
Ferroptosis in Toxicology: Present and Future

Abstract
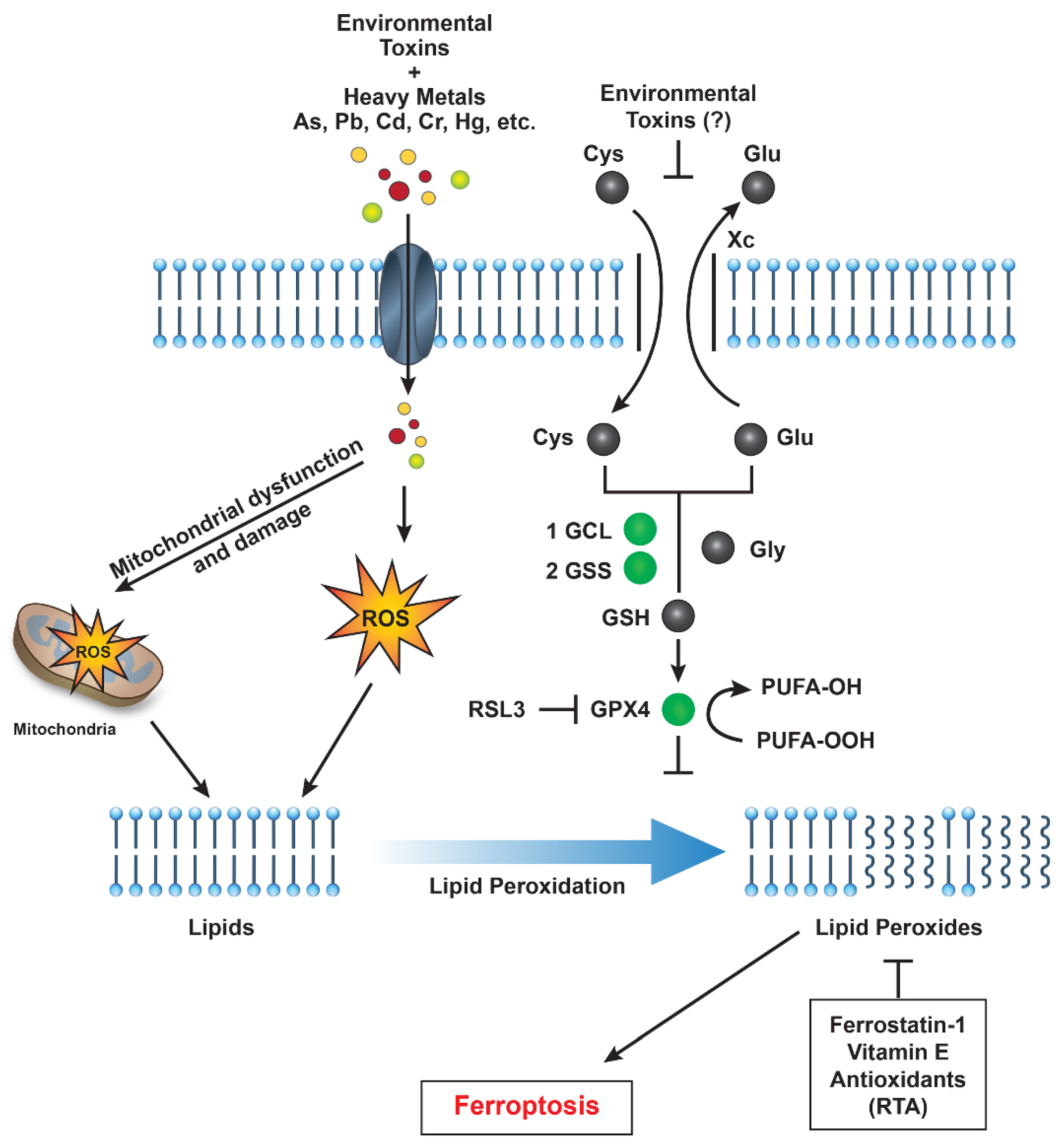
1. Introduction
1.1. Perfluoroalkyl and Polyfluoroalkyl Substances (PFAS)
1.2. Heavy Metals
1.3. Industrial Pollutants and Airborne Particulates
1.4. Pesticides
1.5. Ferroptosis Combined with Apoptosis and Necrosis
1.6. Ferroptosis and Apoptosis Crosslinks
1.7. Ferroptosis–Necrosis Crosslinks
1.8. Biomarkers of Ferroptosis
1.9. Ferroptosis Inhibitors in Therapeutic and Preventive Measures
2. Conclusions and Future Developments
3. Summary
Funding
Acknowledgments
Conflicts of Interest
References
- Sinha, B.K. Roles of free radicals in the toxicity of environmental pollutants and toxicants. J. Clin. Toxicol. 2013, S13, e001. [Google Scholar] [CrossRef]
- Halliwell, B. Commentary for “Oxygen free radicals and iron in relation to biology and medicine: Some problems and concepts”. Arch. Biochem. Biophys. 2022, 718, 109151. [Google Scholar] [CrossRef] [PubMed]
- Sinha, B.K.; Mimnaugh, E.G. Free radicals and anticancer drug resistance: Oxygen free radicals in the mechanisms of drug cytotoxicity and resistance by certain tumors. Free Radic. Biol. Med. 1990, 8, 567–581. [Google Scholar] [CrossRef] [PubMed]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 2014, 3, e02523. [Google Scholar] [CrossRef]
- Aschner, M.; Skalny, A.V.; Martins, A.C.; Sinitskii, A.I.; Farina, M.; Lu, R.; Barbosa, F., Jr.; Gluhcheva, Y.G.; Santamaria, A.; Tinkov, A.A. Ferroptosis as a mechanism of non-ferrous metal toxicity. Arch. Toxicol. 2022, 96, 2391–2417. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Zhao, B.; Zhou, L.; Zhang, Z.; Shen, Y.; Lv, H.; AlQudsy, L.H.H.; Shang, P. Ferroptosis, a novel pharmacological mechanism of anti-cancer drugs. Cancer Lett. 2020, 483, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Reichert, C.O.; de Freitas, F.A.; Sampaio-Silva, J.; Rokita-Rosa, L.; Barros, P.L.; Levy, D.; Bydlowski, S.P. Ferroptosis Mechanisms Involved in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8765. [Google Scholar] [CrossRef] [PubMed]
- Peleman, C.; Francque, S.; Berghe, T.V. Emerging role of ferroptosis in metabolic dysfunction-associated steatotic liver disease: Revisiting hepatic lipid peroxidation. EBioMedicine 2024, 102, 105088. [Google Scholar] [CrossRef]
- Sahoo, K.; Sharma, A. Understanding the mechanistic roles of environmental heavy metal stressors in regulating ferroptosis: Adding new paradigms to the links with diseases. Apoptosis 2023, 28, 277–292. [Google Scholar] [CrossRef]
- Yang, L.; Cai, X.; Li, R. Ferroptosis Induced by Pollutants: An Emerging Mechanism in Environmental Toxicology. Environ. Sci. Technol. 2024, 58, 2166–2184. [Google Scholar] [CrossRef]
