
Biological Modulation of Autophagy by Nanoplastics: A Current Overview

 ,
, 
 ,
,  , ,
, ,  and
and 
Abstract
1. Introduction
2. MPs and NPs: Properties, Environmental Fate, and Biological Impact
2.1. Classification, Characteristics, and Forms of Microplastics

{kind=link}
{kind=link}
{kind=link}
| Category | Description | Common Polymers | Origin and Formation Mechanisms | Morphology and Physical Features | Environmental and Health Implications | References |
|---|---|---|---|---|---|---|
| Definition | Solid plastic particles, insoluble in water, ranging from 1 µm to 5 mm in size | PE, HDPE, LDPE, PP, PS, PVC, PET, PMMA | Primarily derived from petroleum-based feedstocks | Diverse shapes including spheres, fragments, fibers | Persistent pollutants with potential for long-term environmental accumulation | [14,15,16,17] |
| Primary Microplastics | Manufactured intentionally at microscale for specific applications | Microbeads in cosmetics, scrubs, medical devices | Industrial production of microscale plastic particles | Typically spherical, uniform size distribution | Direct environmental release; potential for bioaccumulation in aquatic and terrestrial organisms | [20,21] |
| Secondary Microplastics | Result from the fragmentation and weathering of larger plastic debris | Derived from consumer products such as plastic bags, bottles, fishing nets | Fragmentation driven by photodegradation (UV radiation), mechanical abrasion, and microbial activity | Irregular shapes, rough and heterogeneous surfaces | Act as vectors for toxic chemicals and pathogens; increased bioavailability and toxicity potential | [22,26,27,28,29] |
| Stability and Persistence | Highly resistant to biodegradation and environmental breakdown | All aforementioned polymers | Chemical inertness and polymer structure confer environmental persistence | - | Long residence times in soil, water, and atmospheric compartments; challenges for waste management | [18] |
| Surface Properties | High specific surface area facilitates adsorption of pollutants | - | Surface properties promote sorption of hydrophobic organic contaminants and microbial colonization | - | Enhanced transport of hazardous substances; potential to disrupt biogeochemical cycles | [19] |
2.2. Human Exposure Routes
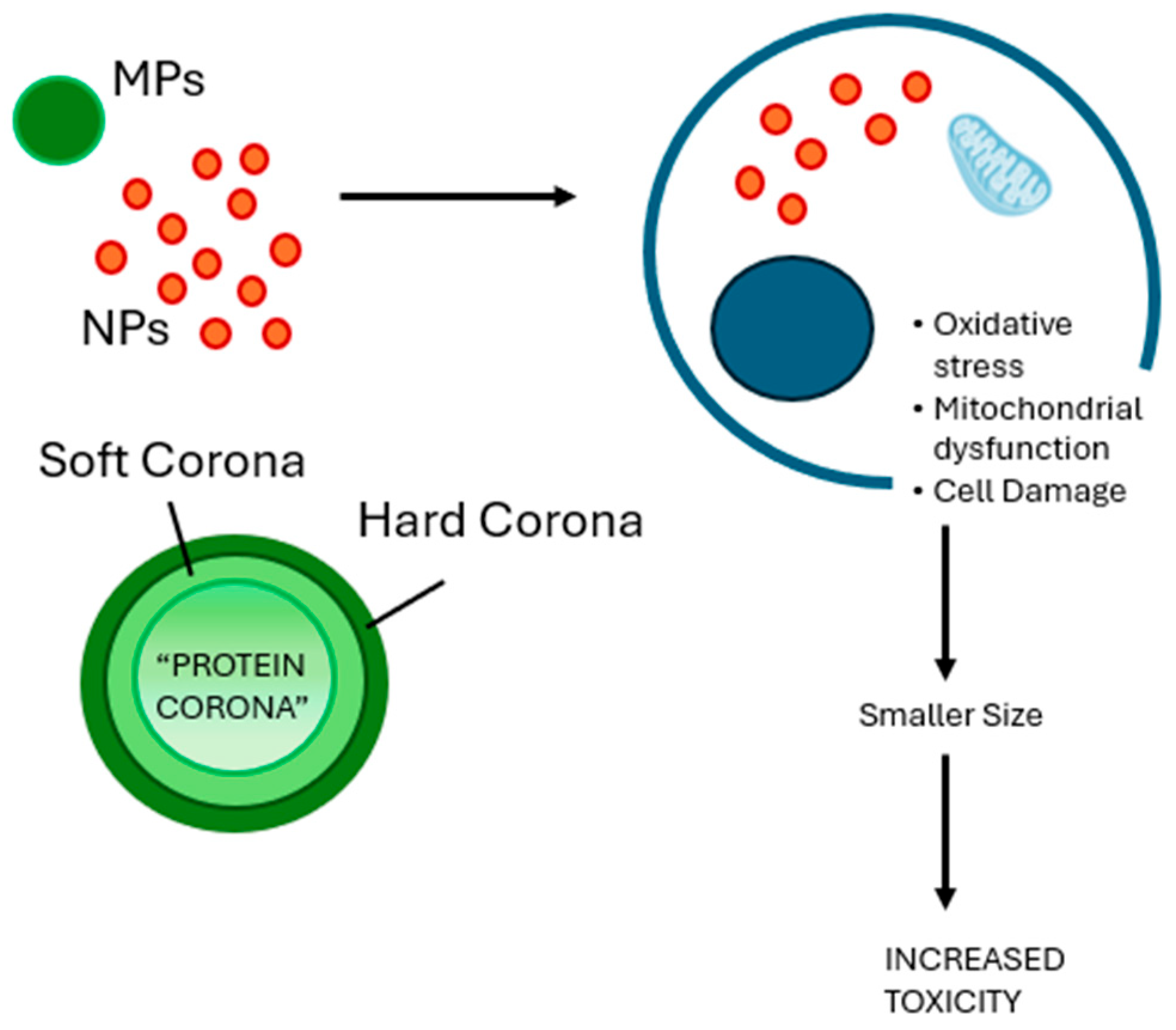
2.3. NP-Specific Toxicity and Cellular Mechanisms: The Importance of Protein Corona and Size
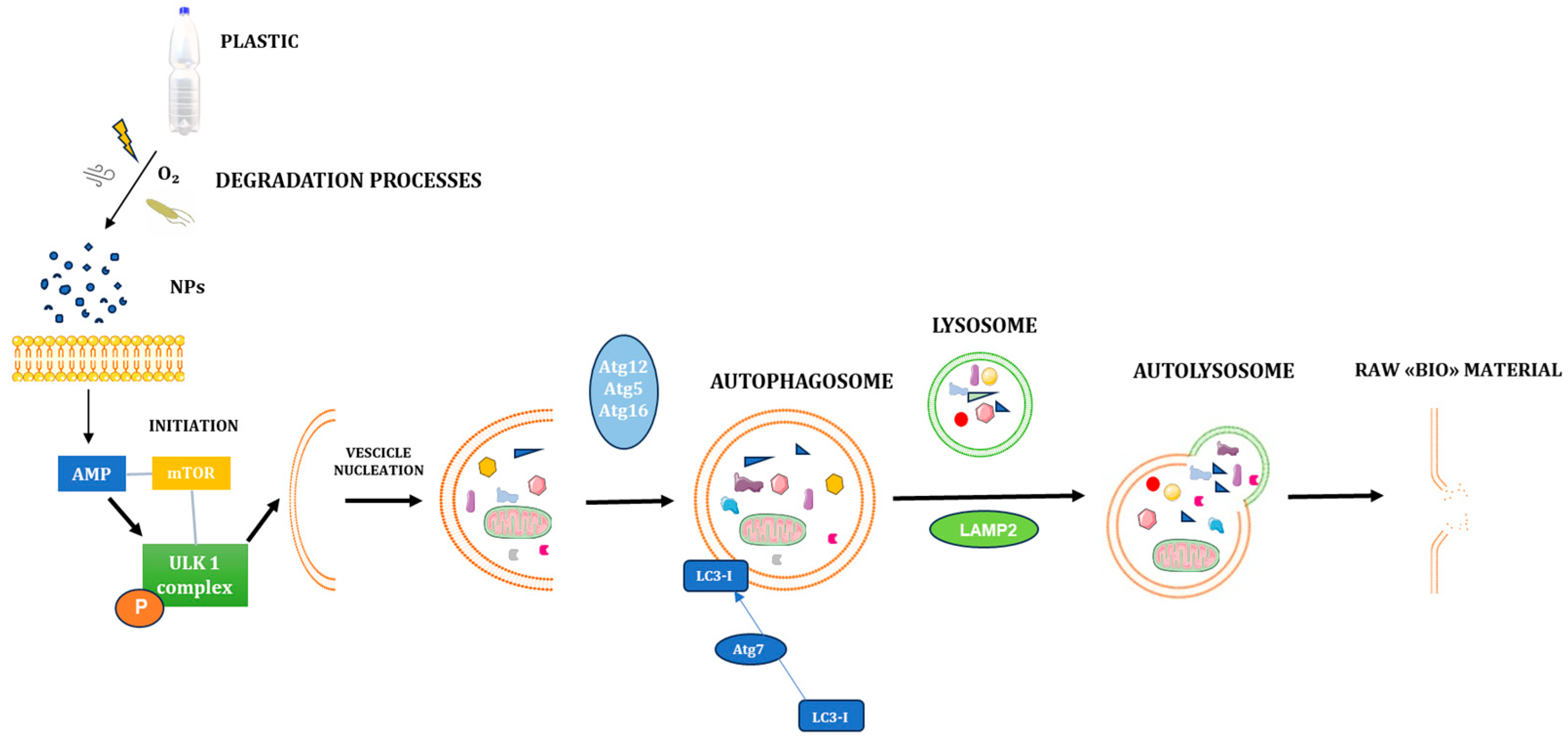
2.4. Autophagy Process
2.5. Autophagy and Biological Systems
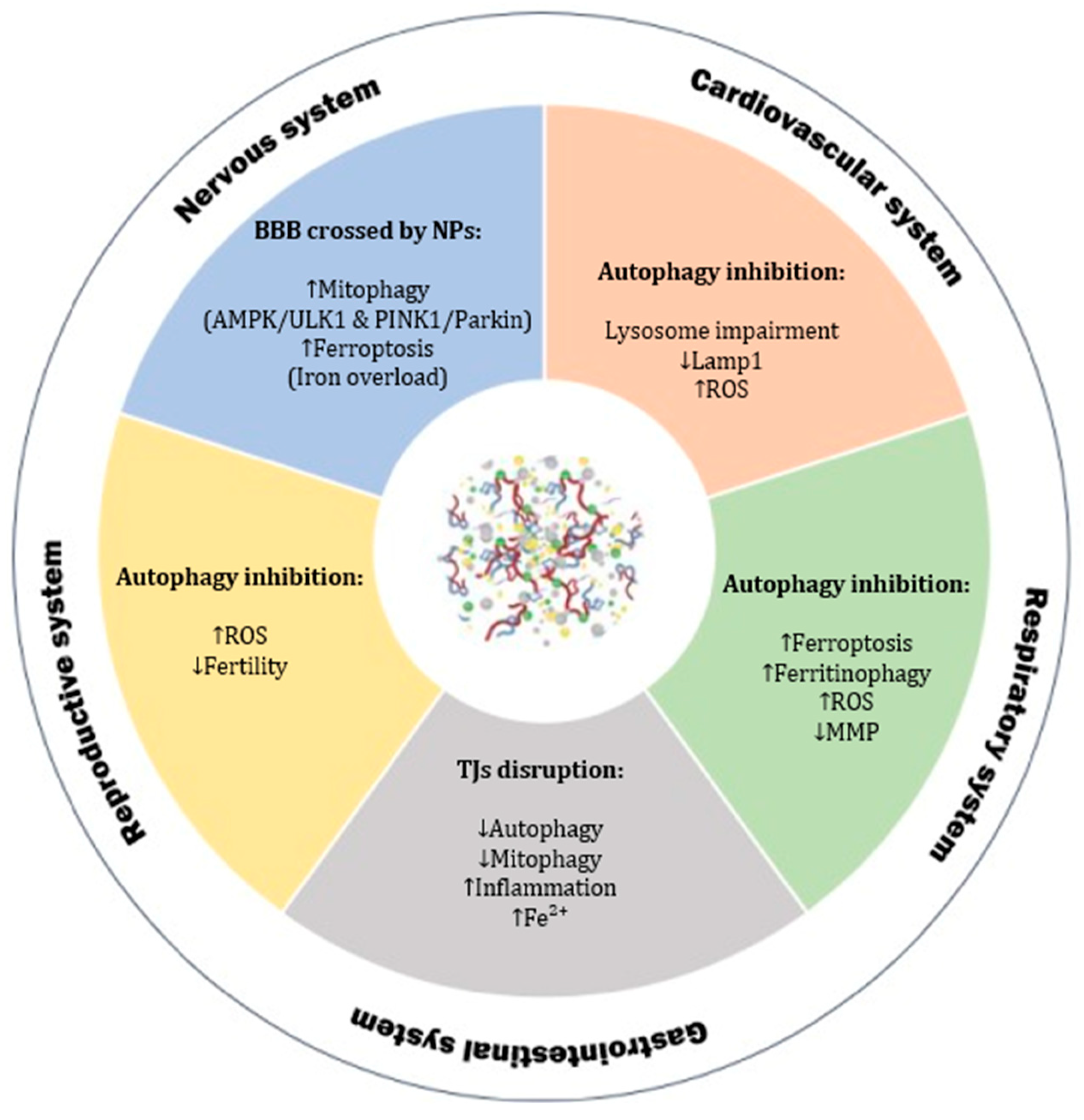
2.6. Nanoplastics and Autophagy
2.6.1. Cardiovascular System
2.6.2. Gastrointestinal System
2.6.3. Nervous System
2.6.4. Reproductive System
2.6.5. Respiratory System
| Model | |||||
|---|---|---|---|---|---|
| Nanoparticles | In Vitro | In Vivo | Effects | System | References |
| PS | hESCs | Zebrafish | Cytotoxicity, affected differentiation, oxidative stress and mitochondrial dysfunction, disturbed autophagy flux, impaired cardiac function. | Cardiovascular | [112] |
| PS | H9C2 | mouse | Oxidative stress, activation of TGF-β1 and autophagy. | Cardiovascular | [113] |
| PS | HUVECs | Cell membrane damage, inducing autophagosome formation and blocking autophagic flux. | Cardiovascular | [114] | |
| PS | CACO-2 | Forty male-ICR mice | Intestinal barrier damage and enterocyte apoptosis, lysosomal dysfunction and autophagic substrate degradation arrest in enterocytes. | GI | [158] |
| PS | L. vannamei | Destruction of hepatopancreas tissue structure, the shedding of microvilli, the increase number of hepatocyte apoptosis and autophagy structure. | GI | [119] | |
| PS | RKO HT-29 HCT-116 HIEC-6 | C57BL/6J mice | Induced autophagy activation, but blocked autophagic flux in human intestinal cells. No major tissue damage observed in vivo. | GI | [118] |
| PS | M. nipponense | Inducing apoptosis and autophagy. | GI | [159] | |
| PS | L. vannamei | Damaging of the intestinal villi, promotion and formation of autophagosomes, increasing of intestinal non-specific immunoenzyme activities, and significantly induction of apoptosis. | GI | [119] | |
| PS | HET-1A HEET | Suppressing mitochondrial autophagy, which exacerbated NP-induced cell inflammation and death. | GI | [122] | |
| PS | HIEC-6 | Inhibition of mitophagy and inducing perturbations in the gut microbiota. | GI | [160] | |
| PS | GES-1 | Decreasing cell proliferation rates and increasing cell apoptosis and autophagy flux. | GI | [161] | |
| PS | SH-SY5Y | C57BL/6 J mice | Activation of the AMPK/ULK1 pathway driving excessive mitochondrial autophagy, death of dopaminergic neuron. | Nervous | [134] |
| PS | Sprague Dawley rats | Induction of autophagy. | Nervous | [162] | |
| PS-NH2 (modified) | bEnd.3 | Inhibition of autophagy pathway in brain endothelial cells, decreased mTOR phosphorylation, altered Beclin-1 and LC3B ratios, and p62 accumulation which contributed to iron overload and subsequent blood–brain barrier disruption. | Nervous | [140] | |
| PS | Swan 71 | C57BL/6 mice | Suppression of ROCK1-mediated migration/invasion and migrasome formation. Activation of autophagy and promotion of the autophagy degradation of SOX2, thus suppressing SOX2-mediated ROCK1 transcription. Damage to trophoblast cells and placenta tissue and induction of miscarriage. | Reproductive | [163] |
| PS | GC-2spd(ts) | Inhibition of cell proliferation and decreasing of cell viability, induction of oxidative stress and autophagy, impairing mitochondrial function of spermatocyte. | Reproductive | [164] | |
| PS | Kunming mice | PS-NPs reduced autophagic flux in offspring undifferentiated spermatogonia, as demonstrated by decreased expression of key markers such as Igbp1/Gabarapl2 and structural disruptions in autophagolysosome. | Reproductive | [145] | |
| PS | KGN | Triggered autophagy, increased LC3B-II/LC3B-I ratio and elevated ATG5 expression, coupled with a reduction in P62 levels. | Reproductive | [150] | |
