
Aβ40 Improves Cerebrovascular Endothelial Function via NOX4-Dependent Hydrogen Peroxide Release

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
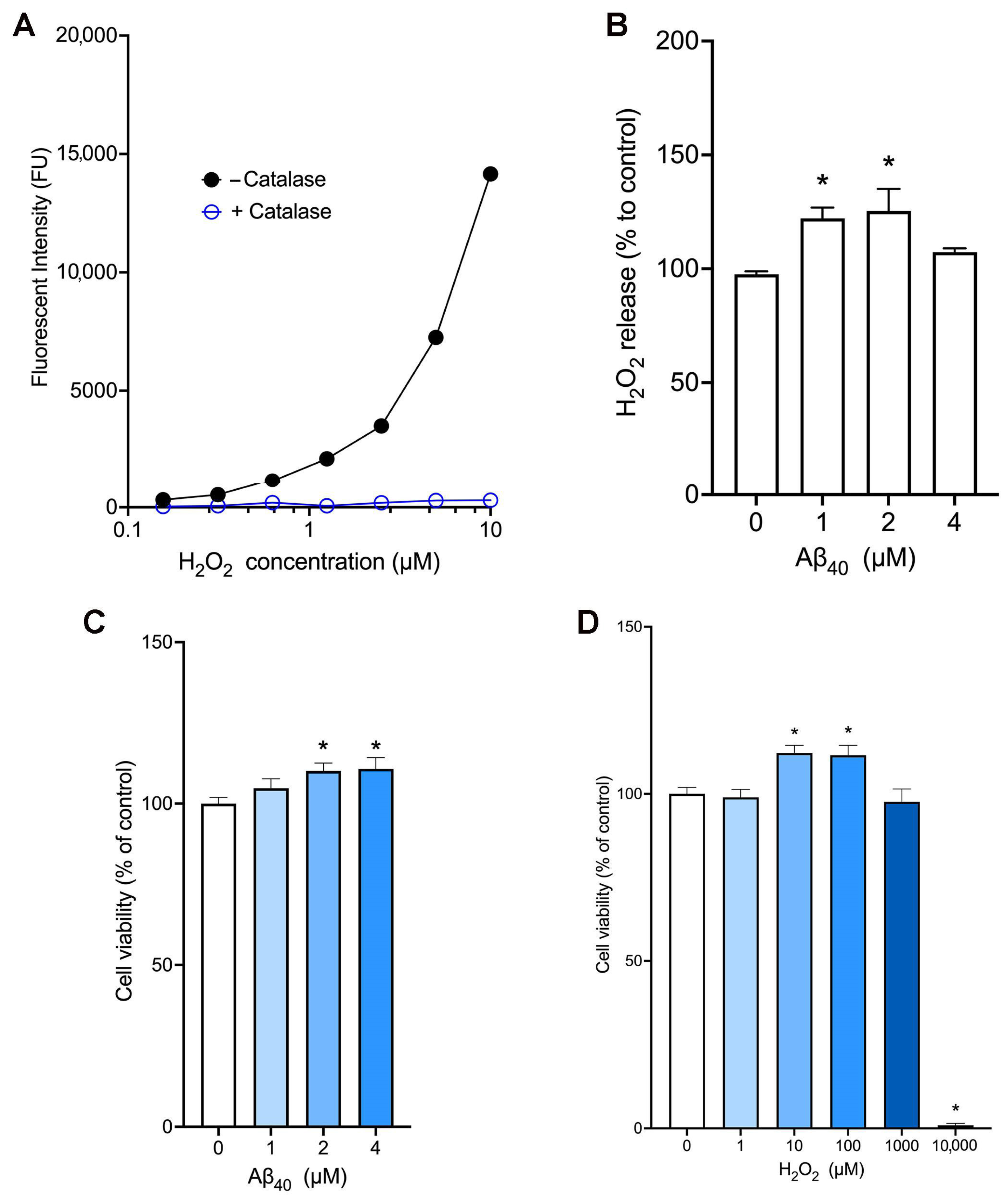
2.1. Aβ40 Increases the Release of Hydrogen Peroxide and Cell Viability in bEnd.3 Cells
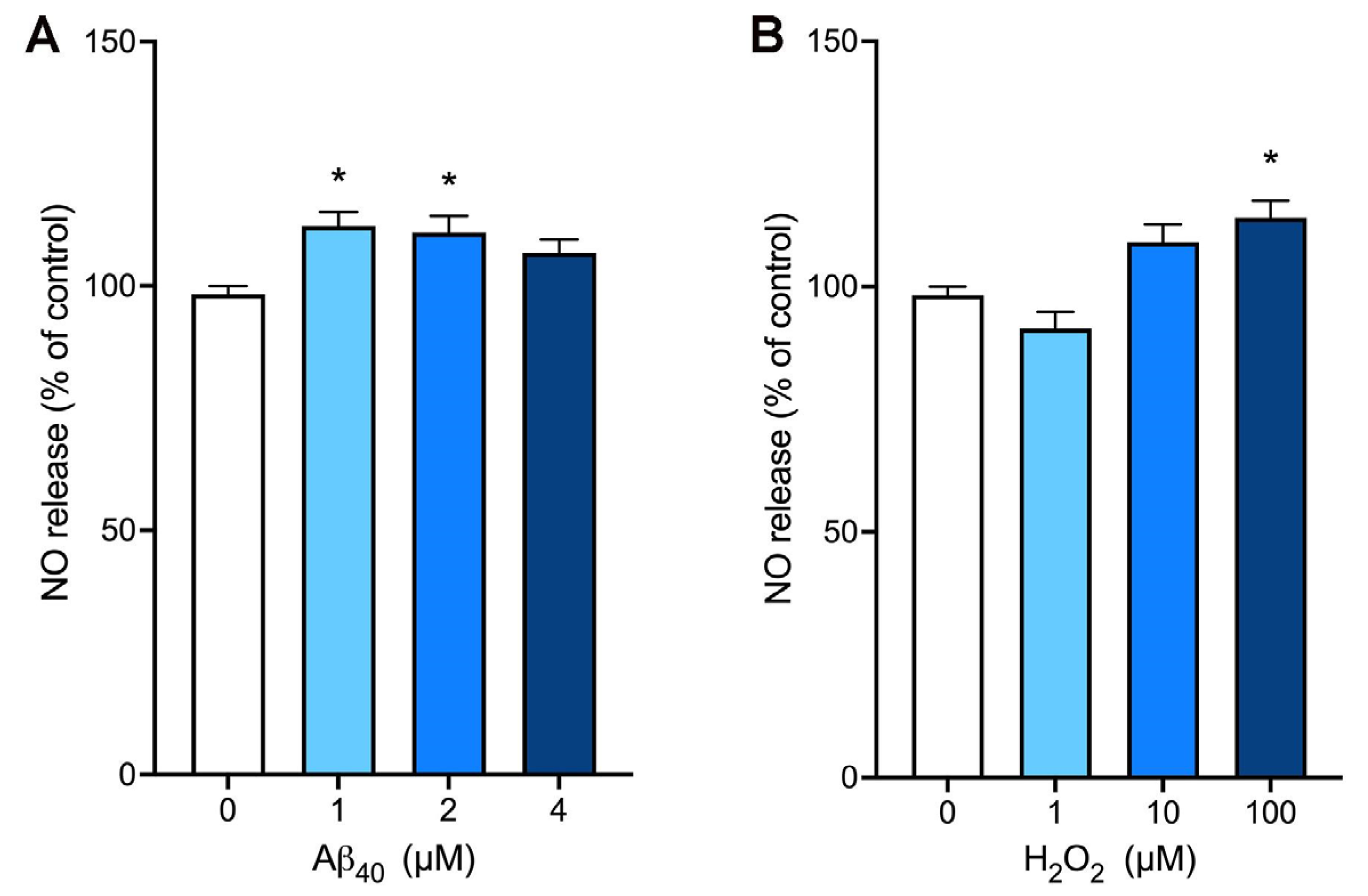
2.2. Aβ40 Enhances the NO Bioavailability in bEnd.3 Cells
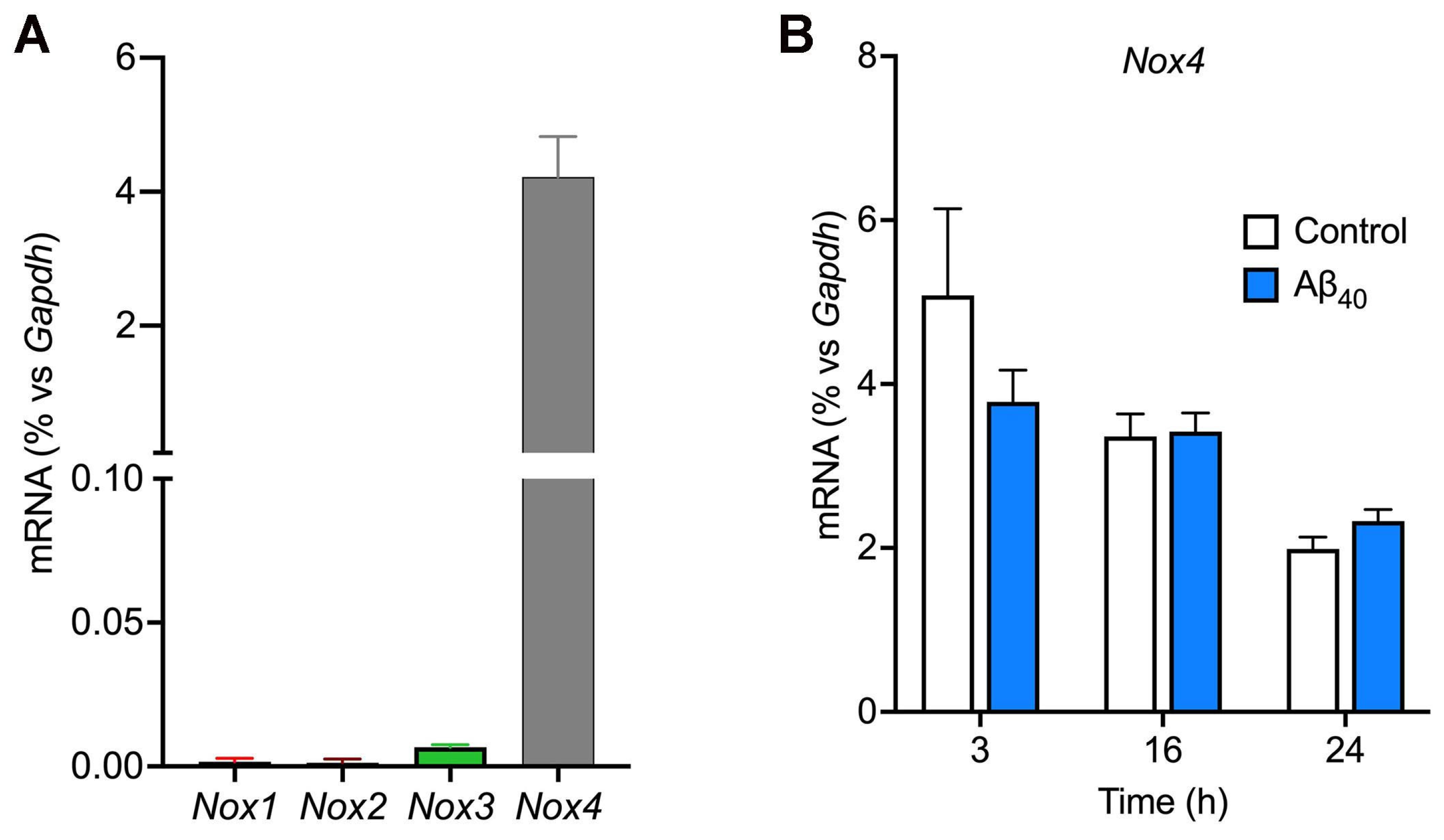
2.3. Nox4 Is the Nox Isoform That Is Predominantly Expressed in bEnd.3 Cells
2.4. NOX4 Is Responsible for Increased Hydrogen Peroxide Production in Response to Aβ40
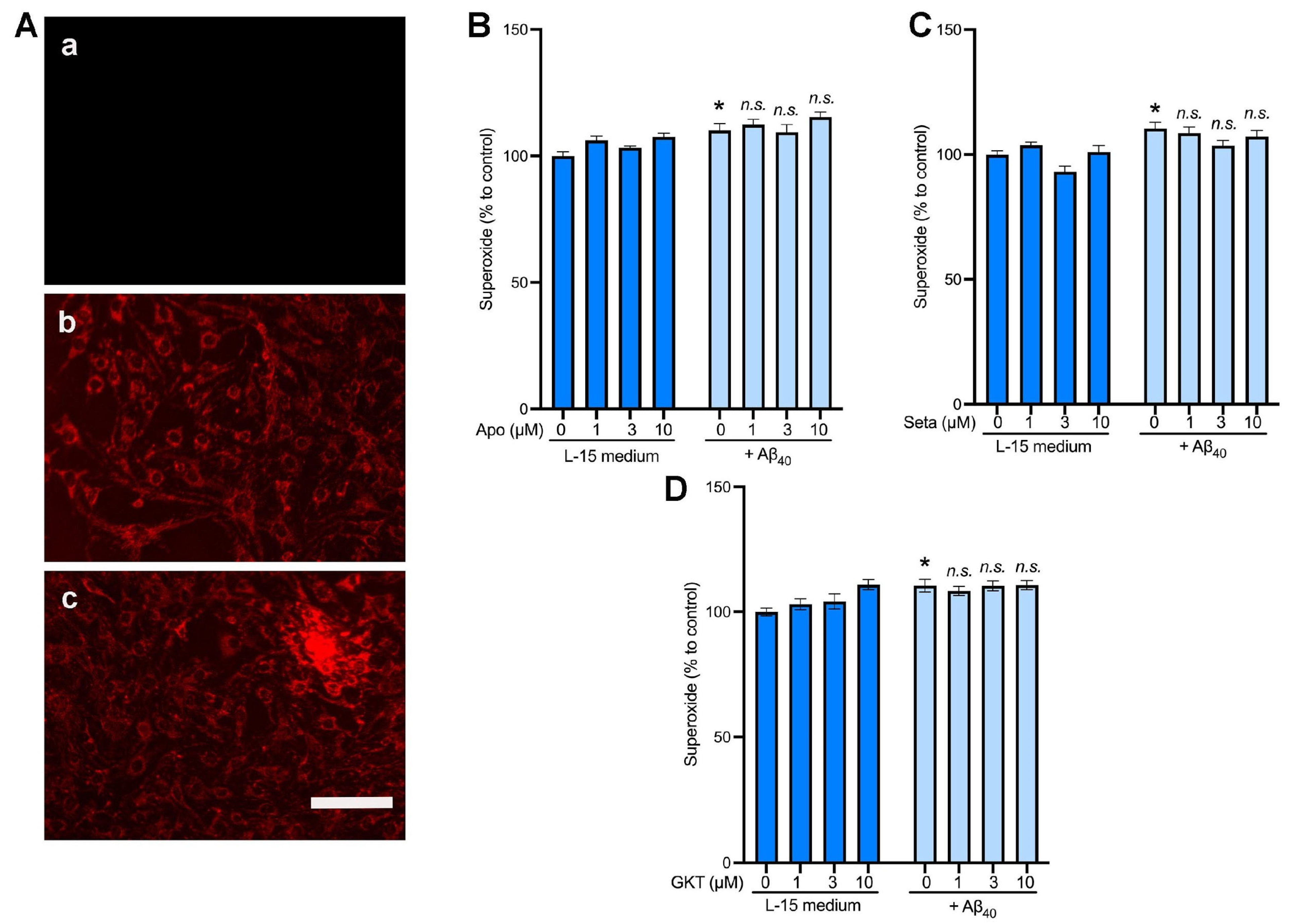
2.5. NOX4 Inhibition Did Not Affect the Intracellular Levels of Superoxide in bEnd.3 Cells
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Detection of Hydrogen Peroxide
4.4. Detection of Nitric Oxide
4.5. Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)
4.6. Cell Viability Assay
4.7. Detection of Superoxide
4.8. 2,2-Diphenyl-1-picrylhydrazyl (DPPH) Assay
4.9. Data Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gale, S.A.; Acar, D.; Daffner, K.R. Dementia. Am. J. Med. 2018, 131, 1161–1169. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Ihara, M. New therapeutic approaches for Alzheimer’s disease and cerebral amyloid angiopathy. Front. Aging Neurosci. 2014, 6, 290. [Google Scholar] [CrossRef]
- Greenberg, S.M.; Bacskai, B.J.; Hernandez-Guillamon, M.; Pruzin, J.; Sperling, R.; van Veluw, S.J. Cerebral amyloid angiopathy and Alzheimer disease—One peptide, two pathways. Nat. Rev. Neurol. 2020, 16, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Wu, J. Amyloid-beta: A double agent in Alzheimer’s disease? Biomed. Pharmacother. 2021, 139, 111575. [Google Scholar] [CrossRef]
