
Chemoresistance Evolution in Ovarian Cancer Delineated by Single-Cell RNA Sequencing

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
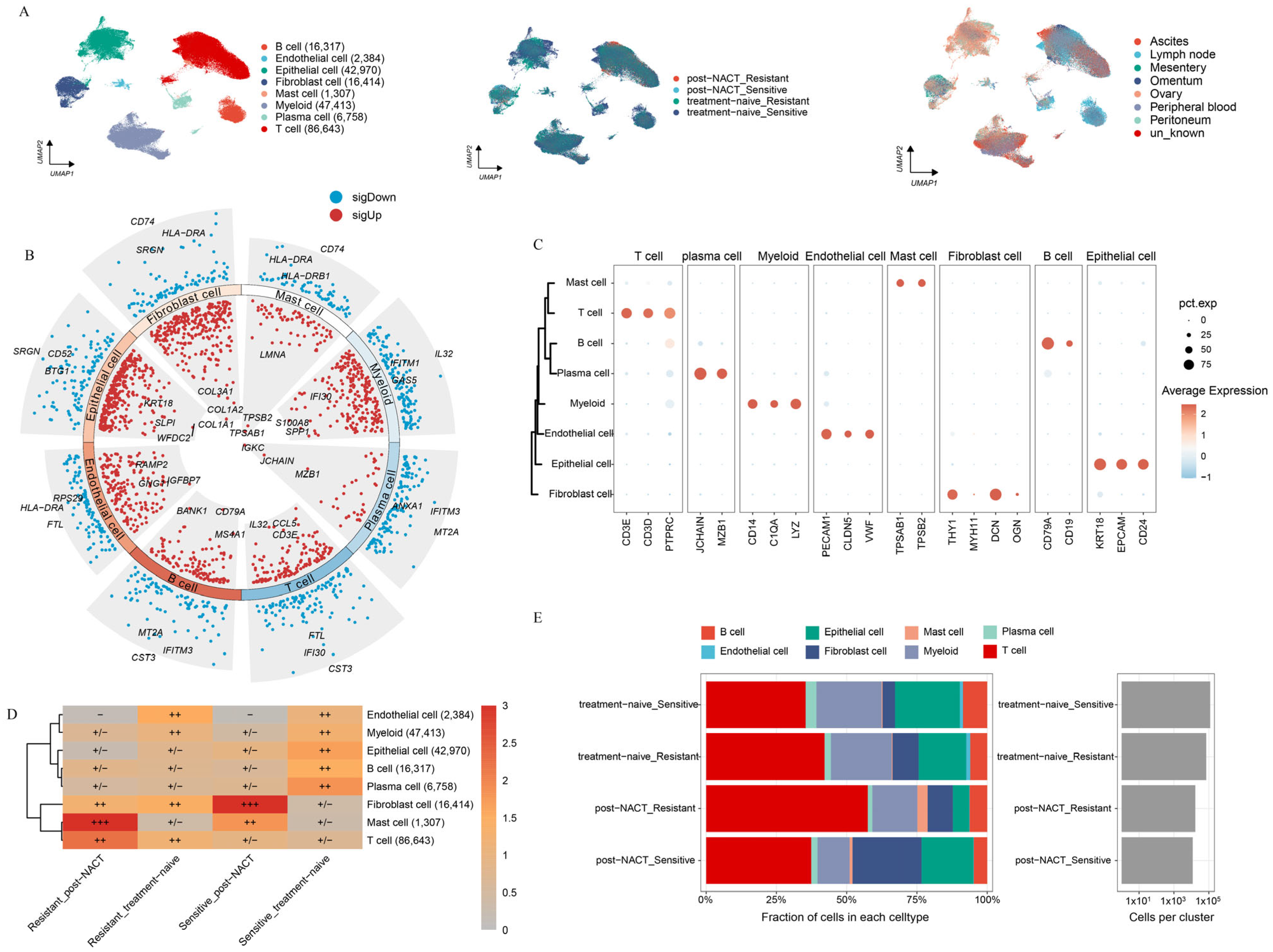
2.1. Landscape of HGSOC by Multiphase scRNA-seq
2.2. Heterogeneity Between Epithelial Cells in HGSOC
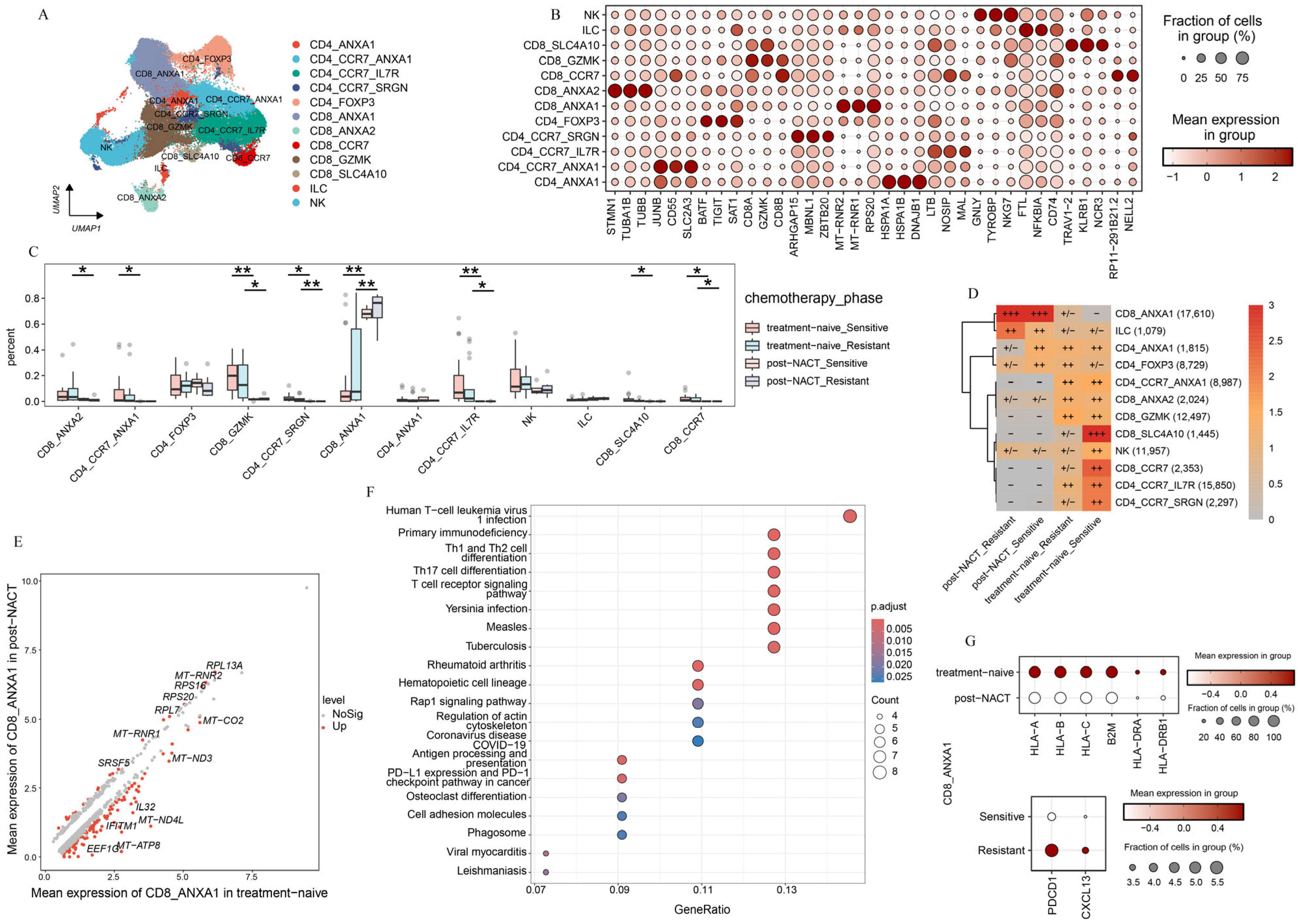
2.3. T-Cell Subsets Show Treatment-Specific Patterns