| PS | BEAS-2B HPAEpiC | Inducing lung epithelial cell ferroptosis and ferritinophagy, disturbing of mitochondrial functions and damage, triggering autophagy. | Respiratory | [156] | |
| PS | HNEPCs | Significant increases in ROS, a decrease in MMP (mitochondrial membrane potential), as well as a greater accumulation of LC3-II and p62. | Respiratory | [155] | |
| PS | HeLa | Activating of autophagic flux. | Respiratory | [165] | |
| PS | BEAS-2B | Inducing of oxidative stress and inhibited cell growth through apoptosis and autophagy. | Respiratory | [166] | |
| PET | HNEpCs | Induction of ROS’s production and alteration of autophagy when LC3-II and p62 protein’s levels increase. | Respiratory | [157] | |
| PS | BEAS-2B | Metabolic alterations related to autophagy and endoplasmic reticulum (ER) stress, such as an increase in amino acids and tricarboxylic acid (TCA) cycle intermediate metabolites. | Respiratory | [167] | |
2.7. Molecular Signaling Pathways Linking NPs to Autophagy Dysregulation
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Choudhury, A.; Simnani, F.Z.; Singh, D.; Patel, P.; Sinha, A.; Nandi, A.; Ghosh, A.; Saha, U.; Kumari, K.; Jaganathan, S.K.; et al. Atmospheric microplastic and nanoplastic: The toxicological paradigm on the cellular system. Ecotoxicol. Environ. Saf. 2023, 259, 115018. [Google Scholar] [CrossRef] [PubMed]
- Kusenberg, M.; Roosen, M.; Zayoud, A.; Djokic, M.R.; Dao Thi, H.; De Meester, S.; Ragaert, K.; Kresovic, U.; Van Geem, K.M. Assessing the feasibility of chemical recycling via steam cracking of untreated plastic waste pyrolysis oils: Feedstock impurities, product yields and coke formation. Waste Manag. 2022, 141, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Taib, N.-A.A.B.; Rahman, M.R.; Huda, D.; Kuok, K.K.; Hamdan, S.; Bakri, M.K.; Julaihi, M.R.M.; Khan, A. A review on poly lactic acid (PLA) as a biodegradable polymer. Polym. Bull. 2023, 80, 1179–1213. [Google Scholar] [CrossRef]
- Hahladakis, J.N.; Velis, C.A.; Weber, R.; Iacovidou, E.; Purnell, P. An overview of chemical additives present in plastics: Migration, release, fate and environmental impact during their use, disposal and recycling. J. Hazard. Mater. 2018, 344, 179–199. [Google Scholar] [CrossRef] [PubMed]
- Abdiev, J.; Safarov, O.; Julanov, H. Study of the properties of polymer composites—Reinforcement based on glass and basalt fibers. Eurasia Sci. Her. 2022, 7, 77–88. [Google Scholar]
- Fried, J.R. Polymer Science and Technology, 3rd ed.; Pearson Education: London, UK, 2014; pp. 1–76. [Google Scholar]
- Lebreton, L.; Andrady, A. Future scenarios of global plastic waste generation and disposal. Palgrave Commun. 2019, 5, 6. [Google Scholar] [CrossRef]
- Alimba, C.G.; Faggio, C. Microplastics in the marine environment: Current trends in environmental pollution and mechanisms of toxicological profile. Environ. Toxicol. Pharmacol. 2019, 68, 61–74. [Google Scholar] [CrossRef] [PubMed]
- García Rellán, A.; Vázquez Ares, D.; Vázquez Brea, C.; Francisco López, A.; Bello Bugallo, P.M. Sources, sinks and transformations of plastics in our oceans: Review, management strategies and modelling. Sci. Total. Environ. 2023, 854, 158745. [Google Scholar] [CrossRef] [PubMed]
- Geyer, R.; Jambeck, J.R.; Law, K.L. Production, use, and fate of all plastics ever made. Sci. Adv. 2017, 3, e1700782. [Google Scholar] [CrossRef] [PubMed]
- Urban-Malinga, B.; Zalewski, M.; Jakubowska, A.; Wodzinowski, T.; Malinga, M.; Pałys, B.; Dąbrowska, A. Microplastics on sandy beaches of the southern Baltic Sea. Mar. Pollut. Bull. 2020, 155, 111170. [Google Scholar] [CrossRef] [PubMed]
- Zaman, A.; Newman, P. Plastics: Are they part of the zero-waste agenda or the toxic-waste agenda? Sustain. Earth 2021, 4, 4. [Google Scholar] [CrossRef]
- Galloway, T.S.; Cole, M.; Lewis, C. Interactions of microplastic debris throughout the marine ecosystem. Nat. Ecol. Evol. 2017, 1, 116. [Google Scholar] [CrossRef] [PubMed]
- Frias, J.P.G.L.; Nash, R. Microplastics: Finding a consensus on the definition. Mar. Pollut. Bull. 2019, 138, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Koelmans, A.A.; Mohamed Nor, N.H.; Hermsen, E.; Kooi, M.; Mintenig, S.M.; De France, J. Microplastics in freshwaters and drinking water: Critical review and assessment of data quality. Water Res. 2019, 155, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Anagnosti, L.; Varvaresou, A.; Pavlou, P.; Protopapa, E.; Carayanni, V. Worldwide actions against plastic pollution from microbeads and microplastics in cosmetics focusing on European policies. Has the issue been handled effectively? Mar. Pollut. Bull. 2021, 162, 111883. [Google Scholar] [CrossRef] [PubMed]
- Schneiderman, D.K.; Hillmyer, M.A. 50th anniversary perspective: There is a great future in sustainable polymers. Macromolecules. 2017, 50, 3733–3749. [Google Scholar] [CrossRef]
- Wright, S.L.; Ulke, J.; Font, A.; Chan, K.L.A.; Kelly, F.J. Atmospheric microplastic deposition in an urban environment and an evaluation of transport. Environ. Int. 2020, 136, 105411. [Google Scholar] [CrossRef] [PubMed]
- Avazzadeh Samani, F.; Meunier, L. Interactions of microplastics with contaminants in freshwater systems: A review of characteristics, bioaccessibility, and environmental factors affecting sorption. J. Environ. Sci. Health Part A Toxic/Hazard. Subst. Environ. Eng. 2023, 58, 222–235. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.; Friot, D. Primary Microplastics in the Oceans: A Global Evaluation of Sources; IUCN: Gland, Switzerland, 2017; Volume 43, p. 8. [Google Scholar]
- Fendall, L.S.; Sewell, M.A. Contributing to marine pollution by washing your face: Microplastics in facial cleansers. Mar. Pollut. Bull. 2009, 58, 1225–1228. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, L.M.R.; Karapanagioti, H.; Álvarez, N.R. Micro(nanoplastics) in the marine environment: Current knowledge and gaps. Curr. Opin. Environ. Sci. Health 2018, 1, 47–51. [Google Scholar] [CrossRef]