- Han, B.H.; Zhou, M.L.; Johnson, A.W.; Singh, I.; Liao, F.; Vellimana, A.K.; Nelson, J.W.; Milner, E.; Cirrito, J.R.; Basak, J.; et al. Contribution of reactive oxygen species to cerebral amyloid angiopathy, vasomotor dysfunction, and microhemorrhage in aged Tg2576 mice. Proc. Natl. Acad. Sci. USA 2015, 112, E881–E890. [Google Scholar] [CrossRef]
- Han, B.H.; Zhou, M.L.; Vellimana, A.K.; Milner, E.; Kim, D.H.; Greenberg, J.K.; Chu, W.; Mach, R.H.; Zipfel, G.J. Resorufin Analogs Preferentially Bind Cerebrovascular Amyloid: Potential Use as Imaging Ligands for Cerebral Amyloid Angiopathy. Mol. Neurodegener. 2011, 6, 86. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.L.; Wang, J.; Zhang, Z.N.; Su, Q.; Guo, J.H. The relationship between amyloid-beta and brain capillary endothelial cells in Alzheimer’s disease. Neural Regen. Res. 2022, 17, 2355–2363. [Google Scholar] [CrossRef]
- Cortes-Canteli, M.; Iadecola, C. Alzheimer’s Disease and Vascular Aging: JACC Focus Seminar. J. Am. Coll. Cardiol. 2020, 75, 942–951. [Google Scholar] [CrossRef]
- Paris, D.; Humphrey, J.; Quadros, A.; Patel, N.; Crescentini, R.; Crawford, F.; Mullan, M. Vasoactive effects of A beta in isolated human cerebrovessels and in a transgenic mouse model of Alzheimer’s disease: Role of inflammation. Neurol. Res. 2003, 25, 642–651. [Google Scholar] [CrossRef]
- Dietrich, H.H.; Xiang, C.; Han, B.H.; Zipfel, G.J.; Holtzman, D.M. Soluble amyloid-beta, effect on cerebral arteriolar regulation and vascular cells. Mol. Neurodegener. 2010, 5, 15. [Google Scholar] [CrossRef]
- Tong, X.K.; Nicolakakis, N.; Kocharyan, A.; Hamel, E. Vascular remodeling versus amyloid beta-induced oxidative stress in the cerebrovascular dysfunctions associated with Alzheimer’s disease. J. Neurosci. 2005, 25, 11165–11174. [Google Scholar] [CrossRef] [PubMed]
