
Transcriptome Analysis Reveals Metabolic Pathways and Key Genes Involved in Oleic Acid Formation of Sunflower (Helianthus annuus L.)


Abstract
1. Introduction
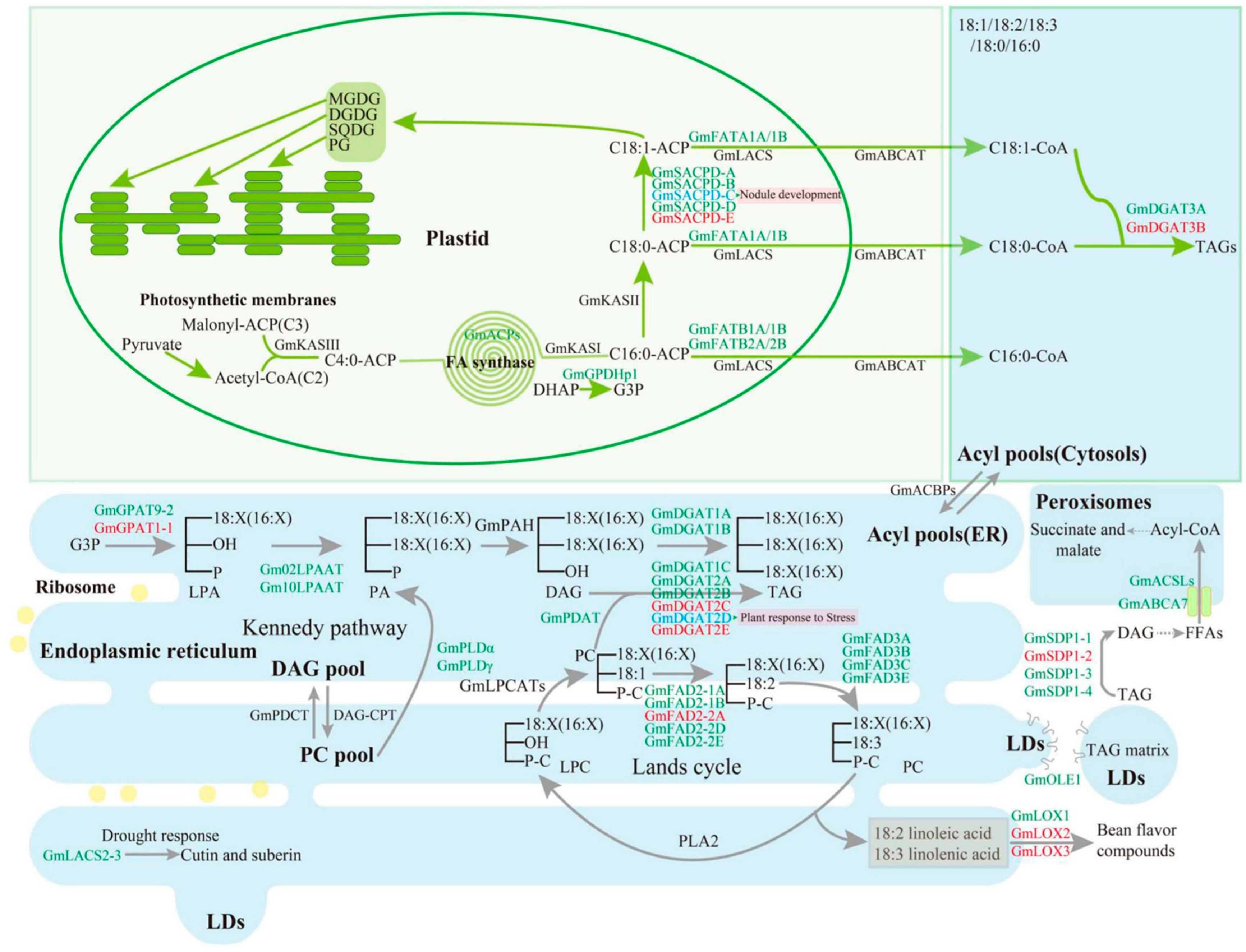
2. Results
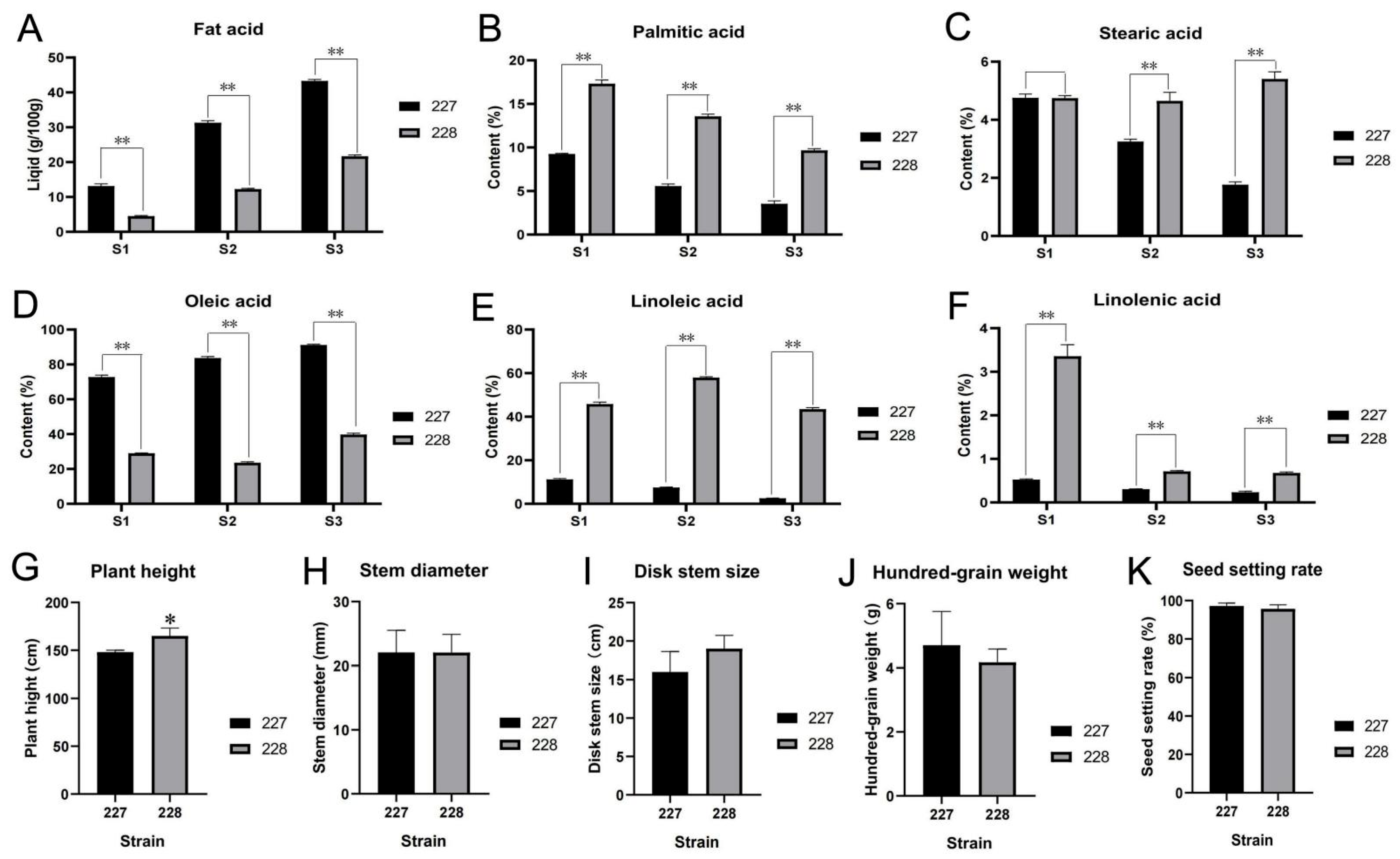
2.1. Analysis of Fatty Acid Content
2.2. Sequencing Quantity, Quality, and New Transcripts
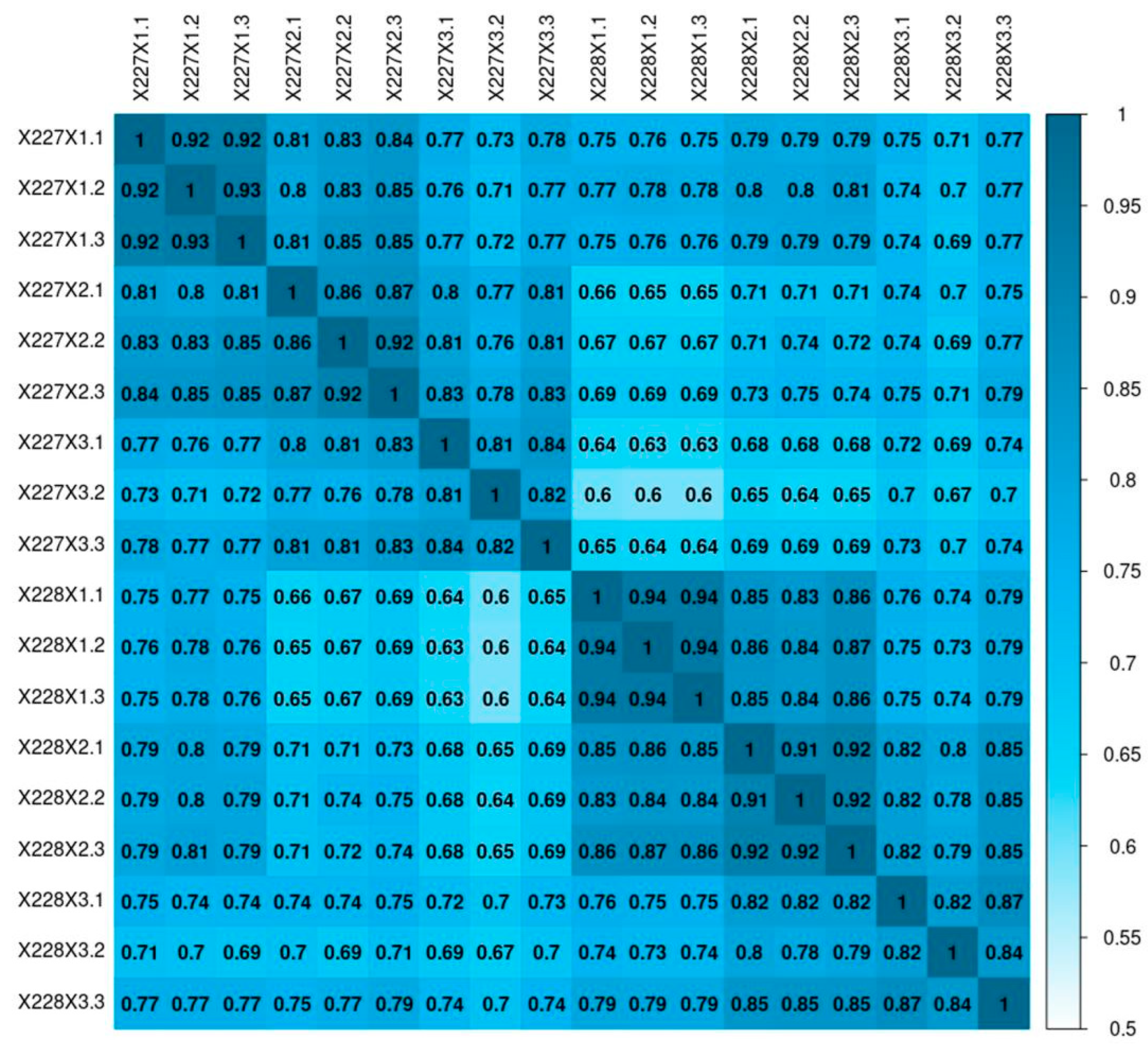
2.3. RNA-Seq Sequencing Repeatability Test
2.4. Gene Expression Level Analysis
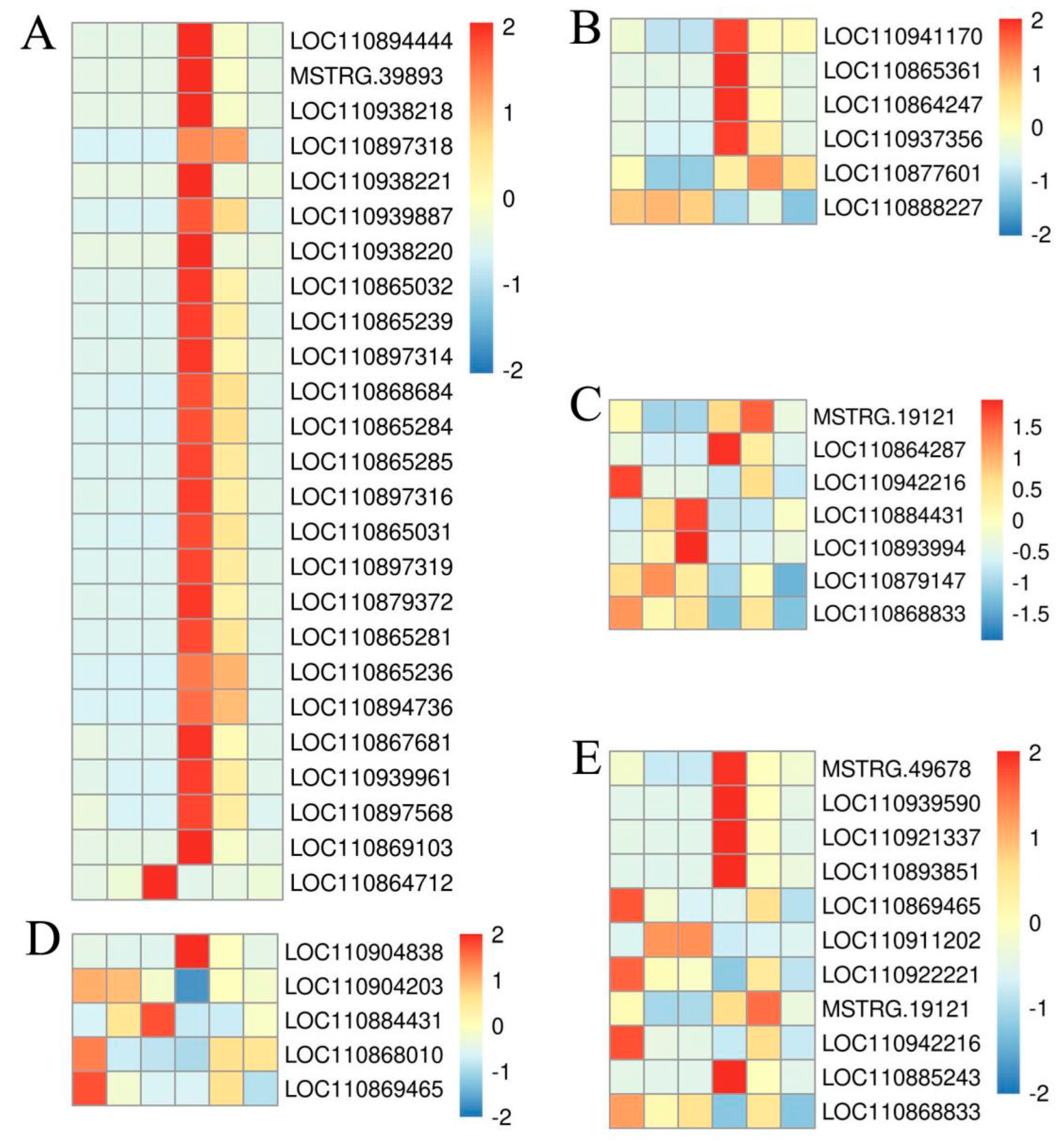
2.5. Detection and Cluster Analysis of Differential Genes
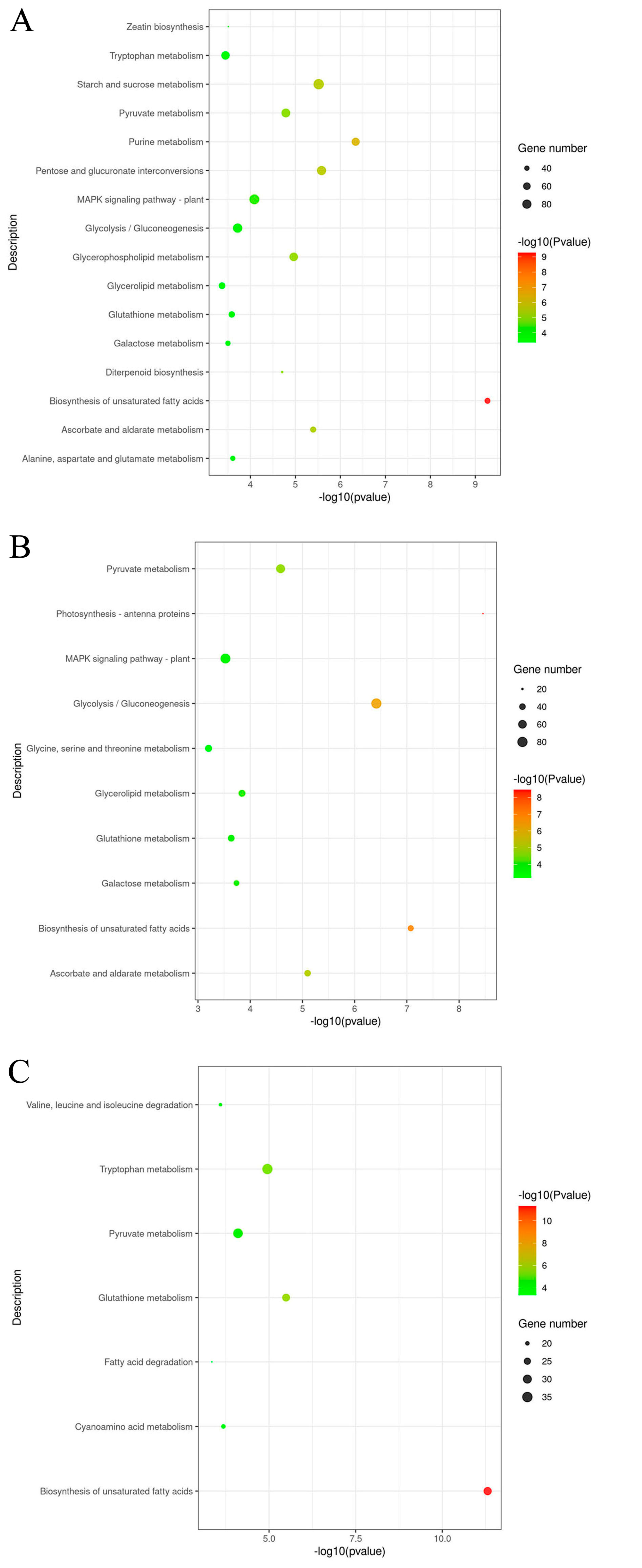
2.6. The Functional Annotation of DEGs
2.7. The Screening and Analysis of DEGs
2.8. The GO Mapping of DEGs Significantly Enriched Pathways
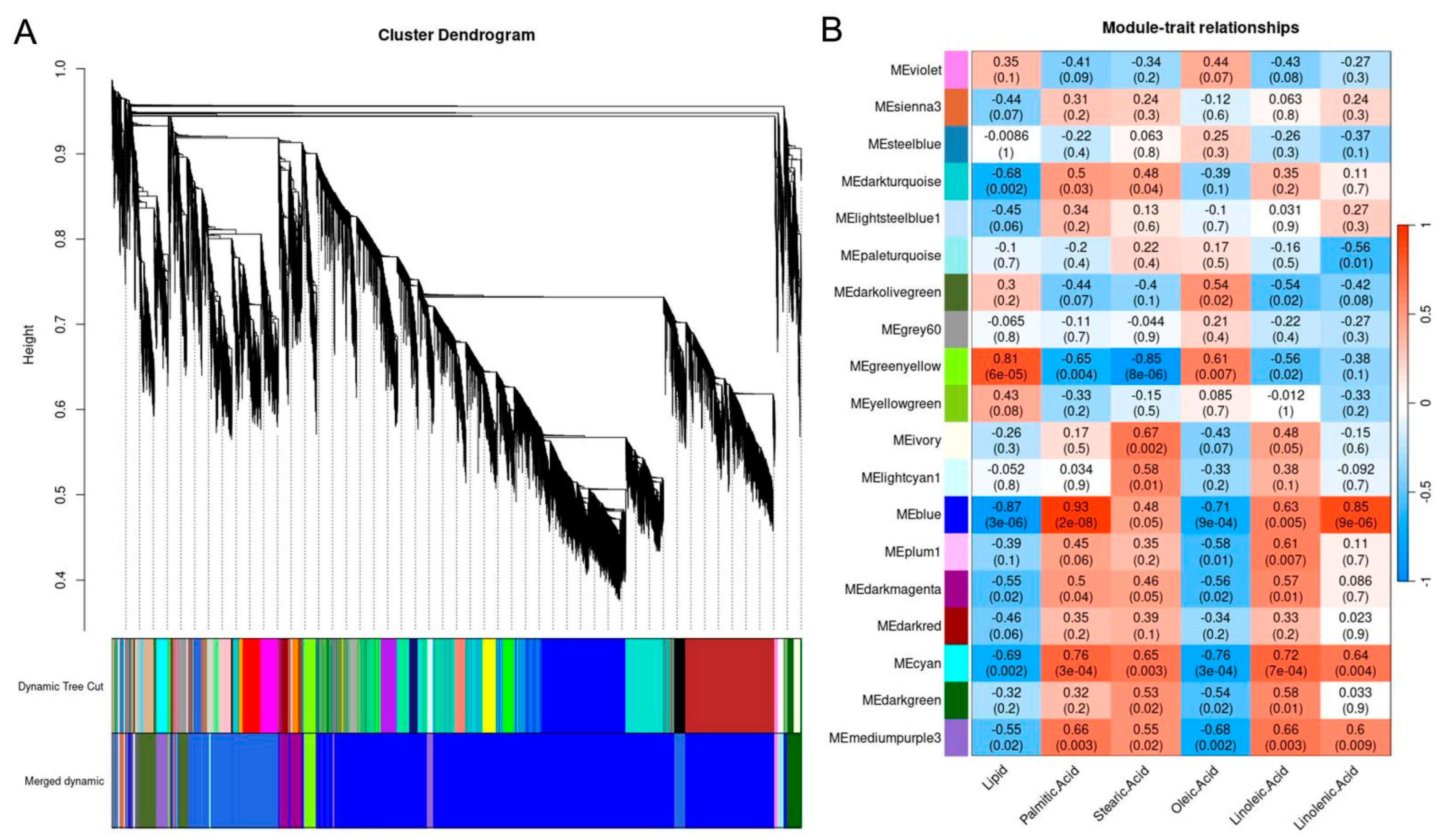
2.9. Co-Expression Network Analysis
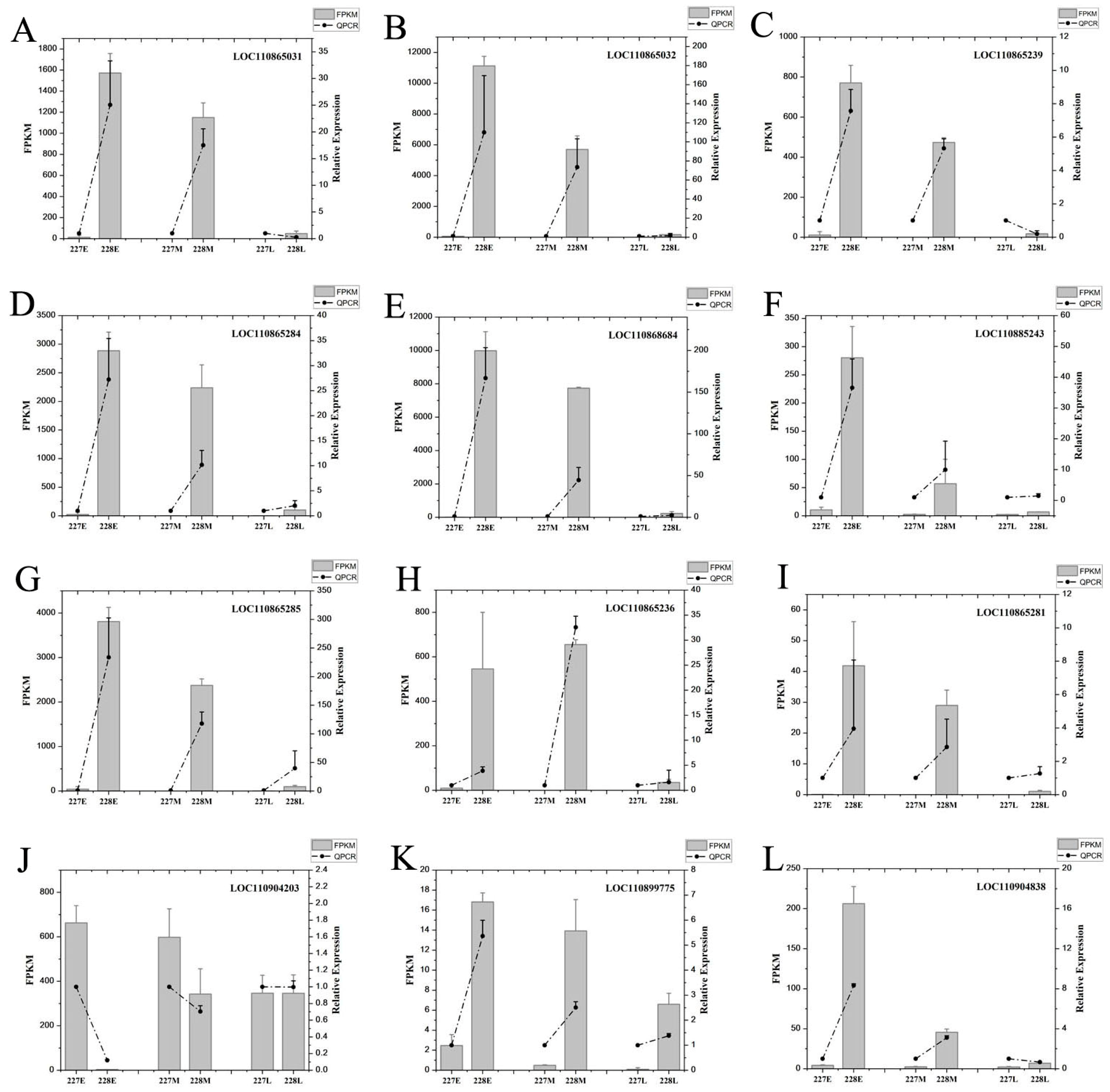
2.10. qRT-PCR Validation
3. Discussion
4. Materials and Methods
4.1. Sunflower Plant Materials
4.2. Fatty Acid Composition and Content Determination
4.3. Sample Collection
4.4. Determination of Fats and Fatty Acids
4.5. RNA-Seq
4.6. RT-qPCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shingfield, K.J.; Reynolds, C.K.; Hervás, G.; Griinari, J.M.; Grandison, A.S.; Beever, D.E. Examination of the persistency of milk fatty acid composition responses to fish oil and sunflower oil in the diet of dairy cows. J. Dairy Sci. 2006, 89, 714–732. [Google Scholar] [CrossRef]
- Zhou, F.; Liu, Y.; Liang, C.; Wang, W.; Li, C.; Guo, Y.; Ma, J.; Yu, Y.; Fan, L.; Yao, Y.; et al. Construction of a high-density genetic linkage map and QTL mapping of oleic acid content and three agronomic traits in sunflower (Helianthus annuus L.) using specific-locus amplified fragment sequencing (SLAF-seq). Breed. Sci. 2018, 68, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Wang, J.; Liu, C.; Lu, X.; Chen, W.; Chen, C.; Yang, J.; Zheng, L. Discrimination of kernel quality characteristics for sunflower seeds based on multispectral imaging approach. Food Anal. Methods 2015, 8, 1629–1636. [Google Scholar] [CrossRef]