- Browne, M.A.; Niven, S.J.; Galloway, T.S.; Rowland, S.J.; Thompson, R.C. Microplastic moves pollutants and additives to worms, reducing functions linked to health and biodiversity. Curr. Biol. 2013, 23, 2388–2392. [Google Scholar] [CrossRef] [PubMed]
- Akhbarizadeh, R.; Dobaradaran, S.; Nabipour, I.; Tangestani, M.; Abedi, D.; Javanfekr, F.; Jeddi, F.; Zendehboodi, A. Abandoned COVID-19 personal protective equipment along the Bushehr shores, the Persian Gulf: An emerging source of secondary microplastics in coastlines. Mar. Pollut. Bull. 2021, 168, 112386. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Liu, Y.; Zhang, T.; Zhang, F.; Ren, H.; Zhang, Y. Analysis of microplastics in human feces reveals a correlation between fecal microplastics and inflammatory bowel disease status. Environ. Sci. Technol. 2022, 56, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Zbyszewski, M.; Corcoran, P.L. Distribution and degradation of fresh water plastic particles along the beaches of Lake Huron, Canada. Water Air Soil Pollut. 2011, 220, 365–372. [Google Scholar] [CrossRef]
- Browne, M.A.; Galloway, T.S.; Thompson, R.C. Spatial Patterns of Plastic Debris along Estuarine Shorelines. Environ. Sci. Technol. 2010, 44, 3404–3409. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Sui, Q.; Du, Y.; Wang, L.; Jing, J.; Zhu, L.; Zhao, X.; Sun, X.; Booth, A.M.; Chen, B.; et al. Secondary PVC microplastics are more toxic than primary PVC microplastics to Oryzias melastigma embryos. J. Hazard. Mater. 2022, 424, 127421. [Google Scholar] [CrossRef] [PubMed]
- Sundt, P.; Schultze, P.-E.; Frode, S. Sources of Microplastics-Pollution to the Marine Environment; Norwegian Environment Agency: Trondheim, Norway, 2014. [Google Scholar]
- Yuan, Z.; Nag, R.; Cummins, E. Human health concerns regarding microplastics in the aquatic environment—From marine to food systems. Sci. Total Environ. 2022, 823, 153730. [Google Scholar] [CrossRef] [PubMed]
- Catarino, A.I.; Macchia, V.; Sanderson, W.G.; Thompson, R.C.; Henry, T.B. Low levels of microplastics (MP) in wild mussels indicate that MP ingestion by humans is minimal compared to exposure via household fibres fallout during a meal. Environ. Pollut. 2018, 237, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Cox, K.D.; Covernton, G.A.; Davies, H.L.; Dower, J.F.; Juanes, F.; Dudas, S.E. Human Consumption of Microplastics. Environ. Sci. Technol. 2019, 53, 7068–7074. [Google Scholar] [CrossRef] [PubMed]
- Lamas, B.; Martins Breyner, N.; Houdeau, E. Impacts of foodborne inorganic nanoparticles on the gut microbiota-immune axis: Potential consequences for host health. Part. Fibre Toxicol. 2020, 17, 19. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Ding, Y.; Cheng, X.; Sheng, D.; Xu, Z.; Rong, Q.; Wu, Y.; Zhao, H.; Ji, X.; Zhang, Y. Polyethylene microplastics affect the distribution of gut microbiota and inflammation development in mice. Chemosphere 2020, 244, 125492. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Tang, Y.; Chen, L.; Lv, S.; Liu, S.; Nie, P.; Aguilar, Z.P.; Xu, H. Restraining the TiO2 nanoparticles-induced intestinal inflammation mediated by gut microbiota in juvenile rats via ingestion of Lactobacillus rhamnosus GG. Ecotoxicol. Environ. Saf. 2020, 206, 111393. [Google Scholar] [CrossRef] [PubMed]
- Bruno, A.; Dovizio, M.; Milillo, C.; Aruffo, E.; Pesce, M.; Gatta, M.; Chiacchiaretta, P.; Di Carlo, P.; Ballerini, P. Orally Ingested Micro- and Nano-Plastics: A Hidden Driver of Inflammatory Bowel Disease and Colorectal Cancer. Cancers 2024, 16, 3079. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.F.; Chen, C.Y.; Lu, T.H.; Liao, C.M. Toxicity-based toxicokinetic/toxicodynamic assessment for bioaccumulation of polystyrene microplastics in mice. J. Hazard. Mater. 2019, 366, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Guilloteau, E.; Djouina, M.; Caboche, S.; Waxin, C.; Deboudt, K.; Beury, D.; Hot, D.; Pichavant, M.; Dubuquoy, L.; Launay, D.; et al. Exposure to atmospheric Ag, TiO2, Ti and SiO2 engineered nanoparticles modulates gut inflammatory response and microbiota in mice. Ecotoxicol. Environ. Saf. 2022, 236, 113442. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Liu, S.; Xu, H. Effects of microplastic and engineered nanomaterials on inflammatory bowel disease: A review. Chemosphere 2023, 326, 138486. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Zhao, L.; Liu, Z.; Zhou, Q.; Zhu, Y.; Zhao, Y.; Yang, X. Long-term exposure to titanium dioxide nanoparticles promotes diet-induced obesity through exacerbating intestinal mucus layer damage and microbiota dysbiosis. Nano Res. 2021, 14, 1512–1522. [Google Scholar] [CrossRef]
- Liu, S.; Zhao, Y.; Liu, Y.; Tang, Y.; Xu, X.; Wang, M.; Tao, X.; Xu, H. Pre-exposure to TiO2-NPs aggravates alcohol-related liver injury by inducing intestinal barrier damage in mice. Toxicol. Sci. 2021, 185, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Remmelts, M. Effect of Nano- and Microplastics on the Human Immune System and Their Influence on Inflammatory Bowel Disease. Bachelor’s Thesis, University of Groningen, Groningen, The Netherlands, 2021. [Google Scholar]
- Wright, S.L.; Kelly, F.J. Plastic and human health: A micro issue? Environ. Sci. Technol. 2017, 51, 6634–6647. [Google Scholar] [CrossRef] [PubMed]
- Bhat, M.A.; Gedik, K.; Gaga, E.O. Atmospheric micro (nano) plastics: Future growing concerns for human health. Air Qual. Atmos. Health 2023, 16, 233–262. [Google Scholar] [CrossRef] [PubMed]
- Reponen, T.; Grinshpun, S.A.; Conwell, K.L.; Wiest, J.; Anderson, M. Aerodynamic versus physical size of spores: Measurement and implication for respiratory deposition. Grana 2001, 40, 119–125. [Google Scholar] [CrossRef]