- Podder, A.; Sadmani, A.; Reinhart, D.; Chang, N.B.; Goel, R. Per and poly-fluoroalkyl substances (PFAS) as a contaminant of emerging concern in surface water: A transboundary review of their occurrences and toxicity effects. J. Hazard. Mater. 2021, 419, 126361. [Google Scholar] [CrossRef]
- Sunderland, E.M.; Hu, X.C.; Dassuncao, C.; Tokranov, A.K.; Wagner, C.C.; Allen, J.G. A review of the pathways of human exposure to poly- and perfluoroalkyl substances (PFASs) and present understanding of health effects. J. Expo. Sci. Environ. Epidemiol. 2019, 29, 131–147. [Google Scholar] [CrossRef]
- Li, M.; Ma, Y.; Cheng, W.; Zhang, L.; Zhou, C.; Zhang, W.; Zhang, W. Association between perfluoroalkyl and polyfluoroalkyl internal exposure and serum alpha-Klotho levels in middle-old aged participants. Front. Public Health 2023, 11, 1136454. [Google Scholar] [CrossRef]
- Zhang, Z.; Sarkar, D.; Biswas, J.K.; Datta, R. Biodegradation of per- and polyfluoroalkyl substances (PFAS): A review. Bioresour. Technol. 2022, 344, 126223. [Google Scholar] [CrossRef]
- Stanifer, J.W.; Stapleton, H.M.; Souma, T.; Wittmer, A.; Zhao, X.; Boulware, L.E. Perfluorinated Chemicals as Emerging Environmental Threats to Kidney Health: A Scoping Review. Clin. J. Am. Soc. Nephrol. 2018, 13, 1479–1492. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Arellano, P.; Lopez-Arellano, K.; Luna, J.; Flores, D.; Jimenez-Salazar, J.; Gavia, G.; Teteltitla, M.; Rodriguez, J.J.; Dominguez, A.; Casas, E.; et al. Perfluorooctanoic acid disrupts gap junction intercellular communication and induces reactive oxygen species formation and apoptosis in mouse ovaries. Environ. Toxicol. 2019, 34, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Li, H.; Lin, C.; Mao, Y.; Rao, J.; Lou, Y.; Yang, X.; Xu, X.; Jin, F. Perfluorooctanoic acid (PFOA) inhibits the gap junction intercellular communication and induces apoptosis in human ovarian granulosa cells. Reprod. Toxicol. 2020, 98, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Evich, M.G.; Davis, M.J.B.; McCord, J.P.; Acrey, B.; Awkerman, J.A.; Knappe, D.R.U.; Lindstrom, A.B.; Speth, T.F.; Tebes-Stevens, C.; Strynar, M.J.; et al. Per- and polyfluoroalkyl substances in the environment. Science 2022, 375, eabg9065. [Google Scholar] [CrossRef]
- Wielsoe, M.; Long, M.; Ghisari, M.; Bonefeld-Jorgensen, E.C. Perfluoroalkylated substances (PFAS) affect oxidative stress biomarkers in vitro. Chemosphere 2015, 129, 239–245. [Google Scholar] [CrossRef]
- Lin, C.Y.; Lee, H.L.; Hwang, Y.T.; Su, T.C. The association between total serum isomers of per- and polyfluoroalkyl substances, lipid profiles, and the DNA oxidative/nitrative stress biomarkers in middle-aged Taiwanese adults. Environ. Res. 2020, 182, 109064. [Google Scholar] [CrossRef]
- Kim, J.H.; Park, H.Y.; Jeon, J.D.; Kho, Y.; Kim, S.K.; Park, M.S.; Hong, Y.C. The modifying effect of vitamin C on the association between perfluorinated compounds and insulin resistance in the Korean elderly: A double-blind, randomized, placebo-controlled crossover trial. Eur. J. Nutr. 2016, 55, 1011–1020. [Google Scholar] [CrossRef]