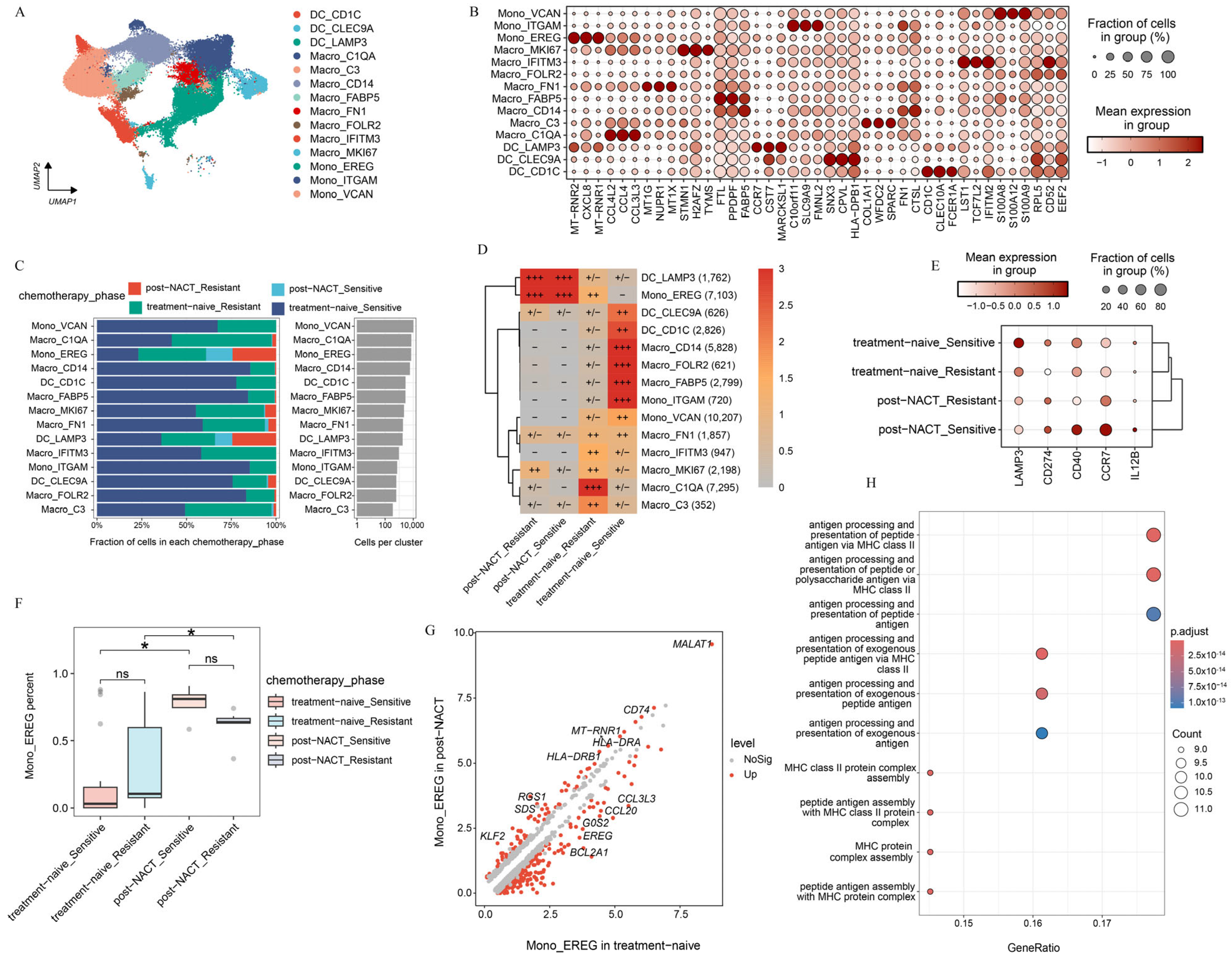
2.4. Myeloid Subsets Show Chemotherapy-Specific Patterns
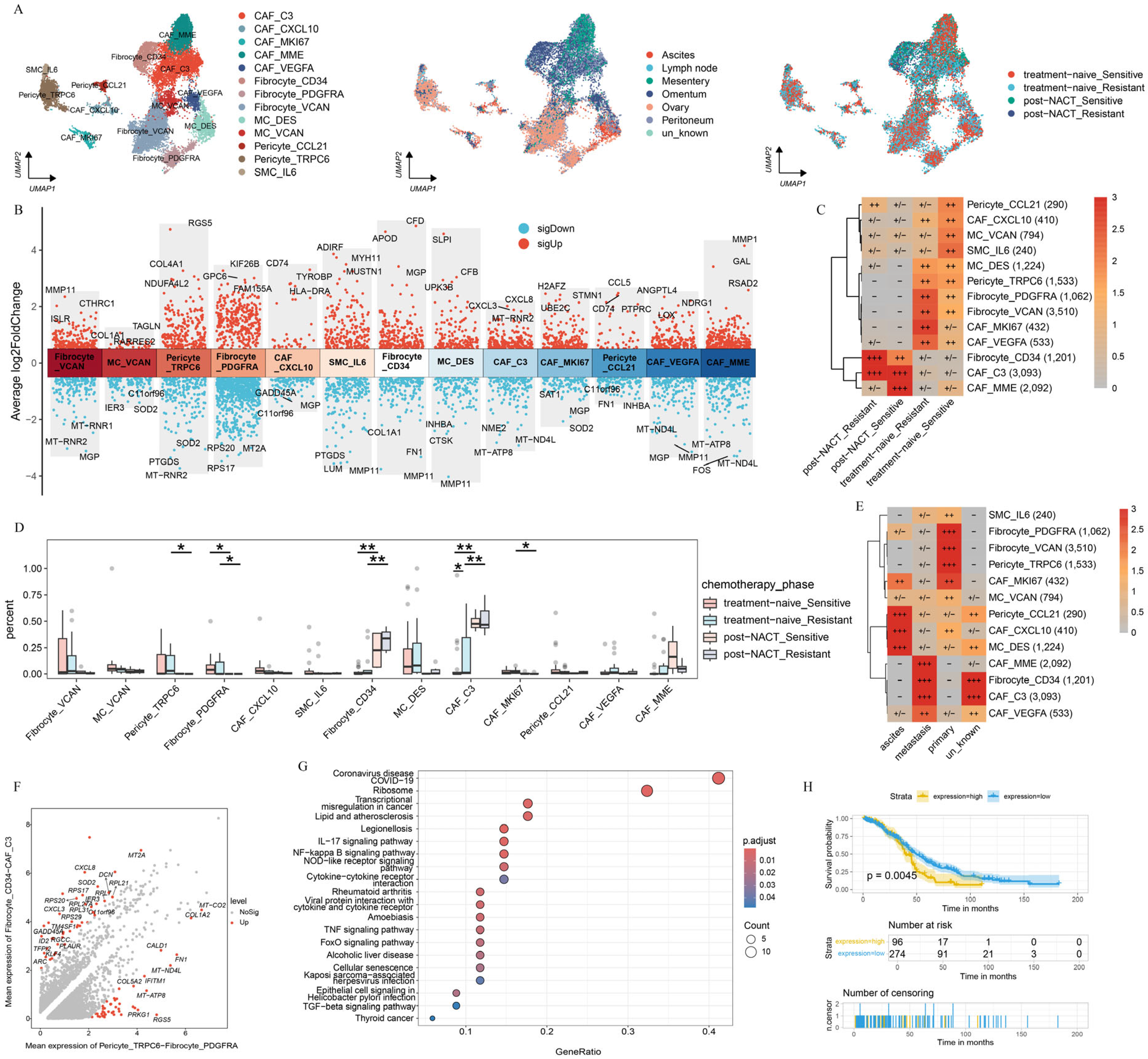
2.5. Chemotherapy Reshapes the Phenotype of Fibroblasts in HGSOC
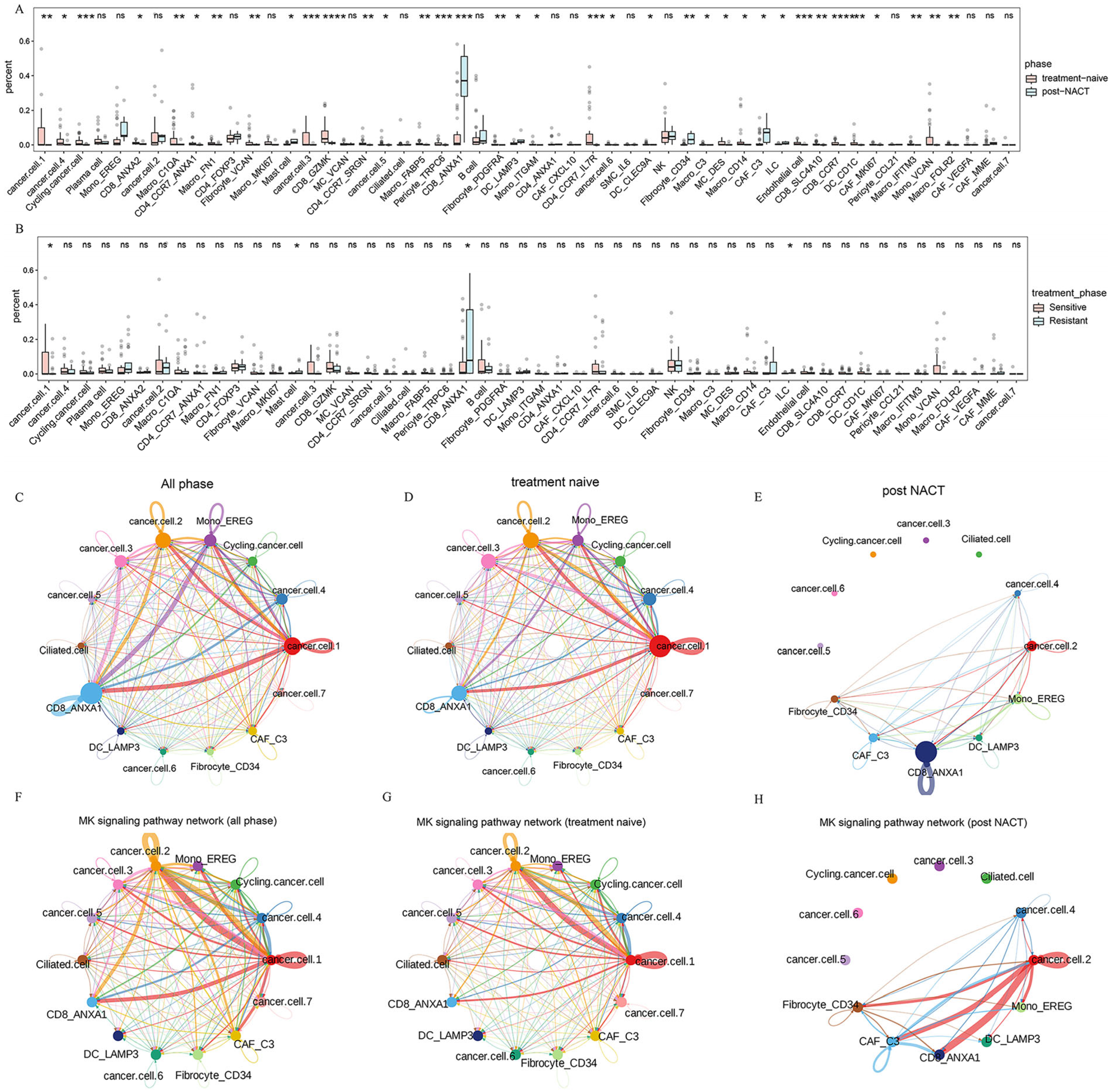
2.6. Transcriptome Reprogramming of Pre-Existing Cell Populations Following Chemotherapy
3. Discussion
4. Materials and Methods
4.1. Public Data Sources
4.2. Single-Cell Data Processing
4.3. Major Cell Type Identification
4.4. Single-Cell Copy Number Variation Analysis
4.5. Pseudotime Analysis
4.6. Differential Expression and Gene Ontology Enrichment Analysis
4.7. Survival Analysis
4.8. Cell–Cell Interaction Analysis
4.9. Statistics and Reproducibility
5. Conclusions
Clinical Perspectives
- A large amount of patients with HGSOC experience recurrence despite initial responsiveness to chemotherapy, suggesting that chemotherapy may reshape the tumor microenvironment.
- Chemotherapy makes a contribution to transcriptome reprogramming of pre-existing cell populations associated with reshaping of the tumor microenvironment.