- Bates, P.D. Understanding the control of acyl flux through the lipid metabolic network of plant oil biosynthesis. Biochim. Biophys. Acta 2016, 1861, 1214–1225. [Google Scholar] [CrossRef]
- Miray, R.; Kazaz, S.; To, A.; Baud, S. Molecular Control of Oil Metabolism in the Endosperm of Seeds. Int. J. Mol. Sci. 2021, 22, 1621. [Google Scholar] [CrossRef]
- Browse, J.; Somerville, C. Glycerolipid Synthesis: Biochemistry and Regulation. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1991, 42, 467–506. [Google Scholar] [CrossRef]
- Román, Á.; Andreu, V.; Hernández, M.L.; Lagunas, B.; Picorel, R.; Martínez-Rivas, J.M.; Alfonso, M. Contribution of the different omega-3 fatty acid desaturase genes to the cold response in soybean. J. Exp. Bot. 2012, 63, 4973–4982. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sicilia, A.; Testa, G.; Santoro, D.F.; Cosentino, S.L.; Lo Piero, A.R. RNASeq analysis of giant cane reveals the leaf transcriptome dynamics under long-term salt stress. BMC Plant Biol. 2019, 19, 1–24. [Google Scholar] [CrossRef]
- Santoro, D.F.; Sicilia, A.; Testa, G.; Cosentino, S.L.; Lo Piero, A.R. Global leaf and root transcriptome in response to cadmium reveals tolerance mechanisms in Arundo donax L. BMC Genom. 2022, 23, 427. [Google Scholar] [CrossRef]