- Han, B.H.; Zhou, M.L.; Abousaleh, F.; Brendza, R.P.; Dietrich, H.H.; Koenigsknecht-Talboo, J.; Cirrito, J.R.; Milner, E.; Holtzman, D.M.; Zipfel, G.J. Cerebrovascular dysfunction in amyloid precursor protein transgenic mice: Contribution of soluble and insoluble amyloid-beta peptide, partial restoration via gamma-secretase inhibition. J. Neurosci. 2008, 28, 13542–13550. [Google Scholar] [CrossRef]
- Niwa, K.; Carlson, G.A.; Iadecola, C. Exogenous A beta1-40 reproduces cerebrovascular alterations resulting from amyloid precursor protein overexpression in mice. J. Cereb. Blood Flow Metab. 2000, 20, 1659–1668. [Google Scholar] [CrossRef]
- Niwa, K.; Younkin, L.; Ebeling, C.; Turner, S.K.; Westaway, D.; Younkin, S.; Ashe, K.H.; Carlson, G.A.; Iadecola, C. Abeta 1-40-related reduction in functional hyperemia in mouse neocortex during somatosensory activation. Proc. Natl. Acad. Sci. USA 2000, 97, 9735–9740. [Google Scholar] [CrossRef] [PubMed]
- Kisler, K.; Nelson, A.R.; Montagne, A.; Zlokovic, B.V. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2017, 18, 419–434. [Google Scholar] [CrossRef]
- Weksler, B.B.; Subileau, E.A.; Perriere, N.; Charneau, P.; Holloway, K.; Leveque, M.; Tricoire-Leignel, H.; Nicotra, A.; Bourdoulous, S.; Turowski, P.; et al. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J. 2005, 19, 1872–1874. [Google Scholar] [CrossRef] [PubMed]
- Vajtr, D.; Benada, O.; Kukacka, J.; Prusa, R.; Houstava, L.; Toupalik, P.; Kizek, R. Correlation of ultrastructural changes of endothelial cells and astrocytes occuring during blood brain barrier damage after traumatic brain injury with biochemical markers of BBB leakage and inflammatory response. Physiol. Res. 2009, 58, 263–268. [Google Scholar] [CrossRef]
