
Repeatome Analysis of Plasma Circulating DNA in Patients with Cardiovascular Disease: Variation with Cell-Free DNA Integrity/Length and Clinical Parameters

 ,
,  , ,
, ,  , , and
, , and
Abstract
1. Introduction
2. Results
2.1. Length Distribution of cfDNA
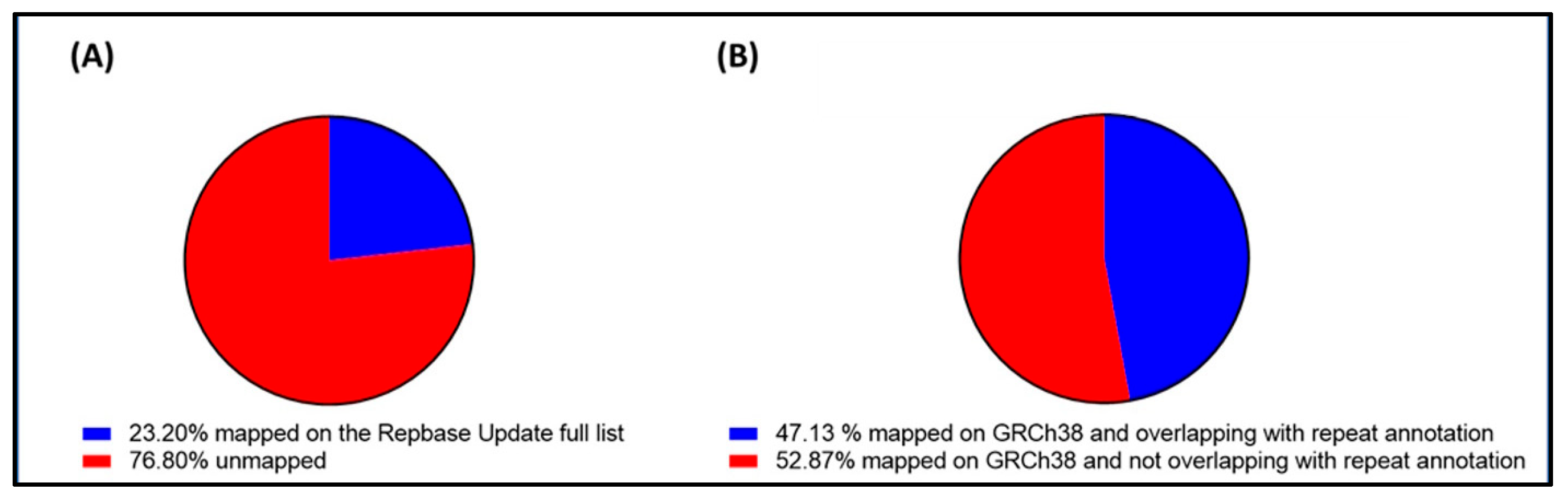
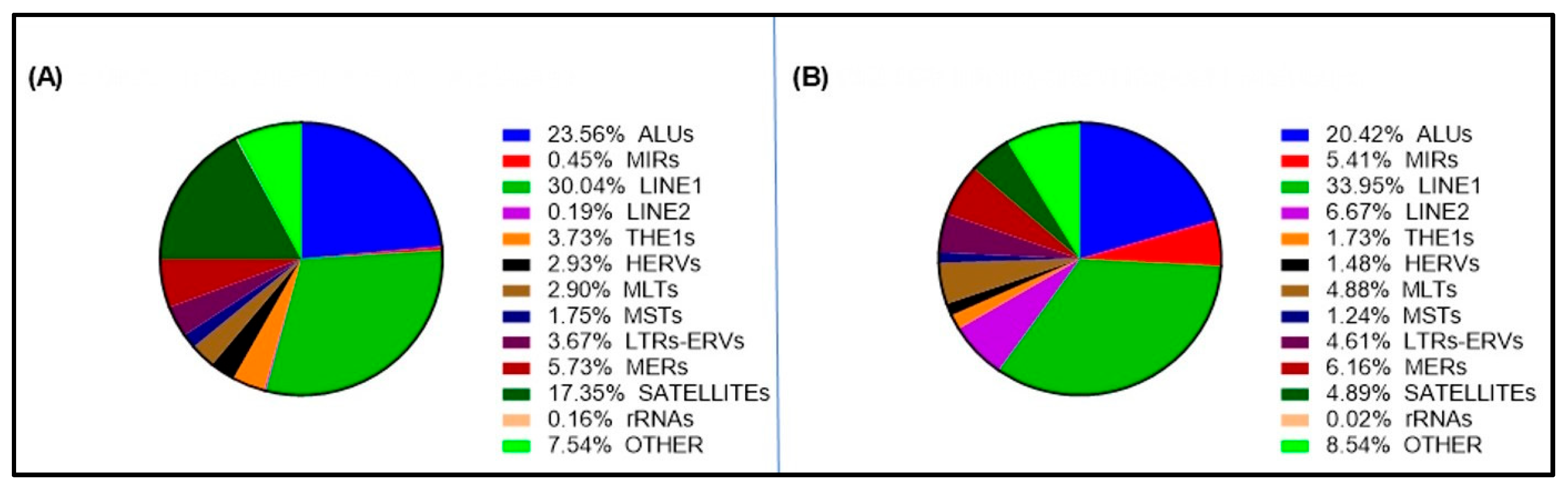
2.2. Overall Abundance and Distribution of Repetitive Elements in Cell-Free DNA
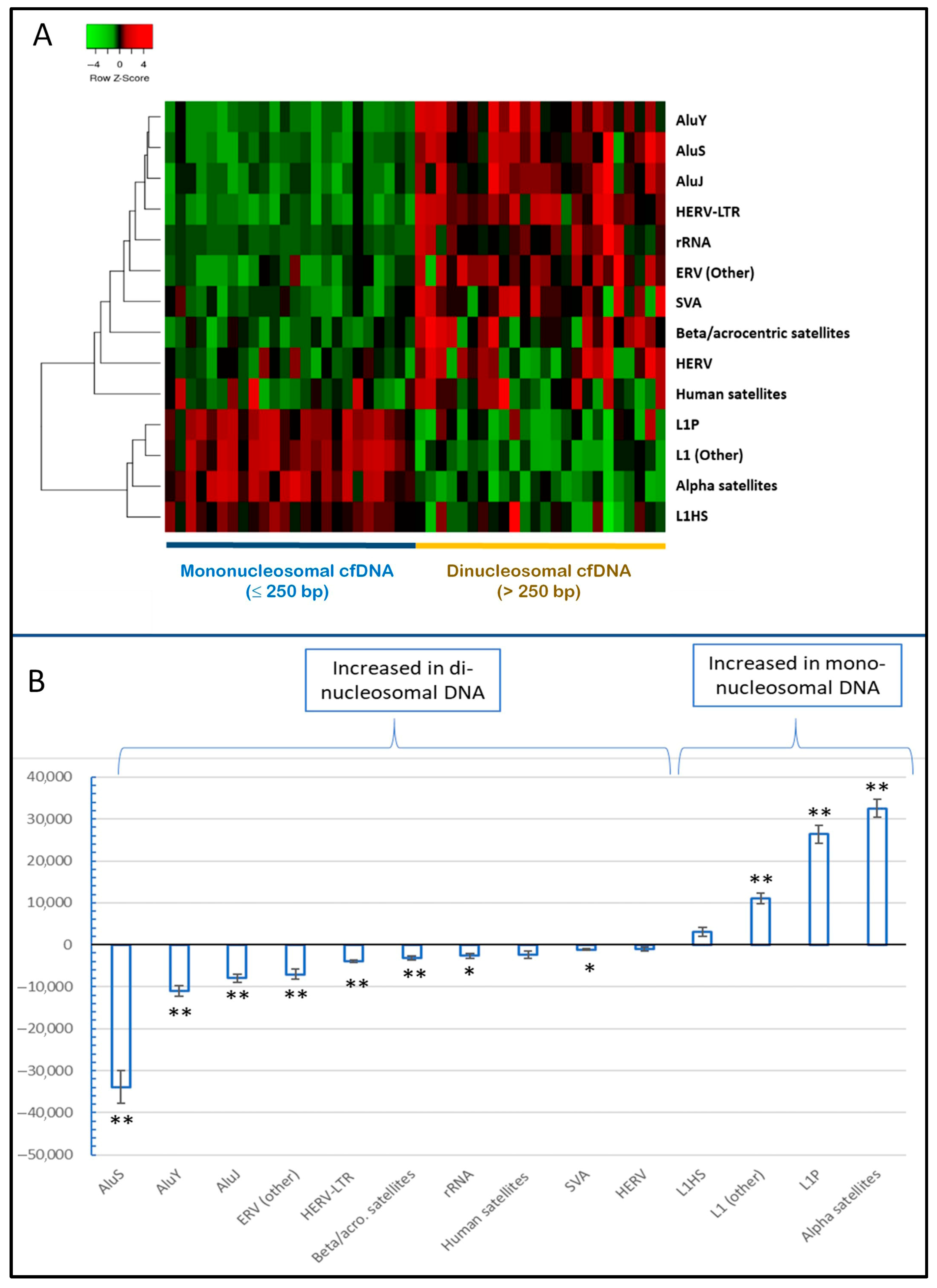
2.3. Variable Distribution of Repetitive Elements in Short and Long cfDNA Fragments and Among Different Samples
2.4. The Relative Abundance of Alu cfDNA Is Inversely Related to cfDNA Integrity
2.5. The Composition of cfDNA Can Vary in Correlation with Clinical Biomarkers
3. Discussion
4. Materials and Methods
4.1. Study Population
4.2. Extraction and Quality Control of cfDNA
4.3. cfDNA Quantification and Integrity Assessment
- (1)
- Samples were analyzed by automated electrophoresis using high-sensitivity D1000 screen tapes on the TapeStation System 4200 (Agilent, Santa Clara, CA, USA); integrity was determined by the ratio of the molar concentration of the longer cfDNA peaks (size range 280–700 bp) to that of the shorter main peak at approximately 170 bp (size range 100–280 bp).
- (2)
- After NGS sequencing, integrity was also calculated as the ratio of the counts of reads in two size ranges (reads > 250 bases; reads ≤ 250 bases).