- Sicilia, A.; Santoro, D.F.; Testa, G.; Cosentino, S.L.; Lo Piero, A.R. Transcriptional response of giant reed (Arundo donax L.) low ecotype to long-term salt stress by unigene-based RNAseq. Phytochemistry 2020, 177, 112436. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, N.; Zhao, L.; Zhu, H.; Tang, C. Transcriptome analysis reveals the defense mechanism of cotton against Verticillium dahliae in the presence of the biocontrol fungus Chaetomium globosum CEF-082. BMC Plant Biol. 2020, 20, 89. [Google Scholar] [CrossRef]
- Sun, Y.; Yang, N.; Li, S.; Chen, F.; Xie, Y.; Tang, C. Mechanism of oxalate decarboxylase Oxd_S12 from Bacillus velezensis BvZ45-1 in defence against cotton verticillium wilt. J. Exp. Bot. 2024, 75, 3500–3520. [Google Scholar] [CrossRef]
- Dai, Y.; Zhang, L.; Sun, X.; Li, F.; Zhang, S.; Zhang, H.; Li, G.; Fang, Z.; Sun, R.; Hou, X.; et al. Transcriptome analysis reveals anthocyanin regulation in Chinese cabbage (Brassica rapa L.) at low temperatures. Sci. Rep. 2022, 12, 6308–6322. [Google Scholar] [CrossRef]
- Song, H.; Taylor, D.C.; Zhang, M. Bioengineering of Soybean Oil and Its Impact on Agronomic Traits. Int. J. Mol. Sci. 2023, 24, 2256. [Google Scholar] [CrossRef]
- Connor, W.E. Importance of n-3 fatty acids in health and disease. Am. J. Clin. Nutr. 2000, 71, 171S–175S. [Google Scholar] [CrossRef]
- Zhao, Y.P.; Liang, W.; Wang, D.; Wang, Y.M.; Liu, Z.J.; Cui, Y.P.; Hua, J.P. Research progress on regulation of plant oil synthesis and genetic improvement. China Agric. Sci. Technol. Guide 2018, 20, 14–24. [Google Scholar]
- Xu, Z.; Li, J.; Guo, X.; Jin, S.; Zhang, X. Metabolic engineering of cottonseed oil biosynthesis pathway via RNA interference. Sci. Rep. 2016, 6, 33342. [Google Scholar] [CrossRef]
- Cui, Y.; Liu, Z.; Zhao, Y.; Wang, Y.; Huang, Y.; Li, L.; Wu, H.; Xu, S.; Hua, J. Overexpression of heteromeric GhACCase subunits enhanced oil accumulation in Upland cotton. Plant Mol. Biol. Rep. 2017, 35, 287–297. [Google Scholar] [CrossRef]
