
Chromosomal Deletion Involving ANKRD26 Leads to Expression of a Fusion Protein Responsible for ANKRD26-Related Thrombocytopenia

 , , ,
, , ,  , , , , , ,
, , , , , ,  add
Show full author list
add
Show full author list
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
Case Report
2. Results
2.1. Molecular Characterization of the Deletion and Identification of an ACBD5/ANKRD26 Fusion Transcript
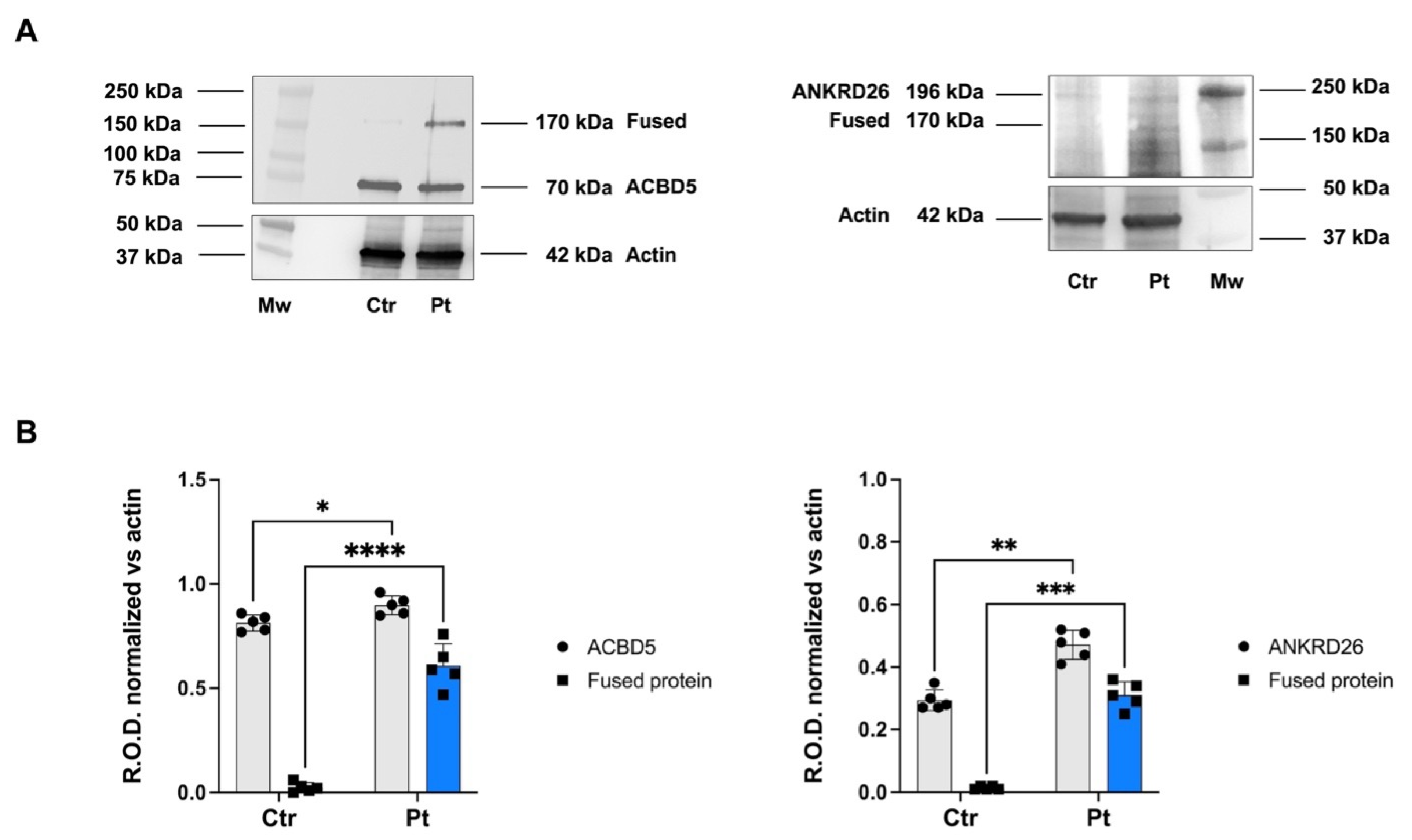
2.2. ACBD5/ANKRD26 Fusion Transcript Is Translated into a Chimeric Protein
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Whole Exome Sequencing
5.2. Sanger Sequencing and Quantitative Polymerase Chain Reaction
5.3. Molecular Characterization of the Deletion
5.4. RNA Isolation and Gene Expression
5.5. Western Blot
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACMG | American College of Medical Genetics |
| AML | Acute Myeloid Leukemia |
| ANKRD26-RT | ANKRD26-related thrombocytopenia |
| cDNA | complementary DNA |
| CML | Chronic Myeloid Leukemia |
| CLL | Chronic Lymphocytic Leukemia |
| CNV | Copy Number Variation |
| IT | Inherited Thrombocytopenia |
| ITP | Immune Thrombocytopenic Purpura |
| MDS | Myelodisplasia |
| PBSC | Peripheral Blood Stem Cell |
| PBS | Phosphate-buffered Saline |
| qPCR | Quantitative Polymerase Chain Reaction |
| SNV | Single Nucleotide Variants |
| TPO | Thrombopoietin |
| UTR | Untranslated Region |
| VUS | Variant of Unknown Significance |
| WB | Western Blot |
| WES | Whole Exome Sequencing |
References
- Palma-Barqueros, V.; Revilla, N.; Sánchez, A.; Zamora Cánovas, A.; Rodriguez-Alén, A.; Marín-Quílez, A.; González-Porras, J.R.; Vicente, V.; Lozano, M.L.; Bastida, J.M.; et al. Inherited Platelet Disorders: An Updated Overview. Int. J. Mol. Sci. 2021, 22, 4521. [Google Scholar] [CrossRef] [PubMed]
- Homan, C.C.; Scott, H.S.; Brown, A.L. Hereditary Platelet Disorders Associated with Germ Line Variants in RUNX1, ETV6, and ANKRD26. Blood 2023, 141, 1533–1543. [Google Scholar] [CrossRef] [PubMed]
- Balduini, A.; Raslova, H.; Di Buduo, C.A.; Donada, A.; Ballmaier, M.; Germeshausen, M.; Balduini, C.L. Clinic, Pathogenic Mechanisms and Drug Testing of Two Inherited Thrombocytopenias, ANKRD26-Related Thrombocytopenia and MYH9-Related Diseases. Eur. J. Med. Genet. 2018, 61, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Almazni, I.; Stapley, R.; Morgan, N.V. Inherited Thrombocytopenia: Update on Genes and Genetic Variants Which May Be Associated With Bleeding. Front. Cardiovasc. Med. 2019, 6, 80. [Google Scholar] [CrossRef]
- Perez Botero, J.; Dugan, S.N.; Anderson, M.W. ANKRD26-Related Thrombocytopenia. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Zaninetti, C.; Santini, V.; Tiniakou, M.; Barozzi, S.; Savoia, A.; Pecci, A. Inherited Thrombocytopenia Caused by ANKRD26 Mutations Misdiagnosed and Treated as Myelodysplastic Syndrome: Report on Two Cases. J. Thromb. Haemost. 2017, 15, 2388–2392. [Google Scholar] [CrossRef]