4.4. Library Preparation and Next Generation Sequencing
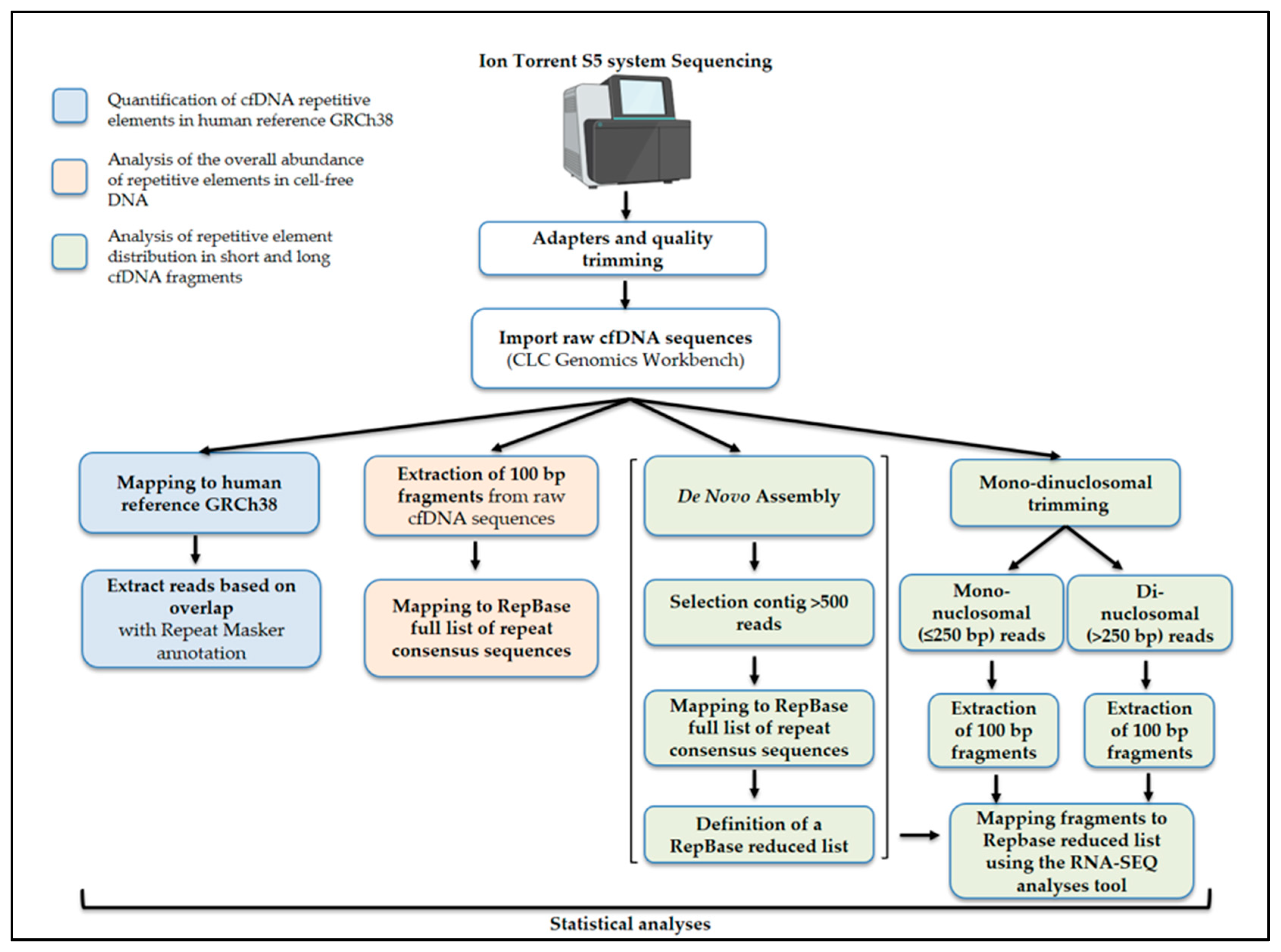
4.5. Bioinformatic Analyses of NGS Data
4.6. Quantification of Repetitive Elements in cfDNA and in the Human Reference Genome
4.7. Analysis of Repeat Family/Subfamily Composition in cfDNA
4.8. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| cfDNA | Circulating cell-free DNA |
| CKD | Chronic kidney disease |
| CPM | Count per million |
| CRP | C-reactive protein |
| eGFR | Estimated glomerular filtration rate |
| ERV | Endogenous retroviruses |
| Hct | Hematocrit |
| HF | Heart failure |
| HERV | Human endogenous retroviruses |
| Hgb | Hemoglobin |
| HMW | High molecular weight |
| LINE-1 | Long interspersed nuclear element-1 |
| LTR | Long terminal repeat |
| MIR | Mammalian-wide interspersed repeats |
| MLT | Mammalian LTR Transposon |
| NETs | Neutrophil extracellular traps |
| Nt-proBNP | N-terminal pro-brain natriuretic peptide |
| NLR | Neutrophil-to-lymphocyte ratio |
| NGS | Next-generation sequencing |
| PLTs | Platelets |
| RBC | Red blood cells |
| SINE | Short interspersed nuclear elements |
| SVA | SINE-R, VNTR, and Alu |
| THE1 | Transposon-like human element-1 |
| WBC | White blood cells |
References
- Hickey, D.A. Selfish DNA: A sexually-transmitted nuclear parasite. Genetics 1982, 101, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Agren, J.A.; Clark, A.G. Selfish genetic elements. PLoS Genet. 2018, 14, e1007700. [Google Scholar] [CrossRef] [PubMed]
- Jagannathan, M.; Cummings, R.; Yamashita, Y.M. A conserved function for pericentromeric satellite DNA. Elife 2018, 7, e34122. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.M.; Yamashita, Y.M. The implications of satellite DNA instability on cellular function and evolution. Semin. Cell Dev. Biol. 2024, 156, 152–159. [Google Scholar] [CrossRef]
- Chattopadhyay, P.; Srinivasa Vasudevan, J.; Pandey, R. Noncoding RNAs: Modulators and modulatable players during infection-induced stress response. Brief. Funct. Genom. 2021, 20, 28–41. [Google Scholar] [CrossRef]
- Cardelli, M. The epigenetic alterations of endogenous retroelements in aging. Mech. Ageing Dev. 2018, 174, 30–46. [Google Scholar] [CrossRef]
- Gorbunova, V.; Seluanov, A.; Mita, P.; McKerrow, W.; Fenyo, D.; Boeke, J.D.; Linker, S.B.; Gage, F.H.; Kreiling, J.A.; Petrashen, A.P.; et al. The role of retrotransposable elements in ageing and age-associated diseases. Nature 2021, 596, 43–53. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef]
- Artner, T.; Sharma, S.; Lang, I.M. Nucleic acid liquid biopsies in cardiovascular disease: Cell-free DNA liquid biopsies in cardiovascular disease. Atherosclerosis 2024, 398, 118583. [Google Scholar] [CrossRef]
- Snyder, M.W.; Kircher, M.; Hill, A.J.; Daza, R.M.; Shendure, J. Cell-free DNA Comprises an In Vivo Nucleosome Footprint that Informs Its Tissues-Of-Origin. Cell 2016, 164, 57–68. [Google Scholar] [CrossRef]
- Vanderstichele, A.; Busschaert, P.; Landolfo, C.; Olbrecht, S.; Coosemans, A.; Froyman, W.; Loverix, L.; Concin, N.; Braicu, E.I.; Wimberger, P.; et al. Nucleosome footprinting in plasma cell-free DNA for the pre-surgical diagnosis of ovarian cancer. NPJ Genom. Med. 2022, 7, 30. [Google Scholar] [CrossRef] [PubMed]
- Medina, J.E.; Dracopoli, N.C.; Bach, P.B.; Lau, A.; Scharpf, R.B.; Meijer, G.A.; Andersen, C.L.; Velculescu, V.E. Cell-free DNA approaches for cancer early detection and interception. J. Immunother. Cancer 2023, 11, e006013. [Google Scholar] [CrossRef] [PubMed]