- Wieland, S.; Balmes, A.; Bender, J.; Kitzinger, J.; Meyer, F.; Ramsperger, A.F.; Roeder, F.; Tengelmann, C.; Wimmer, B.H.; Laforsch, C.; et al. From properties to toxicity: Comparing microplastics to other airborne microparticles. J. Hazard. Mater. 2022, 428, 128151. [Google Scholar] [CrossRef] [PubMed]
- Schirinzi, G.F.; Pérez-Pomeda, I.; Sanchís, J.; Rossini, C.; Farré, M.; Barceló, D. Cytotoxic effects of commonly used nanomaterials and microplastics on cerebral and epithelial human cells. Environ. Res. 2017, 159, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, K.; Loridas, S. Pulmonary oxidative stress, inflammation and cancer: Respirable particulate matter, fibrous dusts and ozone as major causes of lung carcinogenesis through reactive oxygen species mechanisms. Int. J. Environ. Res. Public Health 2013, 10, 3886–3907. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, S. Routes of human exposure to micro(nano)plastics. Curr. Opin. Toxicol. 2021, 27, 41–46. [Google Scholar] [CrossRef]
- Biswas, S.; Bagchi, D.; Ghosh, D. The Effects of (Micro and Nano) Plastics on the Human Body: Nervous System, Respiratory System, Digestive System, Placental Barrier, Skin, and Excretory System. In Assessing the Effects of Emerging Plastics on the Environment and Public Health; Joo, S., Ed.; IGI Global Scientific Publishing: Hershey, PA, USA, 2022; pp. 148–171. [Google Scholar]
- Revel, M.; Châtel, A.; Mouneyrac, C. Micro(nano)plastics: A threat to human health? Curr. Opin. Environ. Sci. Health 2018, 1, 17–23. [Google Scholar] [CrossRef]
- Schneider, M.; Stracke, F.; Hansen, S.; Schaefer, U.F. Nanoparticles and their interactions with the dermal barrier. Dermato-Endocrinol. 2009, 1, 197–206. [Google Scholar] [CrossRef] [PubMed]
- de Souza Machado, A.A.; Kloas, W.; Zarfl, C.; Hempel, S.; Rillig, M.C. Microplastics as an emerging threat to terrestrial ecosystems. Glob. Chang Biol. 2018, 24, 1405–1416. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, M.; Lynch, I.; Ejtehadi, M.R.; Monopoli, M.P.; Bombelli, F.B.; Laurent, S. Protein–nanoparticle interactions: Opportunities and challenges. Chem. Rev. 2011, 111, 5610–5637. [Google Scholar] [CrossRef] [PubMed]
- Corbo, C.; Molinaro, R.; Parodi, A.; Toledano Furman, N.E.; Salvatore, F.; Tasciotti, E. The impact of nanoparticle protein corona on cytotoxicity, immunotoxicity and target drug delivery. Nanomedicine 2016, 11, 81–100. [Google Scholar] [CrossRef] [PubMed]
- Nasser, F.; Lynch, I. Secreted protein eco-corona mediates uptake and impacts of polystyrene nanoparticles on Daphnia magna. J. Proteom. 2016, 137, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Panda, P.K.; Kumari, P.; Patel, P.; Samal, S.K.; Mishra, S.; Tambuwala, M.M.; Dutt, A.; Hilscherová, K.; Mishra, Y.K.; Varma, R.S.; et al. Molecular nanoinformatics approach assessing the biocompatibility of biogenic silver nanoparticles with channelized intrinsic steatosis and apoptosis. Green Chem. 2022, 24, 1190–1210. [Google Scholar] [CrossRef]
- Yu, Y.; Luan, Y.; Dai, W. Time evolution of protein corona formed by polystyrene nanoplastics and urease. Int. J. Biol. Macromol. 2022, 218, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Akhter, M.H.; Khalilullah, H.; Gupta, M.; Alfaleh, M.A.; Alhakamy, N.A.; Riadi, Y.; Md, S. Impact of Protein Corona on the Biological Identity of Nanomedicine: Understanding the Fate of Nanomaterials in the Biological Milieu. Biomedicines 2021, 9, 1496. [Google Scholar] [CrossRef] [PubMed]
- Tenzer, S.; Docter, D.; Kuharev, J.; Musyanovych, A.; Fetz, V.; Hecht, R.; Schlenk, F.; Fischer, D.; Kiouptsi, K.; Reinhardt, C.; et al. Rapid formation of plasma protein corona critically affects nanoparticle pathophysiology. Nat. Nanotechnol. 2013, 8, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.M.; Juettner, V.V.; Hong, S. Biomolecular corona on nanoparticles: A survey of recent literature and its implications in targeted drug delivery. Front. Chem. 2014, 2, 108. [Google Scholar] [CrossRef] [PubMed]
- Saie, A.A.; Ray, M.; Mahmoudi, M.; Rotello, V.M. Engineering the nanoparticle-protein interface for cancer therapeutics. Cancer Treat. Res. 2015, 166, 245–273. [Google Scholar] [PubMed]
- Milani, S.; Bombelli, F.B.; Pitek, A.S.; Dawson, K.A.; Radler, J. Reversible versus irreversible binding of transferrin to polystyrene nanoparticles: Soft and hard corona. ACS Nano 2012, 6, 2532–2541. [Google Scholar] [CrossRef] [PubMed]
- Cedervall, T.; Lynch, I.; Lindman, S. Understanding the nanoparticle–protein corona using methods to quantify exchange rates and affinities of proteins for nanoparticles. Proc. Natl. Acad. Sci. USA 2007, 104, 2050–2055. [Google Scholar] [CrossRef] [PubMed]
- Walczyk, D.; Bombelli, F.B.; Monopoli, M.P.; Lynch, I.; Dawson, K.A. What the cell “sees” in bionanoscience. J. Am. Chem. Soc. 2010, 132, 5761–5768. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Piao, J.; Kang, B.; Eom, Y.; Kim, D.H.; Song, J.S. The toxic effects of polystyrene microplastic/nanoplastic particles on retinal pigment epithelial cells and retinal tissue. Environ. Sci. Pollut. Res. Int. 2024, 31, 54950–54961. [Google Scholar] [CrossRef] [PubMed]
- Rappoport, J.Z. Focusing on clathrin-mediated endocytosis. Biochem. J. 2008, 412, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Rothen-Rutishauser, B.M.; Schürch, S.; Haenni, B.; Kapp, N.; Gehr, P. Interaction of fine particles and nanoparticles with red blood cells visualized with advanced microscopic techniques. Environ. Sci. Technol. 2006, 40, 4353–4359. [Google Scholar] [CrossRef] [PubMed]
- Firdessa, R.; Oelschlaeger, T.A.; Moll, H. Identification of multiple cellular uptake pathways of polystyrene nanoparticles and factors affecting the uptake: Relevance for drug delivery systems. Eur. J. Cell Biol. 2014, 93, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Sorkin, A. Cargo recognition during clathrin-mediated endocytosis: A team effort. Curr. Opin. Cell Biol. 2004, 16, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Rejman, J.; Oberle, V.; Zuhorn, I.S.; Hoekstra, D. Size-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis. Biochem. J. 2004, 377, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Y.; Zhong, Z.; Liu, X.; Li, Z.; Li, J.; Sun, L.; Sen, H. Correlation between cellular uptake and cytotoxicity of polystyrene micro/nanoplastics in HeLa cells: A size-dependent matter. PLoS ONE 2023, 18, e0289473. [Google Scholar]