- Arukwe, A.; Mortensen, A.S. Lipid peroxidation and oxidative stress responses of salmon fed a diet containing perfluorooctane sulfonic- or perfluorooctane carboxylic acids. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2011, 154, 288–295. [Google Scholar] [CrossRef]
- Wen, L.L.; Lin, C.Y.; Chou, H.C.; Chang, C.C.; Lo, H.Y.; Juan, S.H. Perfluorooctanesulfonate Mediates Renal Tubular Cell Apoptosis through PPARgamma Inactivation. PLoS ONE 2016, 11, e0155190. [Google Scholar] [CrossRef]
- Gorrochategui, E.; Lacorte, S.; Tauler, R.; Martin, F.L. Perfluoroalkylated Substance Effects in Xenopus laevis A6 Kidney Epithelial Cells Determined by ATR-FTIR Spectroscopy and Chemometric Analysis. Chem. Res. Toxicol. 2016, 29, 924–932. [Google Scholar] [CrossRef]
- Mohan, V.e.a. Effectivemess of N-acetylcysteine and Taurine in reducing microalbuminuria in diabetic and hypertensive pateints with chronic kidney diseases: A retrospective, observational study. Wolrd J. Pharmeceutical Sci. Res. 2025, 4, 265–273. [Google Scholar] [CrossRef]
- Brown-Leung, J.M.; Cannon, J.R. Neurotransmission Targets of Per- and Polyfluoroalkyl Substance Neurotoxicity: Mechanisms and Potential Implications for Adverse Neurological Outcomes. Chem. Res. Toxicol. 2022, 35, 1312–1333. [Google Scholar] [CrossRef] [PubMed]
- Bharal, B.; Ruchitha, C.; Kumar, P.; Pandey, R.; Rachamalla, M.; Niyogi, S.; Naidu, R.; Kaundal, R.K. Neurotoxicity of per- and polyfluoroalkyl substances: Evidence and future directions. Sci. Total Environ. 2024, 955, 176941. [Google Scholar] [CrossRef] [PubMed]
- Starnes, H.M.; Rock, K.D.; Jackson, T.W.; Belcher, S.M. A Critical Review and Meta-Analysis of Impacts of Per- and Polyfluorinated Substances on the Brain and Behavior. Front. Toxicol. 2022, 4, 881584. [Google Scholar] [CrossRef]
- Zheng, F.; Goncalves, F.M.; Abiko, Y.; Li, H.; Kumagai, Y.; Aschner, M. Redox toxicology of environmental chemicals causing oxidative stress. Redox Biol. 2020, 34, 101475. [Google Scholar] [CrossRef]
- Mansoor, S.; Ali, A.; Kour, N.; Bornhorst, J.; AlHarbi, K.; Rinklebe, J.; Abd El Moneim, D.; Ahmad, P.; Chung, Y.S. Heavy Metal Induced Oxidative Stress Mitigation and ROS Scavenging in Plants. Plants 2023, 12, 3003. [Google Scholar] [CrossRef]
- Xue, Q.; Yan, D.; Chen, X.; Li, X.; Kang, R.; Klionsky, D.J.; Kroemer, G.; Chen, X.; Tang, D.; Liu, J. Copper-dependent autophagic degradation of GPX4 drives ferroptosis. Autophagy 2023, 19, 1982–1996. [Google Scholar] [CrossRef]
- Zhao, T.; Yu, Z.; Zhou, L.; Wang, X.; Hui, Y.; Mao, L.; Fan, X.; Wang, B.; Zhao, X.; Sun, C. Regulating Nrf2-GPx4 axis by bicyclol can prevent ferroptosis in carbon tetrachloride-induced acute liver injury in mice. Cell Death Discov. 2022, 8, 380. [Google Scholar] [CrossRef]