- Tessier, N.P.; Hardy, L.M.; Deleuze, J.F.; How-Kit, A. Circulating cell-free nucleic acids of plasma in human aging, healthy aging and longevity: Current state of knowledge. Front. Genet. 2023, 14, 1321280. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.; Liu, D.; Perry, L.; Zhu, J.; Cid-Serra, X.; Deane, A.; Yeo, C.; Ajani, A. Cell-free DNA as a potential biomarker for acute myocardial infarction: A systematic review and meta-analysis. Int. J. Cardiol. Heart Vasc. 2023, 47, 101246. [Google Scholar] [CrossRef]
- Grabuschnig, S.; Soh, J.; Heidinger, P.; Bachler, T.; Hirschbock, E.; Rosales Rodriguez, I.; Schwendenwein, D.; Sensen, C.W. Circulating cell-free DNA is predominantly composed of retrotransposable elements and non-telomeric satellite DNA. J. Biotechnol. 2020, 313, 48–56. [Google Scholar] [CrossRef]
- Annapragada, A.V.; Niknafs, N.; White, J.R.; Bruhm, D.C.; Cherry, C.; Medina, J.E.; Adleff, V.; Hruban, C.; Mathios, D.; Foda, Z.H.; et al. Genome-wide repeat landscapes in cancer and cell-free DNA. Sci. Transl. Med. 2024, 16, eadj9283. [Google Scholar] [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef]
- Umetani, N.; Kim, J.; Hiramatsu, S.; Reber, H.A.; Hines, O.J.; Bilchik, A.J.; Hoon, D.S. Increased integrity of free circulating DNA in sera of patients with colorectal or periampullary cancer: Direct quantitative PCR for ALU repeats. Clin. Chem. 2006, 52, 1062–1069. [Google Scholar] [CrossRef]
- Waki, K.; Yokomizo, K.; Yoshiyama, K.; Takamori, S.; Komatsu, N.; Yamada, A. Integrity of circulating cell-free DNA as a prognostic biomarker for vaccine therapy in patients with nonsmall cell lung cancer. Immunopharmacol. Immunotoxicol. 2021, 43, 176–182. [Google Scholar] [CrossRef]
- Hussein, N.A.; Mohamed, S.N.; Ahmed, M.A. Plasma ALU-247, ALU-115, and cfDNA Integrity as Diagnostic and Prognostic Biomarkers for Breast Cancer. Appl. Biochem. Biotechnol. 2019, 187, 1028–1045. [Google Scholar] [CrossRef]
- Yuwono, N.L.; Henry, C.E.; Ford, C.E.; Warton, K. Total and endothelial cell-derived cell-free DNA in blood plasma does not change during menstruation. PLoS ONE 2021, 16, e0250561. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.; Li, K.; Lou, X.; Guo, Y.; Wang, Y.; Wu, L.; Zhang, D. Plasma Cell-Free DNA as a Novel Biomarker for the Diagnosis and Monitoring of Atherosclerosis. Cells 2022, 11, 3248. [Google Scholar] [CrossRef] [PubMed]
- Shaban, S.A.; Al-Rahim, A.M.; Suleiman, A.A. ALU repeat as potential molecular marker in the detection and prognosis of different cancer types: A systematic review. Mol. Clin. Oncol. 2022, 16, 86. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Zhang, W.; Liu, H.; Qian, B.; Lai, R.; Yao, Z.; Wang, Y.; Yan, Y.; Yuan, Z. Plasma Cell-Free DNA Is a Potential Biomarker for Diagnosis of Calcific Aortic Valve Disease. Cardiology 2024, 149, 155–162. [Google Scholar] [CrossRef]
- Cardelli, M.; Pierpaoli, E.; Marchegiani, F.; Marcheselli, F.; Piacenza, F.; Giacconi, R.; Recchioni, R.; Casoli, T.; Stripoli, P.; Provinciali, M.; et al. Biomarkers of cell damage, neutrophil and macrophage activation associated with in-hospital mortality in geriatric COVID-19 patients. Immun. Ageing 2022, 19, 65. [Google Scholar] [CrossRef]
- Cardelli, M.; Marchegiani, F.; Stripoli, P.; Piacenza, F.; Recchioni, R.; Di Rosa, M.; Giacconi, R.; Malavolta, M.; Galeazzi, R.; Arosio, B.; et al. Plasma cfDNA abundance as a prognostic biomarker for higher risk of death in geriatric cardiovascular patients. Mech. Ageing Dev. 2024, 219, 111934. [Google Scholar] [CrossRef]