- Liu, L. Gene expression Profile Analysis of Cotton Fiber Secondary Wall Thickening Stage and Functional Study of GhKAR, GhHAD, GhENR. Ph.D. Thesis, China Agricultural University, Beijing, China, 2015; pp. 51–71. [Google Scholar]
- Pidkowich, M.S.; Nguyen, H.T.; Heilmann, I.; Ischebeck, T.; Shanklin, J. Modulating seed beta-ketoacyl-acyl carrier protein synthase II level converts the composition of a temperate seed oil to that of a palm-like tropical oil. Proc. Natl. Acad. Sci. USA 2007, 104, 4742–4747. [Google Scholar] [CrossRef]
- Ozseyhan, M.E.; Li, P.; Na, G.; Li, Z.; Wang, C.; Lu, C. Improved fatty acid profiles in seeds of Camelina sativa by artificial microRNA mediated FATB gene suppression. Biochem. Biophys. Res. Commun. 2018, 503, 621–624. [Google Scholar] [CrossRef]
- Shockey, J.; Regmi, A.; Cotton, K.; Adhikari, N.; Browse, J.; Bates, P.D. Identification of Arabidopsis GPAT9 (At5g60620) as an essential gene involved in triacylglycerol biosynthesis. Plant Physiol. 2016, 170, 163–179. [Google Scholar] [CrossRef] [PubMed]
- Jako, C.; Kumar, A.; Wei, Y.; Zou, J.; Barton, D.L.; Giblin, E.M.; Covello, P.S.; Taylor, D.C. Seed-specific over- expression of an Arabidopsis cDNA encoding a diacylglycerel acyltransferase enhances seed oil content and seed weight. Plant Physiol. 2001, 126, 861–874. [Google Scholar] [CrossRef]
- Zhou, F.; Huang, X.T.; Liang, C.B.; Li, C.; Wang, W.J.; Ma, J.; Liu, Y.; Guo, Y.L. Cloning and Expression Analysis of Fatty Acid Dehydrogenase Gene FAD2 in Sunflowers for Oil Use. Heilongjiang Agric. Sci. 2016, 8–12. [Google Scholar]
- Guan, L.-L.; Xu, Y.-W.; Wang, Y.-B.; Chen, L.; Shao, J.-F.; Wu, W. Isolation and Characterization of a Temperature-Regulated Microsomal Oleate Desaturase Gene (CtFAD2-1) from Safflower (Carthamus tinctorius L.). Plant Mol. Biol. Report. 2011, 30, 391–402. [Google Scholar] [CrossRef]