- Samet, J.M.; Wages, P.A. Oxidative Stress from Environmental Exposures. Curr. Opin. Toxicol. 2018, 7, 60–66. [Google Scholar] [CrossRef]
- Anetor, J.I. Rising environmental cadmium levels in developing countries: Threat to genome stability and health. Niger. J. Physiol. Sci. 2012, 27, 103–115. [Google Scholar] [CrossRef]
- Das, N.; Paul, S.; Chatterjee, D.; Banerjee, N.; Majumder, N.S.; Sarma, N.; Sau, T.J.; Basu, S.; Banerjee, S.; Majumder, P.; et al. Arsenic exposure through drinking water increases the risk of liver and cardiovascular diseases in the population of West Bengal, India. BMC Public Health 2012, 12, 639. [Google Scholar] [CrossRef] [PubMed]
- McCormack, M.C.; Breysse, P.N.; Hansel, N.N.; Matsui, E.C.; Tonorezos, E.S.; Curtin-Brosnan, J.; Williams, D.L.; Buckley, T.J.; Eggleston, P.A.; Diette, G.B. Common household activities are associated with elevated particulate matter concentrations in bedrooms of inner-city Baltimore pre-school children. Environ. Res. 2008, 106, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Surawski, N.C.; Miljevic, B.; Ayoko, G.A.; Elbagir, S.; Stevanovic, S.; Fairfull-Smith, K.E.; Bottle, S.E.; Ristovski, Z.D. Physicochemical characterization of particulate emissions from a compression ignition engine: The influence of biodiesel feedstock. Environ. Sci. Technol. 2011, 45, 10337–10343. [Google Scholar] [CrossRef] [PubMed]
- Leikauf, G.D.; Kim, S.H.; Jang, A.S. Mechanisms of ultrafine particle-induced respiratory health effects. Exp. Mol. Med. 2020, 52, 329–337. [Google Scholar] [CrossRef]
- Doiron, D.; de Hoogh, K.; Probst-Hensch, N.; Fortier, I.; Cai, Y.; De Matteis, S.; Hansell, A.L. Air pollution, lung function and COPD: Results from the population-based UK Biobank study. Eur. Respir. J. 2019, 54, 1802140. [Google Scholar] [CrossRef]
- Hou, L.; Huang, R.; Sun, F.; Zhang, L.; Wang, Q. NADPH oxidase regulates paraquat and maneb-induced dopaminergic neurodegeneration through ferroptosis. Toxicology 2019, 417, 64–73. [Google Scholar] [CrossRef]
- Shukla, S.; Singh, D.; Kumar, V.; Chauhan, A.K.; Singh, S.; Ahmad, I.; Pandey, H.P.; Singh, C. NADPH oxidase mediated maneb- and paraquat-induced oxidative stress in rat polymorphs: Crosstalk with mitochondrial dysfunction. Pestic. Biochem. Physiol. 2015, 123, 74–86. [Google Scholar] [CrossRef]
- Chowdhury, A.R.; Zielonka, J.; Kalyanaraman, B.; Hartley, R.C.; Murphy, M.P.; Avadhani, N.G. Mitochondria-targeted paraquat and metformin mediate ROS production to induce multiple pathways of retrograde signaling: A dose-dependent phenomenon. Redox Biol. 2020, 36, 101606. [Google Scholar] [CrossRef]