- Ungerer, V.; Bronkhorst, A.J.; Uhlig, C.; Holdenrieder, S. Cell-Free DNA Fragmentation Patterns in a Cancer Cell Line. Diagnostics 2022, 12, 1896. [Google Scholar] [CrossRef]
- Che, H.; Jiang, P.; Choy, L.Y.L.; Cheng, S.H.; Peng, W.; Chan, R.W.Y.; Liu, J.; Zhou, Q.; Lam, W.K.J.; Yu, S.C.Y.; et al. Genomic origin, fragmentomics, and transcriptional properties of long cell-free DNA molecules in human plasma. Genome Res. 2024, 34, 189–200. [Google Scholar] [CrossRef]
- Noe, M.; Mathios, D.; Annapragada, A.V.; Koul, S.; Foda, Z.H.; Medina, J.E.; Cristiano, S.; Cherry, C.; Bruhm, D.C.; Niknafs, N.; et al. DNA methylation and gene expression as determinants of genome-wide cell-free DNA fragmentation. Nat. Commun. 2024, 15, 6690. [Google Scholar] [CrossRef]
- van de Wouw, J.; Broekhuizen, M.; Sorop, O.; Joles, J.A.; Verhaar, M.C.; Duncker, D.J.; Danser, A.H.J.; Merkus, D. Chronic Kidney Disease as a Risk Factor for Heart Failure With Preserved Ejection Fraction: A Focus on Microcirculatory Factors and Therapeutic Targets. Front. Physiol. 2019, 10, 1108. [Google Scholar] [CrossRef]
- Tanaka, Y.; Yamashita, R.; Suzuki, Y.; Nakai, K. Effects of Alu elements on global nucleosome positioning in the human genome. BMC Genom. 2010, 11, 309. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, R.; de Llobet Cucalon, L.I.; Di Vona, C.; Le Dilly, F.; Vidal, E.; Lioutas, A.; Oliete, J.Q.; Jochem, L.; Cutts, E.; Dieci, G.; et al. TFIIIC binding to Alu elements controls gene expression via chromatin looping and histone acetylation. Mol. Cell 2020, 77, 475–487.e11. [Google Scholar] [CrossRef] [PubMed]
- Kustanovich, A.; Schwartz, R.; Peretz, T.; Grinshpun, A. Life and death of circulating cell-free DNA. Cancer Biol. Ther. 2019, 20, 1057–1067. [Google Scholar] [CrossRef] [PubMed]
- Celec, P.; Vlkova, B.; Laukova, L.; Babickova, J.; Boor, P. Cell-free DNA: The role in pathophysiology and as a biomarker in kidney diseases. Expert. Rev. Mol. Med. 2018, 20, e1. [Google Scholar] [CrossRef]
- Grover, D.; Mukerji, M.; Bhatnagar, P.; Kannan, K.; Brahmachari, S.K. Alu repeat analysis in the complete human genome: Trends and variations with respect to genomic composition. Bioinformatics 2004, 20, 813–817. [Google Scholar] [CrossRef]
- Dixon, J.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef]
- Luo, X.; Liu, Y.; Dang, D.; Hu, T.; Hou, Y.; Meng, X.; Zhang, F.; Li, T.; Wang, C.; Li, M.; et al. 3D genome of macaque fetal brain reveals evolutionary innovations during primate corticogenesis. Cell 2021, 184, 723–740.e21. [Google Scholar] [CrossRef]
- Dang, D.; Zhang, S.W.; Duan, R.; Zhang, S. Defining the separation landscape of topological domains for decoding consensus domain organization of the 3D genome. Genome Res. 2023, 33, 386–400. [Google Scholar] [CrossRef]
- Salinas-Pena, M.; Serna-Pujol, N.; Jordan, A. Genomic profiling of six human somatic histone H1 variants denotes that H1X accumulates at recently incorporated transposable elements. Nucleic Acids Res. 2024, 52, 1793–1813. [Google Scholar] [CrossRef]
- Xing, J.; Hedges, D.J.; Han, K.; Wang, H.; Cordaux, R.; Batzer, M.A. Alu element mutation spectra: Molecular clocks and the effect of DNA methylation. J. Mol. Biol. 2004, 344, 675–682. [Google Scholar] [CrossRef]
- Babenko, V.N.; Chadaeva, I.V.; Orlov, Y.L. Genomic landscape of CpG rich elements in human. BMC Evol. Biol. 2017, 17, 19. [Google Scholar] [CrossRef] [PubMed]
- Repeat Masker. Human [Homo Sapiens] Genomic Dataset. Available online: https://www.repeatmasker.org/species/hg.html (accessed on 1 December 2024).
