Aplidin™ induces the mitochondrial apoptotic pathway via oxidative stress-mediated JNK and p38 activation and protein kinase C δ. Oncogene 2002, 21, 7533–7544. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Garcıa-Fernández, L.F.; González, L.; Suárez, Y.; Losada, A.; Alcaide, V.; Martınez, T.; Fernández-Sousa, J.M.; Sánchez-Puelles, J.M.; Munoz, A. AplidinTM induces apoptosis in human cancer cells via glutathione depletion and sustained activation of the epidermal growth factor receptor, Src, JNK, and p38 MAPK. J. Biol. Chem. 2003, 278, 241–250. [Google Scholar] [CrossRef]
- Cuadrado, A.; Gonzalez, L.; Suarez, Y.; Martínez, T.; Munoz, A. JNK activation is critical for Aplidin™-induced apoptosis. Oncogene 2004, 23, 4673–4680. [Google Scholar] [CrossRef]
- Gonzalez-Santiago, L.; Suárez, Y.; Zarich, N.; Munoz-Alonso, M.; Cuadrado, A.; Martinez, T.; Goya, L.; Iradi, A.; Saez-Tormo, G.; Maier, J. Aplidin® induces JNK-dependent apoptosis in human breast cancer cells via alteration of glutathione homeostasis, Rac1 GTPase activation, and MKP-1 phosphatase downregulation. Cell Death Differ. 2006, 13, 1968–1981. [Google Scholar] [CrossRef]
- Gajate, C.; An, F.; Mollinedo, F. Rapid and selective apoptosis in human leukemic cells induced by Aplidine through a Fas/CD95-and mitochondrial-mediated mechanism. Clin. Cancer Res. 2003, 9, 1535–1545. [Google Scholar]
- Gajate, C.; Mollinedo, F. Cytoskeleton-mediated death receptor and ligand concentration in lipid rafts forms apoptosis-promoting clusters in cancer chemotherapy. J. Biol. Chem. 2005, 280, 11641–11647. [Google Scholar] [CrossRef]
- Broggini, M.; Marchini, S.; Galliera, E.; Borsotti, P.; Taraboletti, G.; Erba, E.; Sironi, M.; Jimeno, J.; Faircloth, G.; Giavazzi, R. Aplidine, a new anticancer agent of marine origin, inhibits vascular endothelial growth factor (VEGF) secretion and blocks VEGF-VEGFR-1 (flt-1) autocrine loop in human leukemia cells MOLT-4. Leukemia 2003, 17, 52–59. [Google Scholar] [CrossRef]
- Taraboletti, G.; Poli, M.; Dossi, R.; Manenti, L.; Borsotti, P.; Faircloth, G.; Broggini, M.; D’incalci, M.; Ribatti, D.; Giavazzi, R. Antiangiogenic activity of aplidine, a new agent of marine origin. Br. J. Cancer 2004, 90, 2418–2424. [Google Scholar] [CrossRef]
- Morande, P.E.; Zanetti, S.R.; Borge, M.; Nannini, P.; Jancic, C.; Bezares, R.F.; Bitsmans, A.; González, M.; Rodríguez, A.L.; Galmarini, C.M. The cytotoxic activity of Aplidin in chronic lymphocytic leukemia (CLL) is mediated by a direct effect on leukemic cells and an indirect effect on monocyte-derived cells. Investig. New Drugs 2012, 30, 1830–1840. [Google Scholar] [CrossRef]
- Spicka, I.; Ocio, E.M.; Oakervee, H.E.; Greil, R.; Banh, R.H.; Catley, L.; Huang, S.-Y.; D’Rozario, J.M.; Dimopoulos, M.A.; Rodriguez, J. Randomized phase III study (ADMYRE) of plitidepsin in combination with dexamethasone vs. dexamethasone alone in patients with relapsed/refractory multiple myeloma. Blood 2017, 130, 1886. [Google Scholar] [CrossRef]
- Mitsiades, C.S.; Ocio, E.M.; Pandiella, A.; Maiso, P.; Gajate, C.; Garayoa, M.; Vilanova, D.; Montero, J.C.; Mitsiades, N.; McMullan, C.J. Aplidin, a marine organism–derived compound with potent antimyeloma activity in vitro and in vivo. Cancer Res. 2008, 68, 5216–5225. [Google Scholar] [CrossRef] [PubMed]