- Zuo, Y.; Xie, J.; Li, X.; Li, Y.; Thirupathi, A.; Zhang, J.; Yu, P.; Gao, G.; Chang, Y.; Shi, Z. Ferritinophagy-Mediated Ferroptosis Involved in Paraquat-Induced Neurotoxicity of Dopaminergic Neurons: Implication for Neurotoxicity in PD. Oxid. Med. Cell. Longev. 2021, 2021, 9961628. [Google Scholar] [CrossRef]
- Wu, S.F.; Ga, Y.; Ma, D.Y.; Hou, S.L.; Hui, Q.Y.; Hao, Z.H. The role of ferroptosis in environmental pollution-induced male reproductive system toxicity. Environ. Pollut. 2024, 363, 125118. [Google Scholar] [CrossRef]
- Chia, S.J.; Tan, E.K.; Chao, Y.X. Historical Perspective: Models of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 2464. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, E.R.; Ray, A. Paraquat, Parkinson’s Disease, and Agnotology. Mov. Disord. 2023, 38, 949–952. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, U.; Srivastava, M.K.; Trivedi, P.; Garg, V.; Srivastava, L.P. Disposition and acute toxicity of imidacloprid in female rats after single exposure. Food Chem. Toxicol. 2014, 68, 190–195. [Google Scholar] [CrossRef]
- Zhang, D.; Wu, C.; Ba, D.; Wang, N.; Wang, Y.; Li, X.; Li, Q.; Zhao, G. Ferroptosis contribute to neonicotinoid imidacloprid-evoked pyroptosis by activating the HMGB1-RAGE/TLR4-NF-kappaB signaling pathway. Ecotoxicol. Environ. Saf. 2023, 253, 114655. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Sheng, J.; Pei, H.; Sheng, Y.; Wang, J.; Zhou, X.; Li, W.; Cao, C.; Yang, Y. Environmental toxin chlorpyrifos induces liver injury by activating P53-mediated ferroptosis via GSDMD-mtROS. Ecotoxicol. Environ. Saf. 2023, 257, 114938. [Google Scholar] [CrossRef]
- Sen Gupta, P.; Karmakar, S.; Biswas, I.; Ghosal, J.; Banerjee, A.; Roy, S.; Mandal, D.P.; Bhattacharjee, S. Vitamin E alleviates chlorpyrifos induced glutathione depletion, lipid peroxidation and iron accumulation to inhibit ferroptosis in hepatocytes and mitigate toxicity in zebrafish. Chemosphere 2024, 359, 142252. [Google Scholar] [CrossRef]
- Mesmar, F.; Muhsen, M.; Mirchandani, R.; Tourigny, J.P.; Tennessen, J.M.; Bondesson, M. The herbicide acetochlor causes lipid peroxidation by inhibition of glutathione peroxidase activity. Toxicol. Sci. 2024, 202, 302–313. [Google Scholar] [CrossRef]
- Huang, Y.C.; Li, Z.; Harder, S.D.; Soukup, J.M. Apoptotic and inflammatory effects induced by different particles in human alveolar macrophages. Inhal. Toxicol. 2004, 16, 863–878. [Google Scholar] [CrossRef]
- Chopra, M.; Schrenk, D. Dioxin toxicity, aryl hydrocarbon receptor signaling, and apoptosis-persistent pollutants affect programmed cell death. Crit. Rev. Toxicol. 2011, 41, 292–320. [Google Scholar] [CrossRef]
- Kang, R.; Kroemer, G.; Tang, D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic. Biol. Med. 2019, 133, 162–168. [Google Scholar] [CrossRef]