- Liu, L.; Xu, K.; Zhang, B.; Ye, Y.; Zhang, Q.; Jiang, W. Cellular internalization and release of polystyrene microplastics and nanoplastics. Sci. Total Environ. 2021, 779, 146523. [Google Scholar] [CrossRef] [PubMed]
- Panda, P.K.; Verma, S.K.; Suar, M. Nanoparticle-biological interactions: The renaissance of bionomics in the myriad nanomedical technologies. Nanomedicine 2021, 16, 2249–2254. [Google Scholar] [CrossRef] [PubMed]
- Shan, S.; Zhang, Y.; Zhao, H.; Zeng, T.; Zhao, X. Polystyrene nanoplastics penetrate across the blood-brain barrier and induce activation of microglia in the brain of mice. Chemosphere 2022, 298, 134261. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.E.; Bareja, A.; Bartlett, D.B.; White, J.P. Autophagy as a Therapeutic Target to Enhance Aged Muscle Regeneration. Cells 2019, 8, 183. [Google Scholar] [CrossRef] [PubMed]
- Park, S.S.; Seo, Y.K.; Kwon, K.S. Sarcopenia targeting with autophagy mechanism by exercise. BMB Rep. 2019, 52, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Ganley, I.G.; Lam Du, H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef] [PubMed]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.M.; Bernardi, H.; Py, G.; Candau, R.B. Autophagy is essential to support skeletal muscle plasticity in response to endurance exercise. Am. J. Physiol. Integr. Comp. Physiol. 2014, 307, R956–R969. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Sinha, S.C.; Kroemer, G. Bcl-2 family members: Dual regulators of apoptosis and autophagy. Autophagy 2008, 4, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Morell, C.; Bort, A.; Vara-Ciruelos, D.; Ramos-Torres, A.; Altamirano-Dimas, M.; Diaz-Laviada, I.; Rodriguez-Henche, N. Up-regulated expression of LAMP2 and autophagy activity during neuroendocrine differentiation of prostate cancer LNCaP cells. PLoS ONE 2016, 11, e0162977. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.; Cuervo, A.M. Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci. 2010, 13, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Frankel, L.B.; Wen, J.; Lees, M.; Hoyer-Hansen, M.; Farkas, T.; Krogh, A.; Jaattela, M.; Lund, A.H. microRNA-101 is a potent inhibitor of autophagy. EMBO J. 2011, 30, 4628–4641. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Tannous, P.; Johnstone, J.L.; Kong, Y.; Shelton, J.M.; Richardson, J.A.; Le, V.; Levine, B.; Rothermel, B.A.; Hill, J.A. Cardiac autophagy is a maladaptive response to hemodynamic stress. J. Clin. Investig. 2007, 117, 1782–1793. [Google Scholar] [CrossRef] [PubMed]
- Sciarretta, S.; Yasuhiro, M.; Daniela, Z.; Junichi, S. Role of autophagy in heart failure and clinical implications. Circ. Res. 2018, 123, 803–824. [Google Scholar]
- Zhu, L.; Liao, Y.; Jiang, B. Role of ROS and autophagy in the pathological process of atherosclerosis. J. Physiol. Biochem. 2024, 80, 743–756. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Sluimer, J.C.; Wang, Y.; Subramanian, M.; Brown, K.; Pattison, J.S.; Robbins, J.; Martinez, J.; Tabas, I. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab. 2012, 15, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Sciarretta, S.; Zhai, P.; Volpe, M.; Sadoshima, J. Pharmacological modulation of autophagy during cardiac stress. J. Cardiovasc. Pharmacol. 2012, 60, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Haq, S.; Grondin, J.; Banskota, S.; Khan, W.I. Autophagy: Roles in intestinal mucosal homeostasis and inflammation. J. Biomed. Sci. 2019, 26, 19. [Google Scholar] [CrossRef] [PubMed]
- Foerster, E.G.; Mukherjee, T.; Cabral-Fernandes, L.; Rocha, J.D.B.; Girardin, S.E.; Philpott, D.J. How autophagy controls the intestinal epithelial barrier. Autophagy 2022, 18, 86–103. [Google Scholar] [CrossRef] [PubMed]
- Alula, K.M.; Theiss, A.L. Autophagy in Crohn’s Disease: Converging on Dysfunctional Innate Immunity. Cells 2023, 12, 1779. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.Z.; Yao, Y.; Zhai, J.S.; Zhu, J.H.; Li, J.P.; Wu, K. The Role of Autophagy in Inflammatory Bowel Disease. Front Physiol. 2021, 12, 621132. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and neurodegeneration: Pathogenic mechanisms and therapeutic opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [PubMed]
- Alirezaei, M.; Kemball, C.C.; Whitton, J.L. Autophagy, inflammation and neurodegenerative disease. Eur. J. Neurosci. 2011, 33, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [PubMed]
- Sung, K.; Jimenez-Sanchez, M. Autophagy in Astrocytes and its Implications in Neurodegeneration. J. Mol. Biol. 2020, 432, 2605–2621. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, J.K.; Paliwal, A.; Saraf, P.; Sachdeva, S.N. Role of autophagy in follicular development and maintenance of primordial follicular pool in the ovary. J. Cell Physiol. 2022, 237, 1157–1170. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Peng, X.; Mei, S. Autophagy in Ovarian Follicular Development and Atresia. Int. J. Biol. Sci. 2019, 15, 726–737. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Wang, H.; Jia, P.; Zhao, H.; Liu, C.; Liu, W.; Song, Z.; Xu, Z.; Yang, L.; Wang, Y.; et al. Autophagy regulates spermatid differentiation via degradation of PDLIM1. Autophagy 2016, 12, 1575–1592. [Google Scholar] [CrossRef] [PubMed]