- The combination of chemotherapy and immunotherapy, such as PD-1/PD-L1 therapy or MDK-NCL signaling suppression therapy, may effectively inhibit tumor progression and hold significant promise for HGSOC treatments.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HGSOC | high-grade serous ovarian cancer |
| TME | tumor microenvironment |
| NACT | Neoadjuvant chemotherapy |
| HLA | human leukocyte antigen |
| OC | Ovarian cancer |
| ScRNA-seq | single-cell RNA sequencing |
| GZMK | Granzyme K |
| DEG | different expressed gene |
| UMAP | uniform manifold approximation and projection |
| CXCL8 | C-X-C Motif Chemokine Ligand |
| PLEK | pleckstrin |
| CNV | cope number variation |
| VEGF | vascular endothelial growth factor |
| ILC | Innate Lymphoid Cell |
| DC | dendritic cell |
| CAF | cancer-associated fibroblast |
| MC | mesothelial cell |
| SMC | vascular smooth muscle cell |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| GSVA | gene set variation analysis |
| GEO | Gene Expression Omnibus |
| TCGA | The Cancer Genome Atlas |
| UMIs | unique molecular identifiers |
| PCs | principal components |
| SNN | shared nearest neighbor |
| Mono | monocytes |
References
- Zheng, X.; Wang, X.; Cheng, X.; Liu, Z.; Yin, Y.; Li, X.; Huang, Z.; Wang, Z.; Guo, W.; Ginhoux, F.; et al. Single-cell analyses implicate ascites in remodeling the ecosystems of primary and metastatic tumors in ovarian cancer. Nat. Cancer 2023, 4, 1138–1156. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Lheureux, S.; Braunstein, M.; Oza, A.M. Epithelial ovarian cancer: Evolution of management in the era of precision medicine. CA Cancer J. Clin. 2019, 69, 280–304. [Google Scholar] [CrossRef] [PubMed]
- McPherson, A.; Roth, A.; Laks, E.; Masud, T.; Bashashati, A.; Zhang, A.W.; Ha, G.; Biele, J.; Yap, D.; Wan, A.; et al. Divergent modes of clonal spread and intraperitoneal mixing in high-grade serous ovarian cancer. Nat. Genet. 2016, 48, 758–767. [Google Scholar] [CrossRef]
- Schwarz, R.F.; Ng, C.K.; Cooke, S.L.; Newman, S.; Temple, J.; Piskorz, A.M.; Gale, D.; Sayal, K.; Murtaza, M.; Baldwin, P.J.; et al. Spatial and temporal heterogeneity in high-grade serous ovarian cancer: A phylogenetic analysis. PLoS Med. 2015, 12, e1001789. [Google Scholar] [CrossRef]
- Izar, B.; Tirosh, I.; Stover, E.H.; Wakiro, I.; Cuoco, M.S.; Alter, I.; Rodman, C.; Leeson, R.; Su, M.J.; Shah, P.; et al. A single-cell landscape of high-grade serous ovarian cancer. Nat. Med. 2020, 26, 1271–1279. [Google Scholar] [CrossRef]
- Zhang, A.W.; McPherson, A.; Milne, K.; Kroeger, D.R.; Hamilton, P.T.; Miranda, A.; Funnell, T.; Little, N.; de Souza, C.P.E.; Laan, S.; et al. Interfaces of Malignant and Immunologic Clonal Dynamics in Ovarian Cancer. Cell 2018, 173, 1755–1769.e22. [Google Scholar] [CrossRef]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef]
- Liu, Y.L.; Selenica, P.; Zhou, Q.; Iasonos, A.; Callahan, M.; Feit, N.Z.; Boland, J.; Vazquez-Garcia, I.; Mandelker, D.; Zehir, A.; et al. BRCA Mutations, Homologous DNA Repair Deficiency, Tumor Mutational Burden, and Response to Immune Checkpoint Inhibition in Recurrent Ovarian Cancer. JCO Precis. Oncol. 2020, 4. [Google Scholar] [CrossRef]
- Monk, B.J.; Colombo, N.; Oza, A.M.; Fujiwara, K.; Birrer, M.J.; Randall, L.; Poddubskaya, E.V.; Scambia, G.; Shparyk, Y.V.; Lim, M.C.; et al. Chemotherapy with or without avelumab followed by avelumab maintenance versus chemotherapy alone in patients with previously untreated epithelial ovarian cancer (JAVELIN Ovarian 100): An open-label, randomised, phase 3 trial. Lancet Oncol. 2021, 22, 1275–1289. [Google Scholar] [CrossRef]
- Moore, K.N.; Bookman, M.; Sehouli, J.; Miller, A.; Anderson, C.; Scambia, G.; Myers, T.; Taskiran, C.; Robison, K.; Maenpaa, J.; et al. Atezolizumab, Bevacizumab, and Chemotherapy for Newly Diagnosed Stage III or IV Ovarian Cancer: Placebo-Controlled Randomized Phase III Trial (IMagyn050/GOG 3015/ENGOT-OV39). J. Clin. Oncol. 2021, 39, 1842–1855. [Google Scholar] [CrossRef] [PubMed]
