
Effectiveness of PROTAC BET Degraders in Combating Cisplatin Resistance in Head and Neck Cancer Cells

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
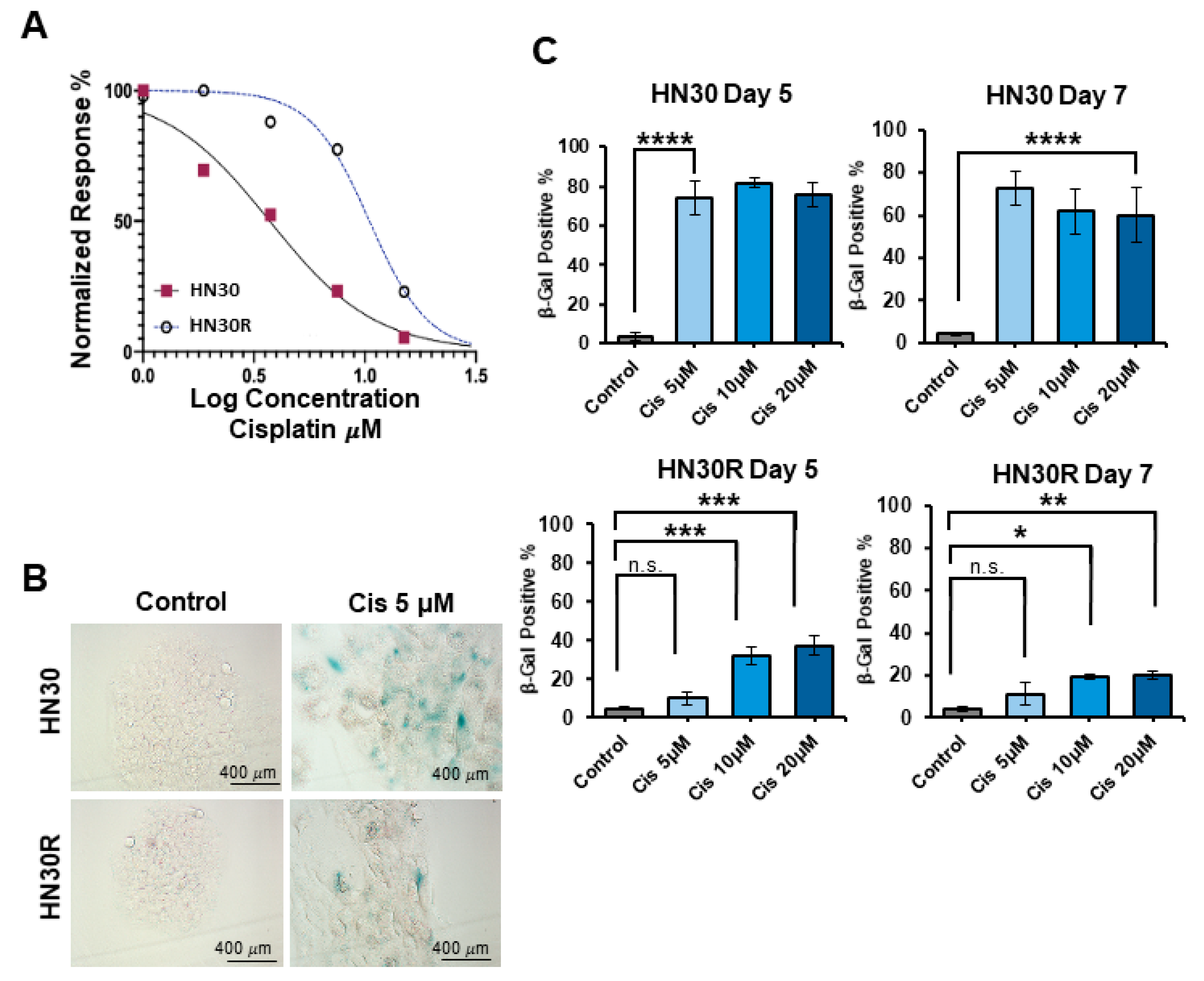