- Gezer, U.; Oberhofer, A.; Worf, K.; Stoetzer, O.; Holdenrieder, S.; Bronkhorst, A. Targeted Sequencing of Human Satellite 2 Repeat Sequences in Plasma cfDNA Reveals Potential Breast Cancer Biomarkers. Diagnostics 2024, 14, 609. [Google Scholar] [CrossRef] [PubMed]
- Gezer, U.; Bronkhorst, A.J.; Holdenrieder, S. The Utility of Repetitive Cell-Free DNA in Cancer Liquid Biopsies. Diagnostics 2022, 12, 1363. [Google Scholar] [CrossRef] [PubMed]
- Bronkhorst, A.J.; Wentzel, J.F.; Ungerer, V.; Peters, D.L.; Aucamp, J.; de Villiers, E.P.; Holdenrieder, S.; Pretorius, P.J. Sequence analysis of cell-free DNA derived from cultured human bone osteosarcoma (143B) cells. Tumor Biol. 2018, 40, 1010428318801190. [Google Scholar] [CrossRef]
- Li, Z.; Liu, B.; Jin, W.; Wu, X.; Zhou, M.; Liu, V.Z.; Goel, A.; Shen, Z.; Zheng, L.; Shen, B. hDNA2 nuclease/helicase promotes centromeric DNA replication and genome stability. EMBO J. 2018, 37, e96729. [Google Scholar] [CrossRef]
- Miga, K.H. Centromeric Satellite DNAs: Hidden Sequence Variation in the Human Population. Genes 2019, 10, 352. [Google Scholar] [CrossRef]
- Nishihara, H. Transposable Elements as Genetic Accelerators of Evolution: Contribution to Genome Size, Gene Regulatory Network Rewiring and Morphological Innovation. Genes Genet. Syst. 2020, 94, 269–281. [Google Scholar] [CrossRef]
- Giordano, J.; Ge, Y.; Gelfand, Y.; Abrusán, G.; Benson, G.; Warburton, P.E. Evolutionary History of Mammalian Transposons Determined by Genome-Wide Defragmentation. PLoS Comput. Biol. 2007, 3, e137. [Google Scholar] [CrossRef]
- DFAM: A Database of Repetitive DNA Families. Available online: https://www.dfam.org (accessed on 1 December 2024).
- Bustacchini, S.; Abbatecola, A.M.; Bonfigli, A.R.; Chiatti, C.; Corsonello, A.; Di Stefano, G.; Galeazzi, R.; Fabbietti, P.; Lisa, R.; Guffanti, E.E.; et al. The Report-AGE project: A permanent epidemiological observatory to identify clinical and biological markers of health outcomes in elderly hospitalized patients in Italy. Aging Clin. Exp. Res. 2015, 27, 893–901. [Google Scholar] [CrossRef]
- CDC (Centers for Disease Control and Prevention) Archive. Available online: https://archive.cdc.gov/www_cdc_gov/nchs/icd/icd9cm.htm (accessed on 18 October 2024).
- Schaeffner, E.S.; Ebert, N.; Delanaye, P.; Frei, U.; Gaedeke, J.; Jakob, O.; Kuhlmann, M.K.; Schuchardt, M.; Tolle, M.; Ziebig, R.; et al. Two novel equations to estimate kidney function in persons aged 70 years or older. Ann. Intern. Med. 2012, 157, 471–481. [Google Scholar] [CrossRef]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Real-Time PCR Alu 115 (Plasma Concentration of Total Alu cfDNA) | Real-Time PCR Alu 247 (Plasma Concentration of Long Alu cfDNA) | Real-Time PCR Alu 247/115 (Alu cfDNA Integrity) | cfDNA_Integrity by H.S. Screen Tape a | cfDNA_Integrity by NGS Reads Length | ||
|---|---|---|---|---|---|---|
| CPM in mononucleosomal cfDNA | Alu-J | 0.168 | 0.020 | −0.296 | −0.400 | −0.479 * |
| Alu-S | 0.189 | −0.039 | −0.555 ** | −0.497 * | −0.615 ** | |
| Alu-Y | 0.001 | −0.092 | −0.100 | −0.453 * | −0.325 | |
| Alu (total) | 0.145 | −0.043 | −0.438 * | −0.522 * | −0.571 ** | |
| CPM in dinucleosomal cfDNA | Alu-J | 0.499 * | 0.332 | −0.313 | −0.571 ** | −0.384 |
| Alu-S | 0.257 | 0.053 | −0.408 * | −0.675 ** | −0.590 ** | |
| Alu-Y | 0.356 | 0.151 | −0.348 | −0.792 ** | −0.352 | |
| Alu (total) | 0.398 | 0.174 | −0.434 * | −0.765 ** | −0.625 ** | |