- Ferri-Borgogno, S.; Zhu, Y.; Sheng, J.; Burks, J.K.; Gomez, J.A.; Wong, K.K.; Wong, S.T.C.; Mok, S.C. Spatial Transcriptomics Depict Ligand-Receptor Cross-talk Heterogeneity at the Tumor-Stroma Interface in Long-Term Ovarian Cancer Survivors. Cancer Res. 2023, 83, 1503–1516. [Google Scholar] [CrossRef] [PubMed]
- Cress, R.D.; Chen, Y.S.; Morris, C.R.; Petersen, M.; Leiserowitz, G.S. Characteristics of Long-Term Survivors of Epithelial Ovarian Cancer. Obstet. Gynecol. 2015, 126, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Hoppenot, C.; Eckert, M.A.; Tienda, S.M.; Lengyel, E. Who are the long-term survivors of high grade serous ovarian cancer? Gynecol. Oncol. 2018, 148, 204–212. [Google Scholar] [CrossRef]
- Baslan, T.; Hicks, J. Unravelling biology and shifting paradigms in cancer with single-cell sequencing. Nat. Rev. Cancer 2017, 17, 557–569. [Google Scholar] [CrossRef]
- Liu, B.; Hu, X.; Feng, K.; Gao, R.; Xue, Z.; Zhang, S.; Zhang, Y.; Corse, E.; Hu, Y.; Han, W.; et al. Temporal single-cell tracing reveals clonal revival and expansion of precursor exhausted T cells during anti-PD-1 therapy in lung cancer. Nat. Cancer 2022, 3, 108–121. [Google Scholar] [CrossRef]
- Zheng, L.; Qin, S.; Si, W.; Wang, A.; Xing, B.; Gao, R.; Ren, X.; Wang, L.; Wu, X.; Zhang, J.; et al. Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science 2021, 374, abe6474. [Google Scholar] [CrossRef]
- Zhang, K.; Erkan, E.P.; Jamalzadeh, S.; Dai, J.; Andersson, N.; Kaipio, K.; Lamminen, T.; Mansuri, N.; Huhtinen, K.; Carpen, O.; et al. Longitudinal single-cell RNA-seq analysis reveals stress-promoted chemoresistance in metastatic ovarian cancer. Sci. Adv. 2022, 8, eabm1831. [Google Scholar] [CrossRef]
- Kim, C.; Gao, R.; Sei, E.; Brandt, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N.E. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 2018, 173, 879–893.e13. [Google Scholar] [CrossRef]
- Hornburg, M.; Desbois, M.; Lu, S.; Guan, Y.; Lo, A.A.; Kaufman, S.; Elrod, A.; Lotstein, A.; DesRochers, T.M.; Munoz-Rodriguez, J.L.; et al. Single-cell dissection of cellular components and interactions shaping the tumor immune phenotypes in ovarian cancer. Cancer Cell 2021, 39, 928–944.e6. [Google Scholar] [CrossRef]
- Anadon, C.M.; Yu, X.; Hanggi, K.; Biswas, S.; Chaurio, R.A.; Martin, A.; Payne, K.K.; Mandal, G.; Innamarato, P.; Harro, C.M.; et al. Ovarian cancer immunogenicity is governed by a narrow subset of progenitor tissue-resident memory T cells. Cancer Cell 2022, 40, 545–557.e13. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Tayob, N.; Penter, L.; Sellars, M.; Tarren, A.; Chea, V.; Carulli, I.; Huang, T.; Li, S.; Cheng, S.C.; et al. Improved T-cell Immunity Following Neoadjuvant Chemotherapy in Ovarian Cancer. Clin. Cancer Res. 2022, 28, 3356–3366. [Google Scholar] [CrossRef] [PubMed]
- Nath, A.; Cosgrove, P.A.; Mirsafian, H.; Christie, E.L.; Pflieger, L.; Copeland, B.; Majumdar, S.; Cristea, M.C.; Han, E.S.; Lee, S.J.; et al. Evolution of core archetypal phenotypes in progressive high grade serous ovarian cancer. Nat. Commun. 2021, 12, 3039. [Google Scholar] [CrossRef] [PubMed]
- Webb, P.M.; Jordan, S.J. Global epidemiology of epithelial ovarian cancer. Nat. Rev. Clin. Oncol. 2024, 21, 389–400. [Google Scholar] [CrossRef]
- Ahmed, N.; Stenvers, K.L. Getting to know ovarian cancer ascites: Opportunities for targeted therapy-based translational research. Front Oncol. 2013, 3, 256. [Google Scholar] [CrossRef]
- Leung, C.S.; Yeung, T.L.; Yip, K.P.; Wong, K.K.; Ho, S.Y.; Mangala, L.S.; Sood, A.K.; Lopez-Berestein, G.; Sheng, J.; Wong, S.T.; et al. Cancer-associated fibroblasts regulate endothelial adhesion protein LPP to promote ovarian cancer chemoresistance. J. Clin. Investig. 2018, 128, 589–606. [Google Scholar] [CrossRef]