- Suresha, G.S.; Rai, R.D.; Santha, I.M. Molecular cloning, expression analysis and growth temperature dependent regulation of a novel oleate desaturase gene (fad2) homologue from Brassica juncea. Aust. J. Crop Sci. 2012, 6, 296–308. [Google Scholar]
- Bhunia, R.K.; Kaur, R.; Mrinal, K.; Maiti, M.K. Metabolic engineering of fatty acid biosynthetic pathway in sesame (Sesamum indicum L.): Assembling tools to develop nutritionally desirable sesame seed oil. Phytochem. Rev. 2016, 15, 799–811. [Google Scholar] [CrossRef]
- Niu, E.; Gao, S.; Hu, W.; Zhang, C.; Liu, D.; Shen, G.; Zhu, S. Genome-Wide Identification and Functional Differentiation of Fatty Acid Desaturase Genes in Olea europaea L. Plants 2022, 11, 1415. [Google Scholar] [CrossRef]
- Dar, A.A.; Choudhury, A.R.; Kancharla, P.K.; Arumugam, N. The FAD2 Gene in Plants: Occurrence, Regulation, and Role. Front. Plant Sci. 2017, 8, 1789. [Google Scholar] [CrossRef]
- Luisa Hernández, M.; Dolores Sicardo, M.; Arjona, P.M.; Martínez-Rivas, J.M. Specialized Functions of Olive FAD2 Gene Family Members Related to Fruit Development and the Abiotic Stress Response. Plant Cell Physiol. 2020, 61, 427–441. [Google Scholar] [CrossRef]
- Long, W.; Hu, M.; Gao, J.; Chen, S.; Zhang, J.; Cheng, L.; Pu, H. Identification and Functional Analysis of Two New Mutant BnFAD2 Alleles That Confer Elevated Oleic Acid Content in Rapeseed. Front. Genet. 2018, 9, 399. [Google Scholar] [CrossRef]
- Okuzaki, A.; Ogawa, T.; Koizuka, C.; Kaneko, K.; Inaba, M.; Imamura, J.; Koizuka, N. CRISPR/Cas9-mediated genome editing of the fatty acid desaturase 2 gene in Brassica napus. Plant Physiol. Biochem. 2018, 131, 63–69. [Google Scholar] [CrossRef]