| Fold change in di- vs. mononucleosomal cfDNA | Alu-J | 0.493 * | 0.386 | −0.212 | −0.486 * | −0.208 |
| Alu-S | 0.255 | 0.110 | −0.240 | −0.632 ** | −0.357 | |
| Alu-Y | 0.337 | 0.171 | −0.314 | −0.614 ** | −0.163 | |
| Alu (total) | 0.433 * | 0.248 | −0.326 | −0.768 ** | −0.372 |
| NLR | Wbc (×103/µL) | Rbc (×106/µL) | Hgb, (g/dL) | Hct (%) | PLTs (×103/µL) | Creatinine (mg/dL) | eGFR (mL/min) | Troponin (pg/mL) | Nt-Probnp (pg/mL) | CRP (mg/dL) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CPM in mononucleosomal cfDNA | Alu-J | 0.123 | −0.110 | −0.225 | −0.235 | −0.227 | −0.056 | −0.313 | 0.402 | 0.132 | −0.103 | −0.079 |
| Alu-S | 0.003 | 0.056 | −0.315 | −0.329 | −0.311 | −0.147 | −0.201 | 0.242 | 0.090 | −0.103 | −0.004 | |
| Alu-Y | −0.279 | −0.003 | −0.001 | −0.157 | −0.083 | 0.139 | −0.418 * | 0.451 * | 0.083 | −0.247 | −0.193 | |
| Alu (total) | −0.094 | −0.003 | −0.175 | −0.255 | −0.211 | −0.025 | −0.330 | 0.384 | 0.031 | −0.141 | −0.036 | |
| L1HS | 0.158 | 0.121 | 0.276 | 0.362 | 0.381 | −0.001 | −0.215 | 0.142 | −0.316 | −0.141 | 0.218 | |
| L1P | 0.012 | −0.111 | 0.070 | 0.398 | 0.342 | −0.180 | −0.178 | 0.105 | −0.249 | −0.282 | 0.114 | |
| Other L1 ^ | 0.070 | −0.050 | 0.010 | 0.255 | 0.223 | −0.241 | −0.108 | 0.004 | −0.283 | −0.391 | 0.071 | |
| SVA | 0.171 | 0.128 | 0.029 | −0.059 | −0.010 | −0.144 | −0.252 | 0.346 | −0.047 | −0.056 | 0.168 | |
| HERV | 0.163 | 0.380 | −0.093 | −0.036 | −0.065 | 0.097 | −0.055 | 0.015 | −0.178 | −0.271 | 0.361 | |
| LTR ° | −0.044 | 0.193 | −0.097 | −0.382 | −0.298 | 0.253 | −0.213 | 0.309 | −0.040 | −0.206 | −0.104 | |
| Other ERV_like | 0.364 | 0.170 | −0.258 | −0.151 | −0.163 | 0.063 | −0.064 | 0.018 | −0.147 | 0.018 | 0.607 * | |
| ALR | −0.463 * | −0.169 | 0.091 | 0.141 | 0.095 | 0.141 | −0.020 | 0.049 | −0.058 | −0.462 | −0.100 | |
| 6kb_BSR | 0.074 | −0.074 | 0.270 | 0.400 | 0.318 | 0.031 | 0.107 | −0.136 | 0.018 | 0.168 | −0.232 | |
| HSAT | 0.352 | −0.147 | 0.058 | −0.042 | −0.013 | 0.183 | 0.223 | −0.229 | 0.369 | 0.488 | −0.029 | |
| rRNA | 0.063 | −0.185 | −0.092 | −0.232 | −0.188 | 0.078 | −0.204 | 0.094 | 0.085 | 0.121 | 0.246 | |
| CPM in dinucleosomal cfDNA | Alu-J | 0.173 | 0.129 | 0.096 | 0.074 | 0.062 | 0.171 | −0.492 * | 0.393 | −0.121 | 0.021 | 0.354 |
| Alu-S | −0.021 | −0.107 | 0.114 | 0.064 | 0.107 | −0.056 | −0.526 ** | 0.578 ** | 0.076 | 0.026 | −0.204 | |
| Alu-Y | −0.088 | 0.019 | −0.134 | −0.121 | −0.116 | −0.210 | −0.504 * | 0.449 * | −0.086 | −0.376 | −0.229 | |
| Alu (total) | 0.023 | 0.033 | 0.045 | 0.036 | 0.060 | −0.085 | −0.594 ** | 0.590 ** | 0.038 | −0.124 | −0.089 | |
| L1HS | −0.172 | 0.030 | 0.059 | 0.299 | 0.239 | −0.035 | −0.395 | 0.368 | −0.575 ** | −0.447 | −0.011 | |
| L1P | −0.103 | −0.333 | 0.110 | 0.340 | 0.283 | −0.289 | 0.070 | 0.028 | −0.126 | 0.335 | −0.389 | |
| Other L1 ^ | 0.256 | 0.097 | −0.068 | 0.127 | 0.074 | −0.296 | 0.194 | −0.211 | 0.046 | −0.106 | −0.071 | |
| SVA | −0.270 | −0.256 | 0.104 | 0.227 | 0.205 | 0.166 | 0.024 | −0.077 | −0.047 | 0.153 | 0.400 | |
| HERV | 0.126 | 0.230 | −0.053 | −0.042 | −0.066 | 0.061 | −0.207 | 0.281 | 0.426 | −0.082 | −0.107 | |
| LTR ° | 0.060 | 0.203 | −0.200 | −0.268 | −0.264 | 0.265 | −0.246 | 0.180 | 0.014 | −0.541 * | 0.196 | |
| Other ERV_like | 0.140 | 0.392 | 0.107 | −0.002 | −0.013 | 0.107 | 0.050 | −0.033 | −0.002 | −0.179 | 0.429 | |
| ALR | −0.432 * | 0.107 | −0.039 | 0.019 | −0.010 | 0.040 | 0.053 | −0.016 | −0.194 | −0.709 ** | 0.136 | |