- Losada, A.; Munoz-Alonso, M.J.; Martinez-Diez, M.; Gago, F.; Dominguez, J.M.; Martinez-Leal, J.F.; Galmarini, C.M. Binding of eEF1A2 to the RNA-dependent protein kinase PKR modulates its activity and promotes tumor cell survival. Br. J. Cancer 2018, 119, 1410–1420. [Google Scholar] [CrossRef] [PubMed]
- Losada, A.; Berlanga, J.J.; Molina-Guijarro, J.M.; Jimenez-Ruiz, A.; Gago, F.; Aviles, P.; de Haro, C.; Martinez-Leal, J.F. Generation of endoplasmic reticulum stress and inhibition of autophagy by plitidepsin induces proteotoxic apoptosis in cancer cells. Biochem. Pharmacol. 2020, 172, 113744. [Google Scholar] [CrossRef] [PubMed]
- Potts, M.B.; McMillan, E.A.; Rosales, T.I.; Kim, H.S.; Ou, Y.-H.; Toombs, J.E.; Brekken, R.A.; Minden, M.D.; MacMillan, J.B.; White, M.A. Mode of action and pharmacogenomic biomarkers for exceptional responders to didemnin B. Nat. Chem. Biol. 2015, 11, 401–408. [Google Scholar] [CrossRef]
- Kakeya, H.; Zhang, H.-P.; Kobinata, K.; Onose, R.; Onozawa, C.; Kudo, T.; Osada, H. Cytotrienin A, a novel apoptosis inducer in human leukemia HL-60 cells. J. Antibiot. 1997, 50, 370–372. [Google Scholar] [CrossRef]
- Lindqvist, L.; Robert, F.; Merrick, W.; Kakeya, H.; Fraser, C.; Osada, H.; Pelletier, J. Inhibition of translation by cytotrienin A--a member of the ansamycin family. RNA 2010, 16, 2404–2413. [Google Scholar] [CrossRef]
- Watabe, M.; Kakeya, H.; Onose, R.; Osada, H. Activation of MST/Krs and c-Jun N-terminal kinases by different signaling pathways during cytotrienin A-induced apoptosis. J. Biol. Chem. 2000, 275, 8766–8771. [Google Scholar] [CrossRef]
- Kakeya, H.; Onose, R.; Osada, H. Caspase-mediated activation of a 36-kDa myelin basic protein kinase during anticancer drug-induced apoptosis. Cancer Res. 1998, 58, 4888–4894. [Google Scholar]
- Sugita, M.; Natori, Y.; Sasaki, T.; Furihata, K.; Shimazu, A.; Seto, H.; Otake, N. Studies on mycotrienin antibiotics, a novel class of ansamycins I. Taxonomy, fermentation, isolation and properties of mycotrienins I and II. J. Antibiot. 1982, 35, 1460–1466. [Google Scholar] [CrossRef]
- Yamada, Y.; Tashiro, E.; Taketani, S.; Imoto, M.; Kataoka, T. Mycotrienin II, a translation inhibitor that prevents ICAM-1 expression induced by pro-inflammatory cytokines. J. Antibiot. 2011, 64, 361–366. [Google Scholar] [CrossRef]
- Shimokawa, K.; Mashima, I.; Asai, A.; Yamada, K.; Kita, M.; Uemura, D. (−)-Ternatin, a highly N-methylated cyclic heptapeptide that inhibits fat accumulation: Structure and synthesis. Tetrahedron Lett. 2006, 47, 4445–4448. [Google Scholar] [CrossRef]
- Carelli, J.D.; Sethofer, S.G.; Smith, G.A.; Miller, H.R.; Simard, J.L.; Merrick, W.C.; Jain, R.K.; Ross, N.T.; Taunton, J. Ternatin and improved synthetic variants kill cancer cells by targeting the elongation factor-1A ternary complex. Elife 2015, 4, e10222. [Google Scholar] [CrossRef]
- Juette, M.F.; Carelli, J.D.; Rundlet, E.J.; Brown, A.; Shao, S.; Ferguson, A.; Wasserman, M.R.; Holm, M.; Taunton, J.; Blanchard, S.C. Didemnin B and ternatin-4 differentially inhibit conformational changes in eEF1A required for aminoacyl-tRNA accommodation into mammalian ribosomes. Elife 2022, 11, e81608. [Google Scholar] [CrossRef]
- Oltion, K.; Carelli, J.D.; Yang, T.; See, S.K.; Wang, H.-Y.; Kampmann, M.; Taunton, J. An E3 ligase network engages GCN1 to promote the degradation of translation factors on stalled ribosomes. Cell 2023, 186, 346–362. [Google Scholar] [CrossRef]