- Do, P.T.; Nguyen, C.X.; Bui, H.T.; Tran, L.T.N.; Stacey, G.; Gillman, J.D.; Zhang, Z.J.; Stacey, M.G. Demonstration of highly efficient dual gRNA CRISPR/Cas9 editing of the homeologous GmFAD2-1A and GmFAD2-1B genes to yield a high oleic, low linoleic and α-linolenic acid phenotype in soybean. BMC Plant Biol. 2019, 19, 311. [Google Scholar] [CrossRef]
- Lakhssassi, N.; Lopes-Caitar, V.S.; Knizia, D.; Cullen, M.A.; Badad, O.; El Baze, A.; Zhou, Z.; Embaby, M.G.; Meksem, J.; Lakhssassi, A.; et al. TILLING-by-Sequencing+ Reveals the Role of Novel Fatty Acid Desaturases (GmFAD2-2s) in Increasing Soybean Seed Oleic Acid Content. Cells 2021, 10, 1245. [Google Scholar] [CrossRef]
- Lakhssassi, N.; Zhou, Z.; Liu, S.; Colantonio, V.; AbuGhazaleh, A.; Meksem, K. Characterization of the FAD2 Gene Family in Soybean Reveals the Limitations of Gel-Based TILLING in Genes with High Copy Number. Front. Plant Sci. 2017, 8, 324. [Google Scholar] [CrossRef]
- Taylor, D.C.; Zhang, Y.; Kumar, A.; Francis, T.; Giblin, E.M.; Barton, D.L.; Ferrie, J.R.; Laroche, A.; Shah, S.; Zhu, W.; et al. Molecular modification of triacylglycerol accumulation by over-expression of DGAT1 to produce canola with increased seed oil content under field conditions. Botany 2009, 87, 533–543. [Google Scholar] [CrossRef]
- Alagna, F.; D’Agostino, N.; Torchia, L.; Servili, M.; Rao, R.; Pietrella, M.; Giuliano, G.; Chiusano, M.L.; Baldoni, L.; Perrotta, G. Comparative 454 pyrosequencing of transcripts from two olive genotypes during fruit development. BMC Genom. 2009, 10, 399. [Google Scholar] [CrossRef]
- Bourgis, F.; Kilaru, A.; Cao, X.; Ngando-Ebongue, G.F.; Drira, N.; Ohlrogge, J.B.; Arondel, V. Comparative transcriptome and metabolite analysis of oil palm and date palm mesocarp that differ dramatically in carbon partitioning. Proc. Natl. Acad. Sci. USA 2011, 108, 12527–12532. [Google Scholar] [CrossRef]