- Gawriluk, T.R.; Hale, A.N.; Flaws, J.A.; Dillon, C.P.; Green, D.R.; Rucker, E.B. Autophagy is a cell survival program for female germ cells in the murine ovary. Reproduction 2011, 141, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, S.; Kuma, A.; Murakami, M.; Kishi, C.; Yamamoto, A.; Mizushima, N. Autophagy is essential for preimplantation development of mouse embryos. Science 2008, 321, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J.; Baker, J.; Donnelly, L.E. Autophagy in asthma and chronic obstructive pulmonary disease. Clin. Sci. 2022, 136, 733–746. [Google Scholar] [CrossRef] [PubMed]
- Milillo, C.; Falcone, L.; Di Carlo, P.; Aruffo, E.; Del Boccio, P.; Cufaro, M.C.; Patruno, A.; Pesce, M.; Ballerini, P. Ozone effect on the inflammatory and proteomic profile of human macrophages and airway epithelial cells. Respir. Physiol. Neurobiol. 2023, 307, 103979. [Google Scholar] [CrossRef] [PubMed]
- Beiras, R.; Bellas, J.; Cachot, J.; Cormier, B.; Cousin, X.; Engwall, M.; Gambardella, C.; Garaventa, F.; Keiter, S.; Le Bihanic, F.; et al. Ingestion and contact with polyethylene microplastics does not cause acute toxicity on marine zooplankton. J. Hazard. Mater. 2018, 360, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Muñiz-González, A.B.; Silva, C.J.; Silva, A.L.P.; Campos, D.; Pestana, J.L.; Martínez-Guitarte, J.L. Suborganismal responses of the aquatic midge Chironomus riparius to polyethylene microplastics. Sci. Total Environ. 2021, 783, 146981. [Google Scholar] [CrossRef] [PubMed]
- Castro, G.B.; Bernegossi, A.C.; Felipe, M.C.; Corbi, J.J. Is the development of Daphnia magna neonates affected by short-term exposure to polyethylene microplastics? J. Environ. Sci. Health A 2020, 55, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Patrick, S. PVC Compounds and Processing. Rapra. Technol. 2004, 15, 3. [Google Scholar]
- Fernández-González, V.; Andrade-Garda, J.M.; López-Mahía, P.; Muniategui-Lorenzo, S. Misidentification of PVC microplastics in marine environment samples. TrAC Trends Anal. Chem. 2022, 153, 116649. [Google Scholar] [CrossRef]
- Li, J.; Weng, H.; Liu, S.; Li, F.; Xu, K.; Wen, S.; Chen, X.; Li, C.; Nie, Y.; Liao, B.; et al. Embryonic exposure of polystyrene nanoplastics affects cardiac development. Sci. Total Environ. 2024, 906, 167406. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Xu, T.; Fan, X.; Chi, Q.; Tang, X.; Li, Z.; Bai, Z.; Li, S. Polystyrene nanoplastics exacerbate lipopolysaccharide-induced myocardial fbrosis and autophagy in mice via ROS/TGF-β1/Smad. Toxicology 2022, 480, 153338. [Google Scholar]
- Lu, Y.-Y.; Li, H.; Ren, H.; Zhang, X.; Huang, F.; Zhang, D.; Huang, Q.; Zhang, X. Size-dependent effects of polystyrene nanoplastics on autophagy response in human umbilical vein endothelial cells. J. Hazard. Mater. 2022, 421, 126770. [Google Scholar] [CrossRef] [PubMed]
- Saha, K.; Subramenium Ganapathy, A.; Wang, A.; Morris, N.M.; Suchanec, E.; Ding, W.; Yochum, G.; Koltun, W.; Nighot, M.; Ma, T.; et al. Autophagy reduces the degradation and promotes membrane localization of occludin to enhance the intestinal epithelial tight junction barrier against paracellular macromolecule flux. J. Crohns. Colitis 2023, 17, 433–449. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.F.; Li, J.J.; Liu, T.; Wei, C.Q.; Ma, L.W.; Nikolenko, V.N.; Chang, W.L. Morphological and biochemical characteristics associated with autophagy in gastrointestinal diseases. World J. Gastroenterol. 2024, 30, 1524–1532. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.F.; Jin, W.T.; Sun, H.N.; Li, C.J.; Jia, J.B. Perturbation of autophagy: An intrinsic toxicity mechanism of nanoparticles. Sci. Total Environ. 2022, 823, 153629. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Feng, Y.; Han, C.; Yao, Z.; Liu, Y.; Luo, C.; Sheng, J. Autophagic response of intestinal epithelial cells exposed to polystyrene nanoplastics. Environ. Toxicol. 2023, 38, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ye, Y.; Yuan, H.; Rihan, N.; Han, M.; Liu, X.; Zhu, T.; Zhao, Y.; Che, X. Exposure to polystyrene nanoplastics induces apoptosis, autophagy, histopathological damage, and intestinal microbiota dysbiosis of the Pacific whiteleg shrimp (Litopenaeus vannamei). Sci. Total Environ. 2024, 919, 170924. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Gao, M.; Liu, B.; Qin, Y.; Chen, L.; Liu, H.; Gong, G. Mitochondrial autophagy: Molecular mechanisms and implications for cardiovascular disease. Cell Death Dis. 2022, 13, 444. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lai, M.; Zhang, X.; Li, Z.; Yang, D.; Zhao, M.; Wang, D.; Sun, Z.; Ehsan, S.; Li, W.; et al. PINK1-parkin-mediated neuronal mitophagy deficiency in prion disease. Cell Death Dis. 2022, 13, 162. [Google Scholar] [CrossRef] [PubMed]
- Guanglin, L.; Shuqin, W. Polystyrene nanoplastics exposure causes inflammation and death of esophageal cell. Ecotoxicol. Environ. Saf. 2024, 269, 115819. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Kang, J.H.; Lee, S. Autophagy in neurodegenerative diseases: A hunter for aggregates. Int. J. Mol. Sci. 2020, 21, 3369. [Google Scholar] [CrossRef] [PubMed]
- Stavoe, A.K.H.; Holzbaur, E.L.F. Autophagy in neurons. Annu. Rev. Cell Dev. Biol. 2019, 35, 477–500. [Google Scholar] [CrossRef] [PubMed]
- Eshraghi, M.; Adlimoghaddam, A.; Mahmoodzadeh, A.; Sharifzad, F.; Yasavoli-Sharahi, H.; Lorzadeh, S.; Albensi, B.C.; Ghavami, S. Alzheimer’s Disease Pathogenesis: Role of Autophagy and Mitophagy Focusing in Microglia. Int. J. Mol. Sci. 2021, 22, 3330. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Oliver, D.M. Amyloid beta and phosphorylated tau-induced defective autophagy and mitophagy in Alzheimer’s disease. Cells 2019, 8, 488. [Google Scholar] [CrossRef] [PubMed]
- Lizama, B.N.; Chu, C.T. Neuronal autophagy and mitophagy in Parkinson’s disease. Mol. Aspects Med. 2021, 82, 100972. [Google Scholar] [CrossRef] [PubMed]