- Sun, C.; Liu, R.; Xia, M.; Hou, Y.; Wang, X.; Lu, J.-j.; Liu, B.; Chen, X. Nannocystin Ax, a natural elongation factor 1α inhibitor from Nannocystis sp., suppresses epithelial-mesenchymal transition, adhesion and migration in lung cancer cells. Toxicol. Appl. Pharmacol. 2021, 420, 115535. [Google Scholar] [CrossRef]
- Zhang, H.; Tian, Y.; Yuan, X.; Xie, F.; Yu, S.; Cai, J.; Sun, B.; Shan, C.; Zhang, W. Site-directed Late-Stage Diversification of Macrocyclic Nannocystins Facilitating Anticancer SAR and Mode of Action Studies. RSC Med. Chem. 2023, 14, 299–312. [Google Scholar] [CrossRef]
- Gemmer, M.; Chaillet, M.L.; van Loenhout, J.; Cuevas Arenas, R.; Vismpas, D.; Gröllers-Mulderij, M.; Koh, F.A.; Albanese, P.; Scheltema, R.A.; Howes, S.C.; et al. Visualization of translation and protein biogenesis at the ER membrane. Nature 2023, 614, 160–167. [Google Scholar] [CrossRef]
- Kashyap, V.K.; Sharma, B.P.; Pandey, D.; Singh, A.K.; Peasah-Darkwah, G.; Singh, B.; Roy, K.K.; Yallapu, M.M.; Chauhan, S.C. Small Molecule with Big Impact: Metarrestin Targets the Perinucleolar Compartment in Cancer Metastasis. Cells 2024, 13, 2053. [Google Scholar] [CrossRef]
- Norton, J.T.; Titus, S.A.; Dexter, D.; Austin, C.P.; Zheng, W.; Huang, S. Automated High-Content Screening for Compounds That Disassemble the Perinucleolar Compartment. J. Biomol. Screen. 2009, 14, 1045–1053. [Google Scholar] [CrossRef]
- Vilimas, T.; Wang, A.Q.; Patnaik, S.; Hughes, E.A.; Singleton, M.D.; Knotts, Z.; Li, D.; Frankowski, K.; Schlomer, J.J.; Guerin, T.M.; et al. Pharmacokinetic evaluation of the PNC disassembler metarrestin in wild-type and Pdx1-Cre;LSL-KrasG12D/+;Tp53R172H/+ (KPC) mice, a genetically engineered model of pancreatic cancer. Cancer Chemother. Pharmacol. 2018, 82, 1067–1080. [Google Scholar] [CrossRef]
- Padilha, E.C.; Shah, P.; Wang, A.Q.; Singleton, M.D.; Hughes, E.A.; Li, D.; Rice, K.A.; Konrath, K.M.; Patnaik, S.; Marugan, J. Metabolism and pharmacokinetics characterization of metarrestin in multiple species. Cancer Chemother. Pharm. 2020, 85, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Bourdi, M.; Rudloff, U.; Patnaik, S.; Marugan, J.; Terse, P.S. Safety assessment of metarrestin in dogs: A clinical candidate targeting a subnuclear structure unique to metastatic cancer cells. Regul. Toxicol. Pharm. 2020, 116, 104716. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Kabir, M.; Sun, N.; Kaniskan, H.U. Preparation of Heterobifunctional Compounds as Degraders of eEF1A2. WO2022159650A1, 28 July 2022. [Google Scholar]
- Villadsen, N.L.; Jacobsen, K.M.; Keiding, U.B.; Weibel, E.T.; Christiansen, B.; Vosegaard, T.; Bjerring, M.; Jensen, F.; Johannsen, M.; Tørring, T.; et al. Synthesis of ent-BE-43547A1 reveals a potent hypoxia-selective anticancer agent and uncovers the biosynthetic origin of the APD-CLD natural products. Nat. Chem. 2017, 9, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, K.M.; Villadsen, N.L.; Toerring, T.; Nielsen, C.B.; Salomon, T.; Nielsen, M.M.; Tsakos, M.; Sibbersen, C.; Scavenius, C.; Nielsen, R.; et al. APD-Containing Cyclolipodepsipeptides Target Mitochondrial Function in Hypoxic Cancer Cells. Cell Chem. Biol. 2018, 25, 1337–1349. [Google Scholar] [CrossRef]
- Liu, C.; Liu, C.; Liu, G.-j.; Wang, M.-m.; Jiao, Y.; Sun, Y.-j.; Guo, H.; Wang, L.; Lu, Y.-x.; Chen, Y. BE-43547A2 exerts hypoxia-selective inhibition on human pancreatic cancer cells through targeting eEF1A1 and disrupting its association with FoxO1. Acta Pharmacol. Sin. 2025, 46, 1433–1444. [Google Scholar] [CrossRef]