- Oakes, J.; Brackenridge, D.; Colletti, R.; Daley, M.; Hawkins, D.J.; Xiong, H.; Mai, J.; Screen, S.E.; Val, D.; Lardizabal, K.; et al. Expression of fungal diacylglycerol acyltransferase2 genes to increase kernel oil in maize. Plant Physiol. 2011, 155, 1146–1157. [Google Scholar] [CrossRef]
- Singer, S.D.; Chen, G.; Mietkiewska, E.; Tomasi, P.; Jayawardhane, K.; Dyer, J.M.; Weselake, R.J. Arabidopsis GPAT9 contributes to synthesis of intracellular glycerolipids but not surface lipids. J. Exp. Bot. 2016, 67, 4627–4638. [Google Scholar] [CrossRef]
- Wallis, J.G.; Bengtsson, J.D.; Browse, J. Molecular Approaches Reduce Saturates and Eliminate trans Fats in Food Oils. Front. Plant Sci. 2022, 13, 908608. [Google Scholar] [CrossRef]
- Bai, Y.; Shen, Y.; Zhang, Z.; Jia, Q.; Xu, M.; Zhang, T.; Fang, H.; Yu, X.; Li, L.; Liu, D.; et al. A GPAT1 Mutation in Arabidopsis Enhances Plant Height but Impairs Seed Oil Biosynthesis. Int. J. Mol. Sci. 2021, 22, 785. [Google Scholar] [CrossRef] [PubMed]
- Aznar, M.J.A.; Durrett, T.P. Simultaneous Targeting of Multiple Gene Homeologs to Alter Seed Oil Production in Camelina sativa. Plant Cell Physiol. 2017, 58, 1260–1267. [Google Scholar] [CrossRef] [PubMed]
- Jessen, D.; Olbrich, A.; Knüfer, J.; Krüger, A.; Hoppert, M.; Polle, A.; Fulda, M. Combined activity of LACS1 and LACS4 is required for proper pollen coat formation in Arabidopsis. Plant J. 2011, 68, 715–726. [Google Scholar] [CrossRef]
- Yu, L.; Tan, X.; Jiang, B.; Sun, X.; Gu, S.; Han, T.; Hou, W. A peroxisomal long-chain acyl-CoA synthetase from Glycine max involved in lipid degradation. PLoS ONE 2014, 9, e100144. [Google Scholar] [CrossRef] [PubMed]
- GB5009.6-2016; National Food Safety Standard—Determination of Fat in Foods. China Food and Drug Administration: Beijing, China, 2016.