- Croce, K.R.; Yamamoto, A. A role for autophagy in Huntington’s disease. Neurobiol. Dis. 2019, 122, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhao, Y.; Dou, J.; Hou, Q.; Cheng, J.; Jiang, X. Bioeffects of Inhaled Nanoplastics on Neurons and Alteration of Animal Behaviors through Deposition in the Brain. Nano Lett. 2022, 22, 1091–1099. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.R.; Chesselet, M.-F. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog. Neurobiol. 2013, 106–107, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Lou, G.; Palikaras, K.; Lautrup, S.; Scheibye-Knudsen, M.; Tavernarakis, N.; Fang, E.F. Mitophagy and neuroprotection. Trends Mol. Med. 2020, 26, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Zhao, Y.; Jiang, Z.; Chen, J. Acetaldehyde induces cytotoxicity via triggering mitochondrial dysfunction and overactive mitophagy. Mol. Neurobiol. 2022, 59, 3933–3946. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liang, B.; Li, Z.; Zhong, Y.; Wang, B.; Zhang, B.; Du, J.; Ye, R.; Xian, H.; Min, W.; et al. Polystyrene nanoplastic exposure induces excessive mitophagy by activating AMPK/ULK1 pathway in differentiated SH-SY5Y cells and dopaminergic neurons in vivo. Part. Fibre Toxicol. 2023, 20, 44. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.A.; Kim, D.; Kim, J.H.; Shin, Y.J.; Kim, E.S.; Akram, M.; Kim, E.H.; Majid, A.; Baek, S.H.; Bae, O.N. Autophagy-mediated occludin degradation contributes to blood- brain barrier disruption during ischemia in bEnd.3 brain endothelial cells and rat ischemic stroke models. Fluids Barriers CNS 2020, 17, 21. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, W.; Sun, Y.; Han, F.; Hu, C.A.; Wu, Z. Amino acid deprivation disrupts barrier function and induces protective autophagy in intestinal porcine epithelial cells. Amino Acids 2015, 47, 2177–2184. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L.; Huang, Y.J.; Cui, J.T.; Song, N.; Xie, J. Iron dysregulation in Parkinson’s Disease: Focused on the Autophagy-Lysosome Pathway. ACS Chem. Neurosci. 2019, 10, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Hu, R.; Yu, R.; Tang, Y.; Li, J. A narrative review of mechanisms of ferroptosis in cancer: New challenges and opportunities. Ann. Transl. Med. 2021, 9, 1599. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Liu, Y.; Xu, Y.; Li, K.; Zhou, L.; Qiao, H.; Xu, Q.; Zhao, J. The role of ferroptosis in blood-brain barrier injury. Cell. Mol. Neurobiol. 2023, 43, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.H.; Baek, S.M.; Park, H.J.; Bian, Y.; Chung, H.Y.; Bae, O.N. Polystyrene nanoplastics promote the blood-brain barrier dysfunction through autophagy pathway and excessive erythrophagocytosis. Ecotoxicol. Environ. Saf. 2025, 289, 117471. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Yin, Q.; Wei, D.; Yang, Z.; Du, Y.; Ma, Y. Autophagy in male reproduction. Syst. Biol. Reprod. Med. 2019, 65, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Peters, A.E.; Mihalas, B.P.; Bromfield, E.G.; Roman, S.D.; Nixon, B.; Sutherland, J.M. Autophagy in female fertility: A role in oxidative stress and aging. Antioxid. Redox Signal. 2020, 32, 550–568. [Google Scholar] [CrossRef] [PubMed]
- Wick, P.; Malek, A.; Manser, P.; Meili, D.; Maeder-Althaus, X.; Diener, L.; Diener, P.A.; Zisch, A.; Krug, H.F.; von Mandach, U. Barrier capacity of human placenta for nanosized materials. Environ. Health Perspect. 2010, 118, 432–436. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Chen, L.; Chen, B.; Tang, Y.; Zhao, Y.; Liu, S.; Xu, H. Toxic effects of TiO2 NPs in the blood-milk barrier of the maternal dams and growth of offspring. Ecotoxicol. Environ. Saf. 2021, 15, 111762. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Li, S.; Jiang, S.; Wang, L.; Zhan, D.; Xiong, M.; Jiang, Y.; Huang, Q.; Kui, H.; Li, X. Maternal exposure to polystyrene nanoplastics disrupts spermatogenesis in mouse offspring by inducing Prdm14 overexpression in undifferentiated spermatogonia. ACS Nano 2025, 19, 2148–2161. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Zhao, J.; Li, L.; Wang, Y.; Dai, X.; Yu, F.; Ma, J. Interfacial interaction between micro/nanoplastics and typical PPCPs and nanoplastics removal via electrosorption from an aqueous solution. Water Res. 2020, 184, 116100. [Google Scholar] [CrossRef] [PubMed]
- Montagnini, B.G.; Forcato, S.; Pernoncine, K.V.; Monteiro, M.C.; Pereira, M.; Costa, N.O.; Moreira, E.G.; Anselmo-Franci, J.A.; Gerardin, D. Developmental and reproductive outcomes in male rats exposed to triclosan: Two-generation study. Front. Endocrinol. 2021, 12, 738980. [Google Scholar] [CrossRef] [PubMed]
- Maksymowicz, M.; Reka, G.; Machowiec, P.; Piecewicz-Szczesna, H. Impact of triclosan on female and male reproductive system and its consequences on fertility: A literature review. J. Fam. Reprod. Health 2022, 16, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Arriagada, D.; Miranda-Rojas, S.; Camarada, M.B.; Ortega, D.E.; Alarcon-Palacio, V.B. The interaction mechanism of polystyrene microplastics with pharmaceuticals and personal care products. Sci. Total Environ. 2023, 861, 160632. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhou, C.; Ma, Z.; Zeng, L.; Wang, H.; Cheng, X.; Zhang, C.; Xue, Y.; Yuan, Y.; Li, J.; et al. Co-exposure to polystyrene nanoplastics and triclosan induces synergistic cytotoxicity in human KGN granulosa cells by promoting reactive oxygen species accumulation. Ecotoxicol. Environ. Saf. 2024, 273, 116121. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.; Xia, P.; Guo, Y.; Ji, P.; Yuan, X.; Wang, L.; Shuang, S.; Zhou, L.; Tong, R.; Zhang, L.; et al. Effects of polystyrene microparticles exposures on spermatogenic cell differentiation and reproductive endpoints in male mice. Environ. Pollut. 2025, 373, 126200. [Google Scholar] [CrossRef] [PubMed]
- Lam, H.C.; Cloonan, S.M.; Bhashyam, A.R.; Haspel, J.A.; Singh, A.; Sathirapongsasuti, J.F.; Cervo, M.; Yao, H.; Chung, A.L.; Mizumura, K.; et al. Histone deacetylase 6-mediated selective autophagy regulates COPD-associated cilia dysfunction. J. Clin. Invest 2013, 123, 5212–5230. [Google Scholar] [CrossRef] [PubMed]