- Hayes, G.; Pinto, J.; Sparks, S.N.; Wang, C.; Suri, S.; Bulte, D.P. Vascular smooth muscle cell dysfunction in neurodegeneration. Front. Neurosci. 2022, 16, 1010164. [Google Scholar] [CrossRef]
- Yamada, M.; Huang, Z.; Dalkara, T.; Endres, M.; Laufs, U.; Waeber, C.; Huang, P.L.; Liao, J.K.; Moskowitz, M.A. Endothelial nitric oxide synthase-dependent cerebral blood flow augmentation by L-arginine after chronic statin treatment. J. Cereb. Blood Flow Metab. 2000, 20, 709–717. [Google Scholar] [CrossRef]
- Schulz, E.; Wenzel, P.; Munzel, T.; Daiber, A. Mitochondrial redox signaling: Interaction of mitochondrial reactive oxygen species with other sources of oxidative stress. Antioxid. Redox Signal. 2014, 20, 308–324. [Google Scholar] [CrossRef] [PubMed]
- Brieger, K.; Schiavone, S.; Miller, F.J., Jr.; Krause, K.H. Reactive oxygen species: From health to disease. Swiss Med. Wkly. 2012, 142, w13659. [Google Scholar] [CrossRef]
- Islam, M.T. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res. 2017, 39, 73–82. [Google Scholar] [CrossRef]
- Carvalho, C.; Moreira, P.I. Oxidative Stress: A Major Player in Cerebrovascular Alterations Associated to Neurodegenerative Events. Front. Physiol. 2018, 9, 806. [Google Scholar] [CrossRef]
- Tarafdar, A.; Pula, G. The Role of NADPH Oxidases and Oxidative Stress in Neurodegenerative Disorders. Int. J. Mol. Sci. 2018, 19, 3824. [Google Scholar] [CrossRef]
- Selemidis, S.; Sobey, C.G.; Wingler, K.; Schmidt, H.H.; Drummond, G.R. NADPH oxidases in the vasculature: Molecular features, roles in disease and pharmacological inhibition. Pharmacol. Ther. 2008, 120, 254–291. [Google Scholar] [CrossRef] [PubMed]