- Nguyen, Y.T.M.; Fujisawa, M.; Nguyen, T.B.; Suehara, Y.; Sakamoto, T.; Matsuoka, R.; Abe, Y.; Fukumoto, K.; Hattori, K.; Noguchi, M.; et al. Tet2 deficiency in immune cells exacerbates tumor progression by increasing angiogenesis in a lung cancer model. Cancer Sci. 2021, 112, 4931–4943. [Google Scholar] [CrossRef]
- Ha, H.; Debnath, B.; Neamati, N. Role of the CXCL8-CXCR1/2 Axis in Cancer and Inflammatory Diseases. Theranostics 2017, 7, 1543–1588. [Google Scholar] [CrossRef]
- Kieran, M.W.; Kalluri, R.; Cho, Y.J. The VEGF pathway in cancer and disease: Responses, resistance, and the path forward. Cold Spring Harb. Perspect. Med. 2012, 2, a006593. [Google Scholar] [CrossRef]
- Tao, Y.; Tian, C.; Qi, S.; Jia, Z.; Xu, Z.; Meng, J.; Xu, G.; Hu, H.; Wang, X.; Zhang, T.; et al. Targeting both death and paracaspase domains of MALT1 with antisense oligonucleotides overcomes resistance to immune-checkpoint inhibitors. Nat. Cancer 2025, 6, 702–717. [Google Scholar] [CrossRef]
- Maier, B.; Leader, A.M.; Chen, S.T.; Tung, N.; Chang, C.; LeBerichel, J.; Chudnovskiy, A.; Maskey, S.; Walker, L.; Finnigan, J.P.; et al. A conserved dendritic-cell regulatory program limits antitumour immunity. Nature 2020, 580, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; He, Y.; Luo, N.; Patel, S.J.; Han, Y.; Gao, R.; Modak, M.; Carotta, S.; Haslinger, C.; Kind, D.; et al. Landscape and Dynamics of Single Immune Cells in Hepatocellular Carcinoma. Cell 2019, 179, 829–845.e20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, Z.; Skrzypczynska, K.M.; Fang, Q.; Zhang, W.; O’Brien, S.A.; He, Y.; Wang, L.; Zhang, Q.; Kim, A.; et al. Single-Cell Analyses Inform Mechanisms of Myeloid-Targeted Therapies in Colon Cancer. Cell 2020, 181, 442–459.e29. [Google Scholar] [CrossRef] [PubMed]
- Tait Wojno, E.D.; Hunter, C.A.; Stumhofer, J.S. The Immunobiology of the Interleukin-12 Family: Room for Discovery. Immunity 2019, 50, 851–870. [Google Scholar] [CrossRef]
- Gao, L.; Ye, Z.; Peng, S.; Lei, P.; Song, P.; Li, Z.; Zhou, L.; Hua, Q.; Cheng, L.; Wei, H.; et al. BCL2A1 is associated with tumor-associated macrophages and unfavorable prognosis in human gliomas. Aging 2023, 15, 11611–11638. [Google Scholar] [CrossRef]
- Zhang, R.; Dong, M.; Tu, J.; Li, F.; Deng, Q.; Xu, J.; He, X.; Ding, J.; Xia, J.; Sheng, D.; et al. PMN-MDSCs modulated by CCL20 from cancer cells promoted breast cancer cell stemness through CXCL2-CXCR2 pathway. Signal Transduct. Target. Ther. 2023, 8, 97. [Google Scholar] [CrossRef]
- Cheng, W.L.; Feng, P.H.; Lee, K.Y.; Chen, K.Y.; Sun, W.L.; Van Hiep, N.; Luo, C.S.; Wu, S.M. The Role of EREG/EGFR Pathway in Tumor Progression. Int. J. Mol.Sci. 2021, 22, 12828. [Google Scholar] [CrossRef]
- Bonnin, E.; Rodrigo Riestra, M.; Marziali, F.; Mena Osuna, R.; Denizeau, J.; Maurin, M.; Saez, J.J.; Jouve, M.; Bonte, P.E.; Richer, W.; et al. CD74 supports accumulation and function of regulatory T cells in tumors. Nat. Commun. 2024, 15, 3749. [Google Scholar] [CrossRef]
- Chawla, S.; Rockstroh, A.; Lehman, M.; Ratther, E.; Jain, A.; Anand, A.; Gupta, A.; Bhattacharya, N.; Poonia, S.; Rai, P.; et al. Gene expression based inference of cancer drug sensitivity. Nat. Commun. 2022, 13, 5680. [Google Scholar] [CrossRef]
- Weigert, A.; Zheng, X.; Nenzel, A.; Turkowski, K.; Gunther, S.; Strack, E.; Sirait-Fischer, E.; Elwakeel, E.; Kur, I.M.; Nikam, V.S.; et al. Fibrocytes boost tumor-supportive phenotypic switches in the lung cancer niche via the endothelin system. Nat. Commun. 2022, 13, 6078. [Google Scholar] [CrossRef]
- Sun, X.; He, X.; Zhang, Y.; Hosaka, K.; Andersson, P.; Wu, J.; Wu, J.; Jing, X.; Du, Q.; Hui, X.; et al. Inflammatory cell-derived CXCL3 promotes pancreatic cancer metastasis through a novel myofibroblast-hijacked cancer escape mechanism. Gut 2022, 71, 129–147. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Liao, X.; Qiu, S.; Xu, H.; Zhang, S.; Wang, S.; Ai, J.; Yang, L. CXCL8 in Tumor Biology and Its Implications for Clinical Translation. Front. Mol. Biosci. 2022, 9, 723846. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Xie, L.; Ge, J.; Li, H.; Zhong, S.; Liu, X. Integrating single-cell RNA-seq and spatial transcriptomics reveals MDK-NCL dependent immunosuppressive environment in endometrial carcinoma. Front. Immunol. 2023, 14, 1145300. [Google Scholar] [CrossRef]