- Pang, C.; Wu, X.; Nikuze, L.; Wei, H. Analysis of Clinical Characteristics and Treatment Efficacy in Two Pediatric Cases of ANKRD26-Related Thrombocytopenia. Platelets 2023, 34, 2262607. [Google Scholar] [CrossRef]
- Savoia, A.; Del Vecchio, M.; Totaro, A.; Perrotta, S.; Amendola, G.; Moretti, A.; Zelante, L.; Iolascon, A. An Autosomal Dominant Thrombocytopenia Gene Maps to Chromosomal Region 10p. Am. J. Hum. Genet. 1999, 65, 1401–1405. [Google Scholar] [CrossRef]
- Gandhi, M.J.; Cummings, C.L.; Drachman, J.G. FLJ14813 Missense Mutation: A Candidate for Autosomal Dominant Thrombocytopenia on Human Chromosome 10. Hum. Hered. 2003, 55, 66–70. [Google Scholar] [CrossRef]
- Punzo, F.; Mientjes, E.J.; Rohe, C.F.; Scianguetta, S.; Amendola, G.; Oostra, B.A.; Bertoli-Avella, A.M.; Perrotta, S. A Mutation in the Acyl-coenzyme A Binding Domain-containing Protein 5 Gene (ACBD5) Identified in Autosomal Dominant Thrombocytopenia. J. Thromb. Haemost. 2010, 8, 2085–2087. [Google Scholar] [CrossRef]
- Pippucci, T.; Savoia, A.; Perrotta, S.; Pujol-Moix, N.; Noris, P.; Castegnaro, G.; Pecci, A.; Gnan, C.; Punzo, F.; Marconi, C.; et al. Mutations in the 5′ UTR of ANKRD26, the Ankirin Repeat Domain 26 Gene, Cause an Autosomal-Dominant Form of Inherited Thrombocytopenia, THC2. Am. J. Hum. Genet. 2011, 88, 115–120. [Google Scholar] [CrossRef]
- Wahlster, L.; Verboon, J.M.; Ludwig, L.S.; Black, S.C.; Luo, W.; Garg, K.; Voit, R.A.; Collins, R.L.; Garimella, K.; Costello, M.; et al. Familial Thrombocytopenia Due to a Complex Structural Variant Resulting in a WAC-ANKRD26 Fusion Transcript. J. Exp. Med. 2021, 218, e20210444. [Google Scholar] [CrossRef]
- Manohar, S.; Gofin, Y.; Streff, H.; Vossaert, L.; Camacho, P.; Murali, C.N. A Familial Deletion of 10p12.1 Associated with Thrombocytopenia. Am. J. Med. Genet. A 2024, 194, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Bluteau, D.; Balduini, A.; Balayn, N.; Currao, M.; Nurden, P.; Deswarte, C.; Leverger, G.; Noris, P.; Perrotta, S.; Solary, E.; et al. Thrombocytopenia-Associated Mutations in the ANKRD26 Regulatory Region Induce MAPK Hyperactivation. J. Clin. Investig. 2014, 124, 580–591. [Google Scholar] [CrossRef] [PubMed]
- Minamiguchi, H.; Kimura, T.; Urata, Y.; Miyazaki, H.; Bamba, T.; Abe, T.; Sonoda, Y. Simultaneous Signalling through C-mpl, C-kit and CXCR4 Enhances the Proliferation and Differentiation of Human Megakaryocyte Progenitors: Possible Roles of the PI3-K, PKC and MAPK Pathways. Br. J. Haematol. 2001, 115, 175–185. [Google Scholar] [CrossRef]
- Dunstan-Harrison, C.; Morison, I.M.; Ledgerwood, E.C. Inherited Thrombocytopenia Associated with a Variant in the FLI1 Binding Site in the 5′ UTR of ANKRD26. Clin. Genet. 2024, 106, 315–320. [Google Scholar] [CrossRef]