- GB5009.168-2016; National Food Safety Standard—Determination of Fatty Acids in Foods. National Health and Family Planning Commission of the People’s Republic of China: Beijing, China, 2016.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Ontology | Description | Count | p-Value | Q-Value |
|---|---|---|---|---|---|
| GO:0045485 | MF | omega-6 fatty acid desaturase activity | 18 | 9.66 × 10−11 | 8.67 × 10−8 |
| GO:0035251 | MF | UDP-glucosyltransferase activity | 106 | 3.38 × 10−10 | 1.51 × 10−7 |
| GO:0080043 | MF | quercetin 3-O-glucosyltransferase activity | 75 | 7.74 × 10−10 | 1.74 × 10−7 |
| mGO:0080044 | MF | quercetin 7-O-glucosyltransferase activity | 75 | 7.74 × 10−10 | 1.74 × 10−7 |
| GO:0046527 | MF | glucosyltransferase activity | 126 | 5.69 × 10−9 | 1.02 × 10−6 |
| GO:0008194 | MF | UDP-glycosyltransferase activity | 131 | 5.46 × 10−8 | 8.17 × 10−6 |
| GO:0016705 | MF | oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen | 137 | 2.18 × 10−7 | 2.79 × 10−5 |
| GO:0009739 | BP | response to gibberellin | 110 | 1.59 × 10−8 | 5.59 × 10−5 |
| GO:0080046 | MF | quercetin 4’-O-glucosyltransferase activity | 22 | 1.73 × 10−5 | 0.001939238 |
| GO:0045544 | MF | gibberellin 20-oxidase activity | 11 | 2.26 × 10−5 | 0.002252498 |
| ID | Ontology | Description | Count | p-Value | Q-Value |
|---|---|---|---|---|---|
| GO:0045485 | MF | omega-6 fatty acid desaturase activity | 16 | 5.98 × 10−10 | 5.33 × 10−7 |
| GO:0005875 | CC | microtubule-associated complex | 58 | 1.68 × 10−7 | 7.72 × 10−5 |
| GO:0031409 | MF | pigment binding | 14 | 5.97 × 10−7 | 0.000266049 |
| GO:0009768 | BP | photosynthesis, light harvesting in photosystem I | 15 | 1.35 × 10−7 | 0.000464913 |
| GO:0008194 | MF | UDP-glycosyltransferase activity | 102 | 2.61 × 10−6 | 0.00046543 |
| GO:0016168 | MF | chlorophyll binding | 14 | 2.42 × 10−6 | 0.00046543 |
| GO:0035251 | MF | UDP-glucosyltransferase activity | 78 | 1.71 × 10−6 | 0.00046543 |
| GO:0046527 | MF | glucosyltransferase activity | 95 | 3.33 × 10−6 | 0.000494834 |
| GO:0046906 | MF | tetrapyrrole binding | 24 | 1.13 × 10−5 | 0.001435517 |
| GO:0003777 | MF | microtubule motor activity | 42 | 2.45 × 10−5 | 0.002235062 |
| ID | Ontology | Description | Count | p-Value | Q-Value |
|---|---|---|---|---|---|
| GO:0045485 | MF | omega-6 fatty acid desaturase activity ω-6 | 16 | 3.50 × 10−16 | 2.69 × 10−13 |
| GO:0035251 | MF | UDP-glucosyltransferase activity | 45 | 2.77 × 10−8 | 1.06 × 10−5 |
| GO:0050183 | MF | phosphatidylcholine 12-monooxygenase activity | 8 | 1.52 × 10−7 | 3.90 × 10−5 |
| GO:0016208 | MF | AMP binding | 10 | 3.03 × 10−7 | 5.01 × 10−5 |
| GO:0046527 | MF | glucosyltransferase activity | 51 | 3.26 × 10−7 | 5.01 × 10−5 |
| GO:0008194 | MF | UDP-glycosyltransferase activity | 53 | 7.86 × 10−7 | 0.000100833 |
| GO:0080043 | MF | quercetin 3-O-glucosyltransferase activity | 29 | 6.59 × 10−6 | 0.000633808 |
| GO:0080044 | MF | quercetin 7-O-glucosyltransferase activity | 29 | 6.59 × 10−6 | 0.000633808 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mu, Y.; Sun, Y.; Wu, Y.; Yi, L.; Yu, H.; Zhang, S. Transcriptome Analysis Reveals Metabolic Pathways and Key Genes Involved in Oleic Acid Formation of Sunflower (Helianthus annuus L.). Int. J. Mol. Sci. 2025, 26, 6757. https://doi.org/10.3390/ijms26146757
Mu Y, Sun Y, Wu Y, Yi L, Yu H, Zhang S. Transcriptome Analysis Reveals Metabolic Pathways and Key Genes Involved in Oleic Acid Formation of Sunflower (Helianthus annuus L.). International Journal of Molecular Sciences. 2025; 26(14):6757. https://doi.org/10.3390/ijms26146757
Chicago/Turabian StyleMu, Yingnan, Ying Sun, Yang Wu, Liuxi Yi, Haifeng Yu, and Shaoying Zhang. 2025. "Transcriptome Analysis Reveals Metabolic Pathways and Key Genes Involved in Oleic Acid Formation of Sunflower (Helianthus annuus L.)" International Journal of Molecular Sciences 26, no. 14: 6757. https://doi.org/10.3390/ijms26146757
APA StyleMu, Y., Sun, Y., Wu, Y., Yi, L., Yu, H., & Zhang, S. (2025). Transcriptome Analysis Reveals Metabolic Pathways and Key Genes Involved in Oleic Acid Formation of Sunflower (Helianthus annuus L.). International Journal of Molecular Sciences, 26(14), 6757. https://doi.org/10.3390/ijms26146757