- Qin, Y.; Wang, X.; Zhao, M.; Liu, Y.; Hou, H.; Wang, T.; Pei, Y.; Zhang, J.; Shen, Z.; Wu, F. Therapeutic potential of targeting the NEDD4L-eEF1A1 axis in cancer therapy. Acta Biochim. Biophys. Sin. 2025. [Google Scholar] [CrossRef]
- Li, B.; Zhou, J.; Ding, K.; Liu, S.; Zhang, L.; Zeng, B.; Yuan, H.; Zuo, P.; Wang, C.; Li, S.; et al. Knockdown of the EEF1A2 gene reduces lung cancer brain metastasis by downregulating the BCL10/NFκB pathway. J. Cancer Metastasis Treat. 2025, 11, 1. [Google Scholar] [CrossRef]

{kind=link}
| Feature | eEF1A1 | eEF1A2 |
|---|---|---|
| Gene Location | Chromosome 6 (human) Chromosome 9 (mouse) | Chromosome 20 (human) Chromosome 2 (mouse) |
| Expression Pattern | Ubiquitous; dominant embryonic isoform | Neuron/muscle/cardiomyocyte-specific; adult isoform; overexpressed in cancers |
| Sequence Identity | 92% identity, 98% similarity to eEF1A2 | More conserved across species than eEF1A1 |
| Key Structural Variations |
|
|
| PTM Profiles |
|
|
| Dimerization Interface | Gly70-independent | Gly70 and Ile71 critical (G70S mutation disrupts dimerization) |
| Disease Associations | Overexpression linked to cancer progression | Mutations (e.g., G70S, P333L) cause neurodevelopmental disorders and cardiomyopathy |
| Subcellular Localization | Cytosolic, perinuclear, F-actin bound | Plasma membrane/ER anchored via PE modifications |
| Therapeutic Potential | Targeting oxidative stress vulnerabilities (e.g., via Cys234) | Neurological disorders (mutation-specific inhibitors) and cancers (membrane-localized targets) |
| Compound | Anticancer Activity and Development Status | References |
|---|---|---|
| Didemnins |
| [30,31,33] |
| Tamandarins |
| [33] |
| Cytotrienin A |
| [34,35] |
| Ansatrienin B |
| [36] |
| Narciclasine |
| [37] |
| Synthetic flavonoids |
| [38] |
| Ternatins |
| [39] |
| Nannocystins |
| [40,41,42,43] |
| Metarrestin |
| [44,45] |
| Synthetic quinolinones |
| [46] |
| Cordyheptapeptide A |
| [47] |
| BE-43547A2 |
| [48,49] |
| Compound | Molecular Mechanism of Action |
|---|---|
| Didemnins |
|
| Triene-ansamycins |
|
| Ternatin-4 |
|
| Nannocystins |
|
| Metarrestin |
|
| BE-43547A2 |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Yu, S.; Wang, Y.; Wu, S.; Shan, C.; Zhang, W. Targeting Eukaryotic Elongation Factor 1A: How Small-Molecule Inhibitors Suppress Tumor Growth via Diverse Pathways. Int. J. Mol. Sci. 2025, 26, 7331. https://doi.org/10.3390/ijms26157331
Zhang H, Yu S, Wang Y, Wu S, Shan C, Zhang W. Targeting Eukaryotic Elongation Factor 1A: How Small-Molecule Inhibitors Suppress Tumor Growth via Diverse Pathways. International Journal of Molecular Sciences. 2025; 26(15):7331. https://doi.org/10.3390/ijms26157331
Chicago/Turabian StyleZhang, Han, Siqi Yu, Ying Wang, Shanmei Wu, Changliang Shan, and Weicheng Zhang. 2025. "Targeting Eukaryotic Elongation Factor 1A: How Small-Molecule Inhibitors Suppress Tumor Growth via Diverse Pathways" International Journal of Molecular Sciences 26, no. 15: 7331. https://doi.org/10.3390/ijms26157331
APA StyleZhang, H., Yu, S., Wang, Y., Wu, S., Shan, C., & Zhang, W. (2025). Targeting Eukaryotic Elongation Factor 1A: How Small-Molecule Inhibitors Suppress Tumor Growth via Diverse Pathways. International Journal of Molecular Sciences, 26(15), 7331. https://doi.org/10.3390/ijms26157331