- Kanayama, M.; He, Y.W.; Shinohara, M.L. The lung is protected from spontaneous inflammation by autophagy in myeloid cells. J. Immunol. 2015, 194, 5465–5471. [Google Scholar] [CrossRef] [PubMed]
- Abdel Fattah, E.; Bhattacharya, A.; Herron, A.; Safdar, Z.; Eissa, N.T. Critical role for IL-18 in spontaneous lung inflammation caused by autophagy deficiency. J. Immunol. 2015, 194, 5407–5416. [Google Scholar] [CrossRef] [PubMed]
- Annangi, B.; Villacorta, A.; López-Mesas, M.; Fuentes-Cebrian, V.; Marcos, R.; Hernández, A. Hazard Assessment of Polystyrene Nanoplastics in Primary Human Nasal Epithelial Cells, Focusing on the Autophagic Effects. Biomolecules 2021, 13, 220. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhang, T.; Ge, Y.; Cheng, Y.; Yin, L.; Pu, Y.; Chen, Z.; Liang, G. Ferritinophagy mediated by oxidative stress-driven mitochondrial damage is involved in the polystyrene nanoparticles-induced ferroptosis of lung injury. ACS Nano 2023, 17, 24988–25004. [Google Scholar] [CrossRef] [PubMed]
- Annangi, B.; Villacorta, A.; Vela, L.; Tavakolpournegari, A.; Marcos, R.; Hernández, A. Effects of true-to-life PET nanoplastics using primary human nasal epithelial cells. Environ. Toxicol. Pharmacol. 2023, 100, 104140. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.H.; Hu, J.N.; Zhang, M.; Meng, Z.; Shi, G.P.; Wang, Z.; Li, W. Maltol attenuates polystyrene nanoplastic-induced enterotoxicity by promoting AMPK/mTOR/TFEB-mediated autophagy and modulating gut microbiota. Environ. Pollut. 2023, 322, 121202. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ye, Y.; Zhu, X.; Li, S.; Rihan, N.; Yao, Z.; Sun, Z.; Gao, P.; Zhao, Y.; Lai, Q. Polystyrene nanoplastics induce apoptosis, histopathological damage, and glutathione metabolism disorder in the intestine of juvenile East Asian river prawns (Macrobrachium nipponense). Sci. Total Environ. 2024, 954, 176718. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jia, Z.; Gao, X.; Zhao, J.; Zhang, H. Polystyrene nanoparticles induced mammalian intestine damage caused by blockage of BNIP3/NIX-mediated mitophagy and gut microbiota alteration. Sci. Total Environ. 2024, 907, 168064. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Zhang, R.; Li, B.; Du, Y.; Li, J.; Tong, X.; Wu, Y.; Ji, X.; Zhang, Y. Tissue distribution of polystyrene nanoplastics in mice and their entry, transport, and cytotoxicity to GES-1 cells. Environ. Pollut. 2021, 280, 116974. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yan, L.; Zhang, Y.; Liu, X.; Wei, Y.; Zhao, Y.; Li, K.; Shi, Y.; Liu, H.; Lai, W.; et al. Maternal exposure to nanopolystyrene induces neurotoxicity in offspring through P53-mediated ferritinophagy and ferroptosis in the rat hippocampus. J. Nanobiotechnology 2024, 22, 651. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Wang, X.; Chen, W.; Xu, Z.; Zhao, J.; Huang, W.; Wang, M.; Zhang, H. Polystyrene nanoplastics activate autophagy and suppress trophoblast cell migration/invasion and migrasome formation to induce miscarriage. ACS Nano 2024, 18, 3733–3751. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Zhang, Z.; Xu, B.; Ding, L.; Yang, H.; He, T.; Du, X.; Pei, X.; Fu, X. Integrated transcriptomic and metabolomic analysis reveals the underlying mechanisms for male reproductive toxicity of polystyrene nanoplastics in mouse spermatocyte-derived GC-2spd(ts) cells. Toxicol. In Vitro 2024, 100, 105893. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Popp, L.; Yang, J.; Kumar, A.; Gangoli, V.S.; Segatori, L. The autophagic response to polystyrene nanoparticles is mediated by transcription factor EB and depends on surface charge. J. Nanobiotechnol. 2015, 13, 87. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-C.; Chen, K.-F.; Lin, K.-Y.A.; Tsang, Y.F.; Hsu, Y.-F.; Lin, C.-H. Evaluation of the pulmonary toxicity of PSNPs using a Transwell-based normal human bronchial epithelial cell culture system. Sci. Total Environ. 2023, 895, 165213. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.L.; Ng, C.T.; Zou, L.; Lu, Y.; Chen, J.; Bay, B.H.; Shen, H.M.; Ong, C.N. Targeted metabolomics reveals differential biological effects of nanoplastics and nanoZnO in human lung cells. Nanotoxicology 2019, 13, 1117–1132. [Google Scholar] [CrossRef] [PubMed]
- Milillo, C.; Aruffo, E.; Di Carlo, P.; Patruno, A.; Gatta, M.; Bruno, A.; Dovizio, M.; Marinelli, L.; Dimmito, M.P.; Di Giacomo, V.; et al. Polystyrene nanoplastics mediate oxidative stress, senescence, and apoptosis in a human alveolar epithelial cell line. Front. Public Health 2024, 12, 1385387. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fanghella, F.; Pesce, M.; Franceschelli, S.; Panella, V.; Elsallabi, O.; Lupi, T.; Rizza, B.; Di Battista, M.G.; Bruno, A.; Ballerini, P.; et al. Biological Modulation of Autophagy by Nanoplastics: A Current Overview. Int. J. Mol. Sci. 2025, 26, 7035. https://doi.org/10.3390/ijms26157035
Fanghella F, Pesce M, Franceschelli S, Panella V, Elsallabi O, Lupi T, Rizza B, Di Battista MG, Bruno A, Ballerini P, et al. Biological Modulation of Autophagy by Nanoplastics: A Current Overview. International Journal of Molecular Sciences. 2025; 26(15):7035. https://doi.org/10.3390/ijms26157035
Chicago/Turabian StyleFanghella, Francesco, Mirko Pesce, Sara Franceschelli, Valeria Panella, Osama Elsallabi, Tiziano Lupi, Benedetta Rizza, Maria Giulia Di Battista, Annalisa Bruno, Patrizia Ballerini, and et al. 2025. "Biological Modulation of Autophagy by Nanoplastics: A Current Overview" International Journal of Molecular Sciences 26, no. 15: 7035. https://doi.org/10.3390/ijms26157035
APA StyleFanghella, F., Pesce, M., Franceschelli, S., Panella, V., Elsallabi, O., Lupi, T., Rizza, B., Di Battista, M. G., Bruno, A., Ballerini, P., Patruno, A., & Speranza, L. (2025). Biological Modulation of Autophagy by Nanoplastics: A Current Overview. International Journal of Molecular Sciences, 26(15), 7035. https://doi.org/10.3390/ijms26157035