| 6kb_BSR | 0.127 | 0.052 | 0.136 | 0.105 | 0.072 | 0.187 | −0.177 | 0.161 | 0.010 | −0.068 | −0.057 | |
| HSAT | 0.261 | −0.218 | 0.111 | −0.050 | 0.014 | −0.016 | 0.100 | −0.081 | 0.357 | 0.438 | −0.246 | |
| rRNA | 0.215 | −0.049 | −0.295 | −0.369 | −0.326 | 0.029 | 0.114 | −0.240 | 0.081 | 0.147 | 0.432 |
| Mononucleosomal cfDNA | Dinucleosomal cfDNA | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Repeat Class | CKD (1 = CKD) | Mean [95% C.I.] | Median (IQR) | p-Value (u) | p-Value (a) | Mean [95% C.I.] | Median (IQR) | p-Value (u) | p-Value (a) |
| Alu-J | 0 | 5.84 × 104 [(5.72–5.95) × 104] | 5.86 × 104 (3.27 × 103) | 0.015 | 0.046 | 6.67 × 104 [(6.31–7.03) × 104] | 6.51 × 104 (8.33 × 103) | 0.186 | 0.56 |
| 1 | 5.59 × 104 [(5.46–5.73) × 104] | 5.63 × 104 (3.28 × 103) | 6.33 × 104 [(6.11–6.55) × 104] | 6.39 × 104 (4.88 × 103) | |||||
| Alu-S | 0 | 2.13 × 105 [(2.08–2.18) × 105] | 2.12 × 105 (8.85 × 103) | 0.013 | 0.039 | 2.56 × 105 [(2.43–2.70) × 105] | 2.53 × 105 (2.40 × 104) | 0.00040 | 0.0012 |
| 1 | 2.05 × 105 [(2.02–2.09) × 105] | 2.06 × 105 (9.94 × 103) | 2.28 × 105 [(2.18–2.38) × 105] | 2.26 × 105 (1.86 × 104) | |||||
| Alu-Y | 0 | 5.60 × 104 [(5.45–5.74) × 104] | 5.59 × 104 (3.30 × 103) | 0.022 | 0.065 | 6.80 × 104 [(6.38–7.22) × 104] | 6.66 × 104 (1.08 × 104) | 0.119 | 0.357 |
| 1 | 5.40 × 104 [(5.30–5.50) × 104] | 5.39 × 104 (2.40 × 103) | 6.39 × 104 [(6.05–6.73) × 104] | 6.32 × 104 (8.64 × 103) | |||||
| Alu (total) | 0 | 3.27 × 105 [(3.20–3.34) × 105] | 3.25 × 105 (1.26 × 104) | 0.0025 | - | 3.91 × 105 [(3.73–4.09) × 105] | 3.88 × 105 (4.68 × 104) | 0.0048 | - |
| 1 | 3.15 × 105 [(3.11–3.20) × 105] | 3.15 × 105 (9.22 × 103) | 3.55 × 105 [(3.41–3.70) × 105] | 3.58 × 105 (2.62 × 104) | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fumarola, S.; Cecati, M.; Marchegiani, F.; Francini, E.; Maniscalco, R.; Sabbatinelli, J.; Gasparrini, M.; Lattanzio, F.; Olivieri, F.; Cardelli, M. Repeatome Analysis of Plasma Circulating DNA in Patients with Cardiovascular Disease: Variation with Cell-Free DNA Integrity/Length and Clinical Parameters. Int. J. Mol. Sci. 2025, 26, 6657. https://doi.org/10.3390/ijms26146657
Fumarola S, Cecati M, Marchegiani F, Francini E, Maniscalco R, Sabbatinelli J, Gasparrini M, Lattanzio F, Olivieri F, Cardelli M. Repeatome Analysis of Plasma Circulating DNA in Patients with Cardiovascular Disease: Variation with Cell-Free DNA Integrity/Length and Clinical Parameters. International Journal of Molecular Sciences. 2025; 26(14):6657. https://doi.org/10.3390/ijms26146657
Chicago/Turabian StyleFumarola, Stefania, Monia Cecati, Francesca Marchegiani, Emanuele Francini, Rosanna Maniscalco, Jacopo Sabbatinelli, Massimiliano Gasparrini, Fabrizia Lattanzio, Fabiola Olivieri, and Maurizio Cardelli. 2025. "Repeatome Analysis of Plasma Circulating DNA in Patients with Cardiovascular Disease: Variation with Cell-Free DNA Integrity/Length and Clinical Parameters" International Journal of Molecular Sciences 26, no. 14: 6657. https://doi.org/10.3390/ijms26146657
APA StyleFumarola, S., Cecati, M., Marchegiani, F., Francini, E., Maniscalco, R., Sabbatinelli, J., Gasparrini, M., Lattanzio, F., Olivieri, F., & Cardelli, M. (2025). Repeatome Analysis of Plasma Circulating DNA in Patients with Cardiovascular Disease: Variation with Cell-Free DNA Integrity/Length and Clinical Parameters. International Journal of Molecular Sciences, 26(14), 6657. https://doi.org/10.3390/ijms26146657