- Vyas, H.; Alcheikh, A.; Lowe, G.; Stevenson, W.S.; Morgan, N.V.; Rabbolini, D.J. Prevalence and Natural History of Variants in the ANKRD26 Gene: A Short Review and Update of Reported Cases. Platelets 2022, 33, 1107–1112. [Google Scholar] [CrossRef]
- Murphy, L.; Mead, A.J. Familial Thrombocytopenia: The Long and Short of It. J. Exp. Med. 2021, 218, e20210604. [Google Scholar] [CrossRef]
- Trottier, A.M.; Feurstein, S.; Godley, L.A. Germline Predisposition to Myeloid Neoplasms: Characteristics and Management of High versus Variable Penetrance Disorders. Best. Pract. Res. Clin. Haematol. 2024, 37, 101537. [Google Scholar] [CrossRef]
- Drazer, M.W.; Homan, C.C.; Yu, K.; Cavalcante de Andrade Silva, M.; McNeely, K.E.; Pozsgai, M.J.; Acevedo-Mendez, M.G.; Segal, J.P.; Wang, P.; Feng, J.; et al. Clonal Hematopoiesis in Patients with ANKRD26 or ETV6 Germline Mutations. Blood Adv. 2022, 6, 4357–4359. [Google Scholar] [CrossRef]
- Perez Botero, J.; Ho, T.P.; Hogan, W.J.; Kenderian, S.; Gangat, N.; Tefferi, A.; Abraham, R.S.; Nguyen, P.; Oliveira, J.L.; He, R.; et al. Clinical Spectrum and Clonal Evolution in Germline Syndromes with Predisposition to Myeloid Neoplasms. Br. J. Haematol. 2018, 182, 141–145. [Google Scholar] [CrossRef]
- The University of Chicago Hematopoietic Malignancies Cancer Risk Team. How I Diagnose and Manage Individuals at Risk for Inherited Myeloid Malignancies. Blood 2016, 128, 1800–1813. [Google Scholar] [CrossRef]
- Perez Botero, J.; Oliveira, J.L.; Chen, D.; Reichard, K.K.; Viswanatha, D.S.; Nguyen, P.L.; Pruthi, R.K.; Majerus, J.; Gada, P.; Gangat, N.; et al. ASXL1 Mutated Chronic Myelomonocytic Leukemia in a Patient with Familial Thrombocytopenia Secondary to Germline Mutation in ANKRD26. Blood Cancer J. 2015, 5, e315. [Google Scholar] [CrossRef]
- Martin, E.S.; Ferrer, A.; Mangaonkar, A.A.; Khan, S.P.; Kohorst, M.A.; Joshi, A.Y.; Hogan, W.J.; Olteanu, H.; Moyer, A.M.; Al-Kali, A.; et al. Spectrum of Hematological Malignancies, Clonal Evolution and Outcomes in 144 Mayo Clinic Patients with Germline Predisposition Syndromes. Am. J. Hematol. 2021, 96, 1450–1460. [Google Scholar] [CrossRef]
- Zaninetti, C.; Gresele, P.; Bertomoro, A.; Klersy, C.; De Candia, E.; Veneri, D.; Barozzi, S.; Fierro, T.; Alberelli, M.A.; Musella, V.; et al. Eltrombopag for the Treatment of Inherited Thrombocytopenias: A Phase II Clinical Trial. Haematologica 2020, 105, 820–828. [Google Scholar] [CrossRef]
- Fiore, M.; Saut, N.; Alessi, M.-C.; Viallard, J.-F. Successful Use of Eltrombopag for Surgical Preparation in a Patient with ANKRD26-Related Thrombocytopenia. Platelets 2016, 27, 828–829. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed]