- Cahill-Smith, S.; Li, J.M. Oxidative stress, redox signalling and endothelial dysfunction in ageing-related neurodegenerative diseases: A role of NADPH oxidase 2. Br. J. Clin. Pharmacol. 2014, 78, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Sorce, S.; Stocker, R.; Seredenina, T.; Holmdahl, R.; Aguzzi, A.; Chio, A.; Depaulis, A.; Heitz, F.; Olofsson, P.; Olsson, T.; et al. NADPH oxidases as drug targets and biomarkers in neurodegenerative diseases: What is the evidence? Free Radic. Biol. Med. 2017, 112, 387–396. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Takac, I.; Schroder, K.; Zhang, L.; Lardy, B.; Anilkumar, N.; Lambeth, J.D.; Shah, A.M.; Morel, F.; Brandes, R.P. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J. Biol. Chem. 2011, 286, 13304–13313. [Google Scholar] [CrossRef]
- Schroder, K.; Zhang, M.; Benkhoff, S.; Mieth, A.; Pliquett, R.; Kosowski, J.; Kruse, C.; Luedike, P.; Michaelis, U.R.; Weissmann, N.; et al. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ. Res. 2012, 110, 1217–1225. [Google Scholar] [CrossRef]
- Reynolds, M.R.; Singh, I.; Azad, T.D.; Holmes, B.B.; Verghese, P.B.; Dietrich, H.H.; Diamond, M.; Bu, G.; Han, B.H.; Zipfel, G.J. Heparan sulfate proteoglycans mediate Abeta-induced oxidative stress and hypercontractility in cultured vascular smooth muscle cells. Mol. Neurodegener. 2016, 11, 9. [Google Scholar] [CrossRef] [PubMed]
- Park, L.; Zhou, P.; Pitstick, R.; Capone, C.; Anrather, J.; Norris, E.H.; Younkin, L.; Younkin, S.; Carlson, G.; McEwen, B.S.; et al. Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc. Natl. Acad. Sci. USA 2008, 105, 1347–1352. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, G.; Szyndralewiez, C.; Molango, S.; Carnesecchi, S.; Heitz, F.; Wiesel, P.; Wood, J.M. Therapeutic potential of NADPH oxidase 1/4 inhibitors. Br. J. Pharmacol. 2017, 174, 1647–1669. [Google Scholar] [CrossRef] [PubMed]