- Kim, J.H.; Najy, A.J.; Li, J.; Luo, X.; Kim, H.C.; Choudry, M.H.A.; Lee, Y.J. Involvement of Bid in the crosstalk between ferroptotic agent-induced ER stress and TRAIL-induced apoptosis. J. Cell. Physiol. 2022, 237, 4180–4196. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, N.; Wang, X.; Jin, X.; Du, H.; Peng, G.; Xue, J. Benzo(a)pyrene-7,8-diol-9,10-epoxide induced p53-independent necrosis via the mitochondria-associated pathway involving Bax and Bak activation. Hum. Exp. Toxicol. 2015, 34, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Muthusamy, S.; Peng, C.; Ng, J.C. Effects of binary mixtures of benzo[a]pyrene, arsenic, cadmium, and lead on oxidative stress and toxicity in HepG2 cells. Chemosphere 2016, 165, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Liu, Z.; Zhang, B.; Hu, B. Defense systems of soil microorganisms in response to compound contamination by arsenic and polycyclic aromatic hydrocarbons. Sci. Total Environ. 2024, 950, 175364. [Google Scholar] [CrossRef]
- Ghosh, A.P.; Klocke, B.J.; Ballestas, M.E.; Roth, K.A. CHOP potentially co-operates with FOXO3a in neuronal cells to regulate PUMA and BIM expression in response to ER stress. PLoS ONE 2012, 7, e39586. [Google Scholar] [CrossRef]
- Muller, T.; Dewitz, C.; Schmitz, J.; Schroder, A.S.; Brasen, J.H.; Stockwell, B.R.; Murphy, J.M.; Kunzendorf, U.; Krautwald, S. Necroptosis and ferroptosis are alternative cell death pathways that operate in acute kidney failure. Cell. Mol. Life Sci. 2017, 74, 3631–3645. [Google Scholar] [CrossRef]
- Li, L.; Tong, A.; Zhang, Q.; Wei, Y.; Wei, X. The molecular mechanisms of MLKL-dependent and MLKL-independent necrosis. J. Mol. Cell Biol. 2021, 13, 3–14. [Google Scholar] [CrossRef]
- Su, Q.; Zhou, L.; Zhong, G.; You, Y.; Sun, J.; Wu, Y.; Liao, J.; Tang, Z.; Hu, L. Arsenic induces hepatotoxicity in chickens via PANoptosis pathway. Pestic. Biochem. Physiol. 2024, 204, 106064. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, X.; Deng, L.; Huang, Y.; Mo, Y.; Ye, J.; Liang, R.; Qin, Y.; Zhang, Q.; Wang, S. Arsenic exposure provoked prostatic PANoptosis by inducing mitochondrial dysfunction in mice and WPMY-1 cells. Ecotoxicol. Environ. Saf. 2025, 295, 118139. [Google Scholar] [CrossRef]
- Kirkwood-Donelson, K.I.; Jarmusch, A.K.; Bortner, C.D.; Merrick, B.A.; Sinha, B.K. Metabolic consequences of erastin-induced ferroptosis in human ovarian cancer cells: An untargeted metabolomics study. Front. Mol. Biosci. 2024, 11, 1520876. [Google Scholar] [CrossRef]
- Soga, T.; Baran, R.; Suematsu, M.; Ueno, Y.; Ikeda, S.; Sakurakawa, T.; Kakazu, Y.; Ishikawa, T.; Robert, M.; Nishioka, T.; et al. Differential metabolomics reveals ophthalmic acid as an oxidative stress biomarker indicating hepatic glutathione consumption. J. Biol. Chem. 2006, 281, 16768–16776. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Ren, H.; Wang, J.; Xiao, X.; Zhu, L.; Wang, Y.; Yang, L. CHAC1: A master regulator of oxidative stress and ferroptosis in human diseases and cancers. Front. Cell Dev. Biol. 2024, 12, 1458716. [Google Scholar] [CrossRef] [PubMed]