- Rojek, K.; Nickels, E.; Neistadt, B.; Marquez, R.; Wickrema, A.; Artz, A.; Van Besien, K.; Larson, R.A.; Lee, M.K.; Segal, J.P.; et al. Identifying Inherited and Acquired Genetic Factors Involved in Poor Stem Cell Mobilization and Donor-Derived Malignancy. Biol. Blood Marrow Transplant. 2016, 22, 2100–2103. [Google Scholar] [CrossRef] [PubMed]
- Arai, H.; Matsui, H.; Chi, S.; Utsu, Y.; Masuda, S.; Aotsuka, N.; Minami, Y. Germline Variants and Characteristic Features of Hereditary Hematological Malignancy Syndrome. Int. J. Mol. Sci. 2024, 25, 652. [Google Scholar] [CrossRef]
- Tawana, K.; Brown, A.L.; Churpek, J.E. Integrating Germline Variant Assessment into Routine Clinical Practice for Myelodysplastic Syndrome and Acute Myeloid Leukaemia: Current Strategies and Challenges. Br. J. Haematol. 2022, 196, 1293–1310. [Google Scholar] [CrossRef]
- Cappelli, E.; Cuccarolo, P.; Stroppiana, G.; Miano, M.; Bottega, R.; Cossu, V.; Degan, P.; Ravera, S. Defects in Mitochondrial Energetic Function Compels Fanconi Anaemia Cells to Glycolytic Metabolism. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 1214–1221. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dell’Orso, G.; Passarella, T.; Cappato, S.; Cappelli, E.; Regis, S.; Maffei, M.; Balbi, M.; Ravera, S.; Di Martino, D.; Viaggi, S.; et al. Chromosomal Deletion Involving ANKRD26 Leads to Expression of a Fusion Protein Responsible for ANKRD26-Related Thrombocytopenia. Int. J. Mol. Sci. 2025, 26, 7330. https://doi.org/10.3390/ijms26157330
Dell’Orso G, Passarella T, Cappato S, Cappelli E, Regis S, Maffei M, Balbi M, Ravera S, Di Martino D, Viaggi S, et al. Chromosomal Deletion Involving ANKRD26 Leads to Expression of a Fusion Protein Responsible for ANKRD26-Related Thrombocytopenia. International Journal of Molecular Sciences. 2025; 26(15):7330. https://doi.org/10.3390/ijms26157330
Chicago/Turabian StyleDell’Orso, Gianluca, Tommaso Passarella, Serena Cappato, Enrico Cappelli, Stefano Regis, Massimo Maffei, Matilde Balbi, Silvia Ravera, Daniela Di Martino, Silvia Viaggi, and et al. 2025. "Chromosomal Deletion Involving ANKRD26 Leads to Expression of a Fusion Protein Responsible for ANKRD26-Related Thrombocytopenia" International Journal of Molecular Sciences 26, no. 15: 7330. https://doi.org/10.3390/ijms26157330
APA StyleDell’Orso, G., Passarella, T., Cappato, S., Cappelli, E., Regis, S., Maffei, M., Balbi, M., Ravera, S., Di Martino, D., Viaggi, S., Davì, S., Corsolini, F., Giarratana, M. C., Arcuri, L., Mariani, E., Morini, R., Massaccesi, E., Guardo, D., Calvillo, M., ... Miano, M. (2025). Chromosomal Deletion Involving ANKRD26 Leads to Expression of a Fusion Protein Responsible for ANKRD26-Related Thrombocytopenia. International Journal of Molecular Sciences, 26(15), 7330. https://doi.org/10.3390/ijms26157330