- Han, B.H.; Cofell, B.; Everhart, E.; Humpal, C.; Kang, S.S.; Lee, S.K.; Kim-Han, J.S. Amentoflavone Promotes Cellular Uptake and Degradation of Amyloid-Beta in Neuronal Cells. Int. J. Mol. Sci. 2022, 23, 5885. [Google Scholar] [CrossRef]
- Sun, L.; Sharma, A.K.; Han, B.H.; Mirica, L.M. Amentoflavone: A Bifunctional Metal Chelator that Controls the Formation of Neurotoxic Soluble Abeta(42) Oligomers. ACS Chem. Neurosci. 2020, 11, 2741–2752. [Google Scholar] [CrossRef]
- Craige, S.M.; Chen, K.; Pei, Y.; Li, C.; Huang, X.; Chen, C.; Shibata, R.; Sato, K.; Walsh, K.; Keaney, J.F., Jr. NADPH oxidase 4 promotes endothelial angiogenesis through endothelial nitric oxide synthase activation. Circulation 2011, 124, 731–740. [Google Scholar] [CrossRef]
- O’Neill, K.M.; Campbell, D.C.; Edgar, K.S.; Gill, E.K.; Moez, A.; McLoughlin, K.J.; O’Neill, C.L.; Dellett, M.; Hargey, C.J.; Abudalo, R.A.; et al. NOX4 is a major regulator of cord blood-derived endothelial colony-forming cells which promotes post-ischaemic revascularization. Cardiovasc. Res. 2020, 116, 393–405. [Google Scholar] [CrossRef]
- Alves-Lopes, R.; Lacchini, S.; Neves, K.B.; Harvey, A.; Montezano, A.C.; Touyz, R.M. Vasoprotective effects of NOX4 are mediated via polymerase and transient receptor potential melastatin 2 cation channels in endothelial cells. J. Hypertens. 2023, 41, 1389–1400. [Google Scholar] [CrossRef]
- Gola, L.; Bierhansl, L.; Csatari, J.; Schroeter, C.B.; Korn, L.; Narayanan, V.; Cerina, M.; Abdolahi, S.; Speicher, A.; Hermann, A.M.; et al. NOX4-derived ROS are neuroprotective by balancing intracellular calcium stores. Cell. Mol. Life Sci. 2023, 80, 127. [Google Scholar] [CrossRef]