- Bresnick, A.R.; Weber, D.J.; Zimmer, D.B. S100 proteins in cancer. Nat. Rev. Cancer 2015, 15, 96–109. [Google Scholar] [CrossRef]
- Padilla, L.; Dakhel, S.; Adan, J.; Masa, M.; Martinez, J.M.; Roque, L.; Coll, T.; Hervas, R.; Calvis, C.; Llinas, L.; et al. S100A7: From mechanism to cancer therapy. Oncogene 2017, 36, 6749–6761. [Google Scholar] [CrossRef]
- Shapir Itai, Y.; Barboy, O.; Salomon, R.; Bercovich, A.; Xie, K.; Winter, E.; Shami, T.; Porat, Z.; Erez, N.; Tanay, A.; et al. Bispecific dendritic-T cell engager potentiates anti-tumor immunity. Cell 2024, 187, 375–389.e18. [Google Scholar] [CrossRef]
- Rached, L.; Laparra, A.; Sakkal, M.; Danlos, F.X.; Barlesi, F.; Carbonnel, F.; De Martin, E.; Ducreux, M.; Even, C.; Le Pavec, J.; et al. Toxicity of immunotherapy combinations with chemotherapy across tumor indications: Current knowledge and practical recommendations. Cancer Treat. Rev. 2024, 127, 102751. [Google Scholar] [CrossRef]
- Wang, Z.; Ji, X.; Zhang, Y.; Yang, F.; Su, H.; Zhang, H.; Li, Z.; Zhang, W.; Sun, W. Interactions between LAMP3+ dendritic cells and T-cell subpopulations promote immune evasion in papillary thyroid carcinoma. J. Immunother. Cancer 2024, 12, e008983. [Google Scholar] [CrossRef]
- Yu, X.; Xu, J. TWIST1 Drives Cytotoxic CD8+ T-Cell Exhaustion through Transcriptional Activation of CD274 (PD-L1) Expression in Breast Cancer Cells. Cancers 2024, 16, 1973. [Google Scholar] [CrossRef]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef]
- Filippou, P.S.; Karagiannis, G.S.; Constantinidou, A. Midkine (MDK) growth factor: A key player in cancer progression and a promising therapeutic target. Oncogene 2020, 39, 2040–2054. [Google Scholar] [CrossRef] [PubMed]
- Sordo-Bahamonde, C.; Lorenzo-Herrero, S.; Gonzalez-Rodriguez, A.P.; Martinez-Perez, A.; Rodrigo, J.P.; Garcia-Pedrero, J.M.; Gonzalez, S. Chemo-Immunotherapy: A New Trend in Cancer Treatment. Cancers 2023, 15, 2912. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Korsunsky, I.; Millard, N.; Fan, J.; Slowikowski, K.; Zhang, F.; Wei, K.; Baglaenko, Y.; Brenner, M.; Loh, P.R.; Raychaudhuri, S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 2019, 16, 1289–1296. [Google Scholar] [CrossRef]
- Qiu, X.; Mao, Q.; Tang, Y.; Wang, L.; Chawla, R.; Pliner, H.A.; Trapnell, C. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 2017, 14, 979–982. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Hanzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Jin, S.; Guerrero-Juarez, C.F.; Zhang, L.; Chang, I.; Ramos, R.; Kuan, C.H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021, 12, 1088. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Tang, Z.; Li, H.; Zhou, R.; Wu, H.; Cen, X.; Zhang, Y.; Dong, W.; Yang, H. Chemoresistance Evolution in Ovarian Cancer Delineated by Single-Cell RNA Sequencing. Int. J. Mol. Sci. 2025, 26, 6760. https://doi.org/10.3390/ijms26146760
Wang Y, Tang Z, Li H, Zhou R, Wu H, Cen X, Zhang Y, Dong W, Yang H. Chemoresistance Evolution in Ovarian Cancer Delineated by Single-Cell RNA Sequencing. International Journal of Molecular Sciences. 2025; 26(14):6760. https://doi.org/10.3390/ijms26146760
Chicago/Turabian StyleWang, Yuanmei, Zongfu Tang, Haoyu Li, Run Zhou, Hao Wu, Xiaoping Cen, Yi Zhang, Wei Dong, and Huanming Yang. 2025. "Chemoresistance Evolution in Ovarian Cancer Delineated by Single-Cell RNA Sequencing" International Journal of Molecular Sciences 26, no. 14: 6760. https://doi.org/10.3390/ijms26146760
APA StyleWang, Y., Tang, Z., Li, H., Zhou, R., Wu, H., Cen, X., Zhang, Y., Dong, W., & Yang, H. (2025). Chemoresistance Evolution in Ovarian Cancer Delineated by Single-Cell RNA Sequencing. International Journal of Molecular Sciences, 26(14), 6760. https://doi.org/10.3390/ijms26146760