- Sinha, B.K.; Tokar, E.J.; Bortner, C.D. Molecular Mechanisms of Cytotoxicity of NCX4040, the Non-Steroidal Anti-Inflammatory NO-Donor, in Human Ovarian Cancer Cells. Int. J. Mol. Sci. 2022, 23, 8611. [Google Scholar] [CrossRef] [PubMed]
- Sinha, B.K.; Murphy, C.; Brown, S.M.; Silver, B.B.; Tokar, E.J.; Bortner, C.D. Mechanisms of Cell Death Induced by Erastin in Human Ovarian Tumor Cells. Int. J. Mol. Sci. 2024, 25, 8666. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Shintoku, R.; Takigawa, Y.; Yamada, K.; Kubota, C.; Yoshimoto, Y.; Takeuchi, T.; Koshiishi, I.; Torii, S. Lipoxygenase-mediated generation of lipid peroxides enhances ferroptosis induced by erastin and RSL3. Cancer Sci. 2017, 108, 2187–2194. [Google Scholar] [CrossRef]
- Koppula, P.; Zhuang, L.; Gan, B. Cystine transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 2021, 12, 599–620. [Google Scholar] [CrossRef]
- Chen, X.; Sun, W.; Song, Y.; Wu, S.; Xie, S.; Xiong, W.; Peng, C.; Peng, Y.; Wang, Z.; Lek, S.; et al. Acute waterborne cadmium exposure induces liver ferroptosis in Channa argus. Ecotoxicol. Environ. Saf. 2024, 283, 116947. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Andia, A.A.; Liu, H.; Csuka, J.M.; Hurlocker, B.; Vaiana, C.A.; Heindel, D.W.; Zuckerman, D.S.; Bos, P.H.; Reznik, E.; et al. FINO(2) initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat. Chem. Biol. 2018, 14, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Guo, Y.; Huang, Y.; Wang, Q.; Huang, Y.; Lei, Y.; Liu, Z.; Zhang, L. GPX4 allosteric activators inhibit ferroptosis and exert myocardial protection in doxorubicin-induced myocardial injury mouse model. Eur. J. Med. Chem. 2024, 277, 116721. [Google Scholar] [CrossRef] [PubMed]
- Taufani, I.P.; Tasminatun, S.; Harimurti, S.; Yang, L.Y.; Huang, C.Y.; Situmorang, J.H. Tannic Acid Suppresses Ferroptosis Induced by Iron Salophene Complex in Kidney Cells and Prevents Iron Overload-Induced Liver and Kidney Dysfunction in Rats. Biol. Trace Elem. Res. 2025, 203, 2701–2713. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Wan, Z.; Jiang, B.; Ouyang, Y.; Feng, W.; Zhu, H.; Tan, Y.; He, R.; Xie, L.; Li, Y. Inducing ferroptosis has the potential to overcome therapy resistance in breast cancer. Front. Immunol. 2022, 13, 1038225. [Google Scholar] [CrossRef]
- Costa, I.; Barbosa, D.J.; Benfeito, S.; Silva, V.; Chavarria, D.; Borges, F.; Remiao, F.; Silva, R. Molecular mechanisms of ferroptosis and their involvement in brain diseases. Pharmacol. Ther. 2023, 244, 108373. [Google Scholar] [CrossRef]
- Lesjak, M.; Simin, N.; Srai, S.K.S. Can Polyphenols Inhibit Ferroptosis? Antioxidants 2022, 11, 150. [Google Scholar] [CrossRef]