- Verghese, P.B.; Castellano, J.M.; Garai, K.; Wang, Y.; Jiang, H.; Shah, A.; Bu, G.; Frieden, C.; Holtzman, D.M. ApoE influences amyloid-beta (Abeta) clearance despite minimal apoE/Abeta association in physiological conditions. Proc. Natl. Acad. Sci. USA 2013, 110, E1807–E1816. [Google Scholar] [CrossRef] [PubMed]
- Wirth, K.J.; Fink, E.; Rudolphi, K.; Heitsch, H.; Deutschlander, N.; Wiemer, G. Amyloid beta-(1-40) stimulates cyclic GMP production via release of kinins in primary cultured endothelial cells. Eur. J. Pharmacol. 1999, 382, 27–33. [Google Scholar] [CrossRef]
- Maimaiti, Y.; Su, T.; Zhang, Z.; Ma, L.; Zhang, Y.; Xu, H. NOX4-mediated astrocyte ferroptosis in Alzheimer’s disease. Cell Biosci. 2024, 14, 88. [Google Scholar] [CrossRef] [PubMed]
- Sewell, M.; Fialova, N.; Montagne, A. Unraveling the transcriptomic landscape of brain vascular cells in dementia: A systematic review. Alzheimer’s Dement. 2025, 21, e14512. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Lu, W.; Zhou, X.; Mu, J.; Shen, W. Unraveling Alzheimer’s disease: Insights from single-cell sequencing and spatial transcriptomic. Front. Neurol. 2024, 15, 1515981. [Google Scholar] [CrossRef]
- Vellimana, A.K.; Milner, E.; Azad, T.D.; Harries, M.D.; Zhou, M.L.; Gidday, J.M.; Han, B.H.; Zipfel, G.J. Endothelial nitric oxide synthase mediates endogenous protection against subarachnoid hemorrhage-induced cerebral vasospasm. Stroke 2011, 42, 776–782. [Google Scholar] [CrossRef]
- Naik, R.Y.; Foster, D.; Bray, P.; Chang, Y.; Han, B.H. Monocyte chemotactic protein-1-induced protein 1 contributes to neuronal injury following hypoxic-ischemia in the neonatal mouse brain. Neuroreport 2020, 31, 833–839. [Google Scholar] [CrossRef]