2.1. Development of a Cisplatin-Resistant HNSCC Cell Line
2.2. Cisplatin-Resistant Cells Are Not Sensitized to ABT-263
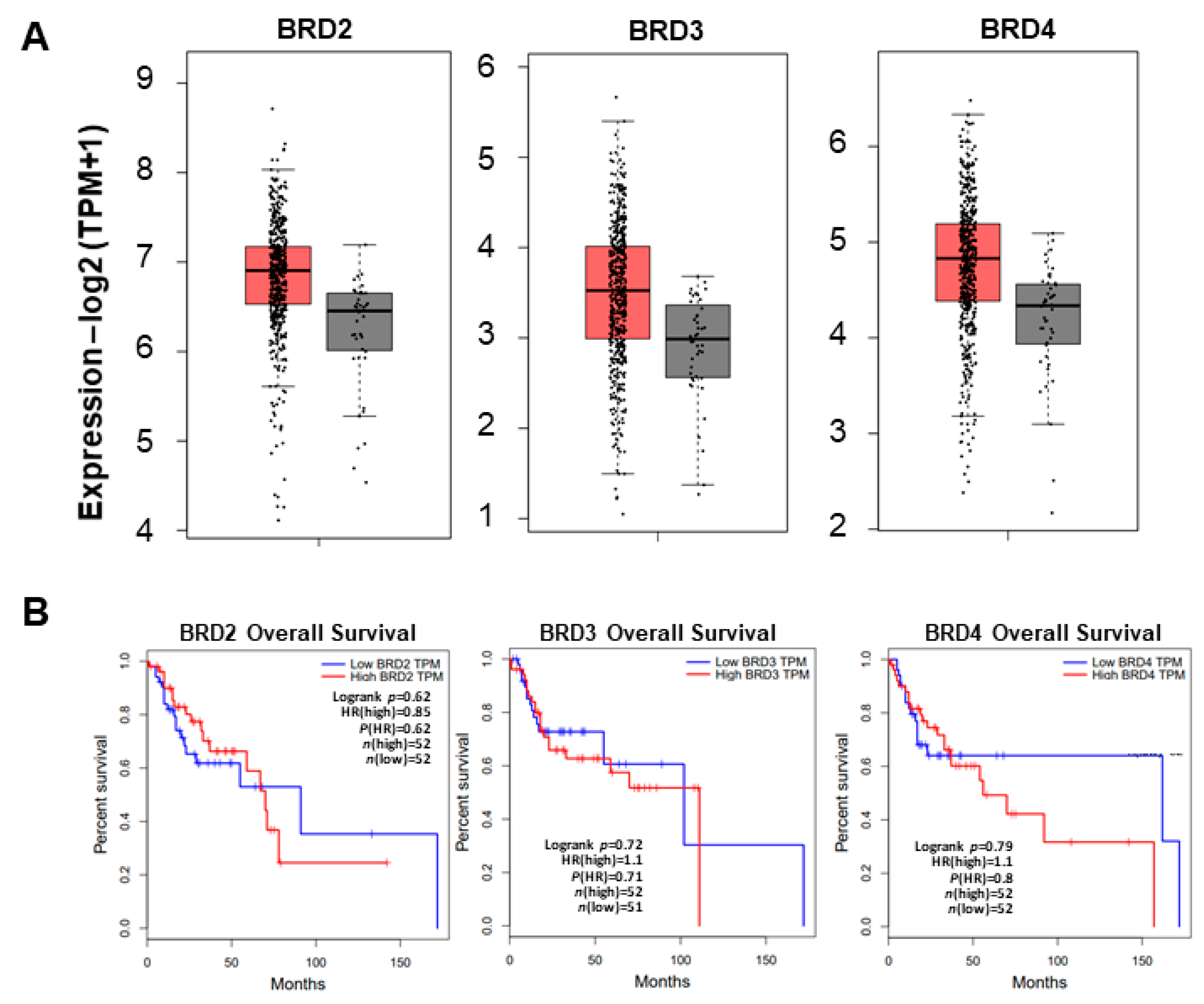