- Meng, Y.; Sun, J.; Zhang, G.; Yu, T.; Piao, H. The three-dimension preclinical models for ferroptosis monitoring. Front. Bioeng. Biotechnol. 2022, 10, 1020971. [Google Scholar] [CrossRef]
- Konstorum, A.; Tesfay, L.; Paul, B.T.; Torti, F.M.; Laubenbacher, R.C.; Torti, S.V. Systems biology of ferroptosis: A modeling approach. J. Theor. Biol. 2020, 493, 110222. [Google Scholar] [CrossRef]
- Lei, G.; Gan, B. Exploring Ferroptosis-Inducing Therapies for Cancer Treatment: Challenges and Opportunities. Cancer Res. 2024, 84, 961–964. [Google Scholar] [CrossRef]
- Huiting, L.N.; Laroche, F.; Feng, H. The Zebrafish as a Tool to Cancer Drug Discovery. Austin J. Pharmacol. Ther. 2015, 3, 1069. [Google Scholar]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422.e421. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef] [PubMed]
- de Souza, I.; Monteiro, L.K.S.; Guedes, C.B.; Silva, M.M.; Andrade-Tomaz, M.; Contieri, B.; Latancia, M.T.; Mendes, D.; Porchia, B.; Lazarini, M.; et al. High levels of NRF2 sensitize temozolomide-resistant glioblastoma cells to ferroptosis via ABCC1/MRP1 upregulation. Cell Death Dis. 2022, 13, 591. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Gene | Function | Expression in Tissue | Function in Ferroptosis |
|---|---|---|---|
| GPX4 | Reduces lipid peroxides | All tissues | Inhibits ferroptosis |
| SLC7A11 | Xc− transporter | Neural, epithelial, immune | GSH synthesis, anti- ferroptosis |
| ACSL4 | PUFA incorporation | Liver, Brain, testis | Sensitizes cells to ferroptosis |
| FTH1 | Ferritin Heavy Chain1, iron storage | Liver, spleen, heart, brain | Sequesters iron, anti-ferroptosis |
| FTL | Ferritin Light Chain | Liver, spleen, heart, brain | Sequesters iron, anti-ferroptosis |
| NCOL4 | Ferritin Cargo Receptor | Liver, gut, kidney, epithelial | Releases iron by ferritin degradation, pro-ferroptosis |
| ALOX15 | Lipoxygenase, catalyzes lipid peroxidation | Lung, brain, Immune | Promotes lipid peroxidation, pro-ferroptosis |
| TFRC | Transferrin Receptor, iron uptake | Gut, kidney, proliferative tissues | Increases iron uptake, pro-ferroptosis |
| NrF2 | Oxidative stress response | Lung, brain, liver | Activates antioxidant genes, anti-ferroptosis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sinha, B.K. Ferroptosis in Toxicology: Present and Future. Int. J. Mol. Sci. 2025, 26, 6658. https://doi.org/10.3390/ijms26146658
Sinha BK. Ferroptosis in Toxicology: Present and Future. International Journal of Molecular Sciences. 2025; 26(14):6658. https://doi.org/10.3390/ijms26146658
Chicago/Turabian StyleSinha, Birandra K. 2025. "Ferroptosis in Toxicology: Present and Future" International Journal of Molecular Sciences 26, no. 14: 6658. https://doi.org/10.3390/ijms26146658
APA StyleSinha, B. K. (2025). Ferroptosis in Toxicology: Present and Future. International Journal of Molecular Sciences, 26(14), 6658. https://doi.org/10.3390/ijms26146658