- Petiti, J.; Caria, S.; Revel, L.; Pegoraro, M.; Divieto, C. Standardized Protocol for Resazurin-Based Viability Assays on A549 Cell Line for Improving Cytotoxicity Data Reliability. Cells 2024, 13, 1959. [Google Scholar] [CrossRef]
- Shin, D.H.; Bae, Y.C.; Kim-Han, J.S.; Lee, J.H.; Choi, I.Y.; Son, K.H.; Kang, S.S.; Kim, W.K.; Han, B.H. Polyphenol amentoflavone affords neuroprotection against neonatal hypoxic-ischemic brain damage via multiple mechanisms. J. Neurochem. 2006, 96, 561–572. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heller, E.; McGurran, L.; Brown, J.K.; Love, K.; Hobbs, M.; Kim-Han, J.S.; Han, B.H. Aβ40 Improves Cerebrovascular Endothelial Function via NOX4-Dependent Hydrogen Peroxide Release. Int. J. Mol. Sci. 2025, 26, 6759. https://doi.org/10.3390/ijms26146759
Heller E, McGurran L, Brown JK, Love K, Hobbs M, Kim-Han JS, Han BH. Aβ40 Improves Cerebrovascular Endothelial Function via NOX4-Dependent Hydrogen Peroxide Release. International Journal of Molecular Sciences. 2025; 26(14):6759. https://doi.org/10.3390/ijms26146759
Chicago/Turabian StyleHeller, Elizabeth, Lindsey McGurran, Joseph K. Brown, Kathleen Love, Matthew Hobbs, Jeong Sook Kim-Han, and Byung Hee Han. 2025. "Aβ40 Improves Cerebrovascular Endothelial Function via NOX4-Dependent Hydrogen Peroxide Release" International Journal of Molecular Sciences 26, no. 14: 6759. https://doi.org/10.3390/ijms26146759
APA StyleHeller, E., McGurran, L., Brown, J. K., Love, K., Hobbs, M., Kim-Han, J. S., & Han, B. H. (2025). Aβ40 Improves Cerebrovascular Endothelial Function via NOX4-Dependent Hydrogen Peroxide Release. International Journal of Molecular Sciences, 26(14), 6759. https://doi.org/10.3390/ijms26146759