2.3. BRD4 Overexpression Decreases Overall Survival in Head and Neck Cancer Patients
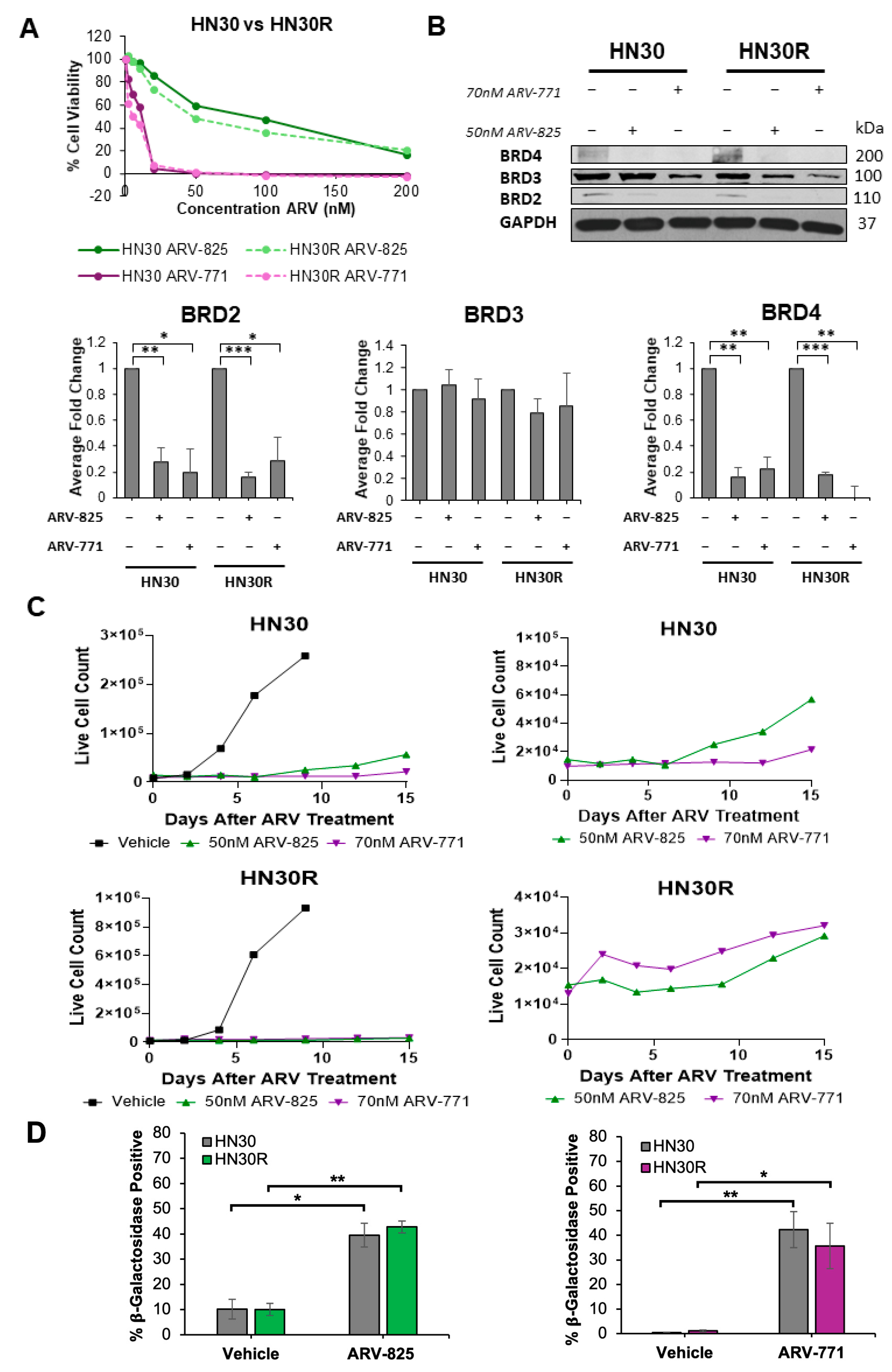
2.4. PROTAC BET Degraders Induce Senescence in the Parental and Resistant HNSCC Cells
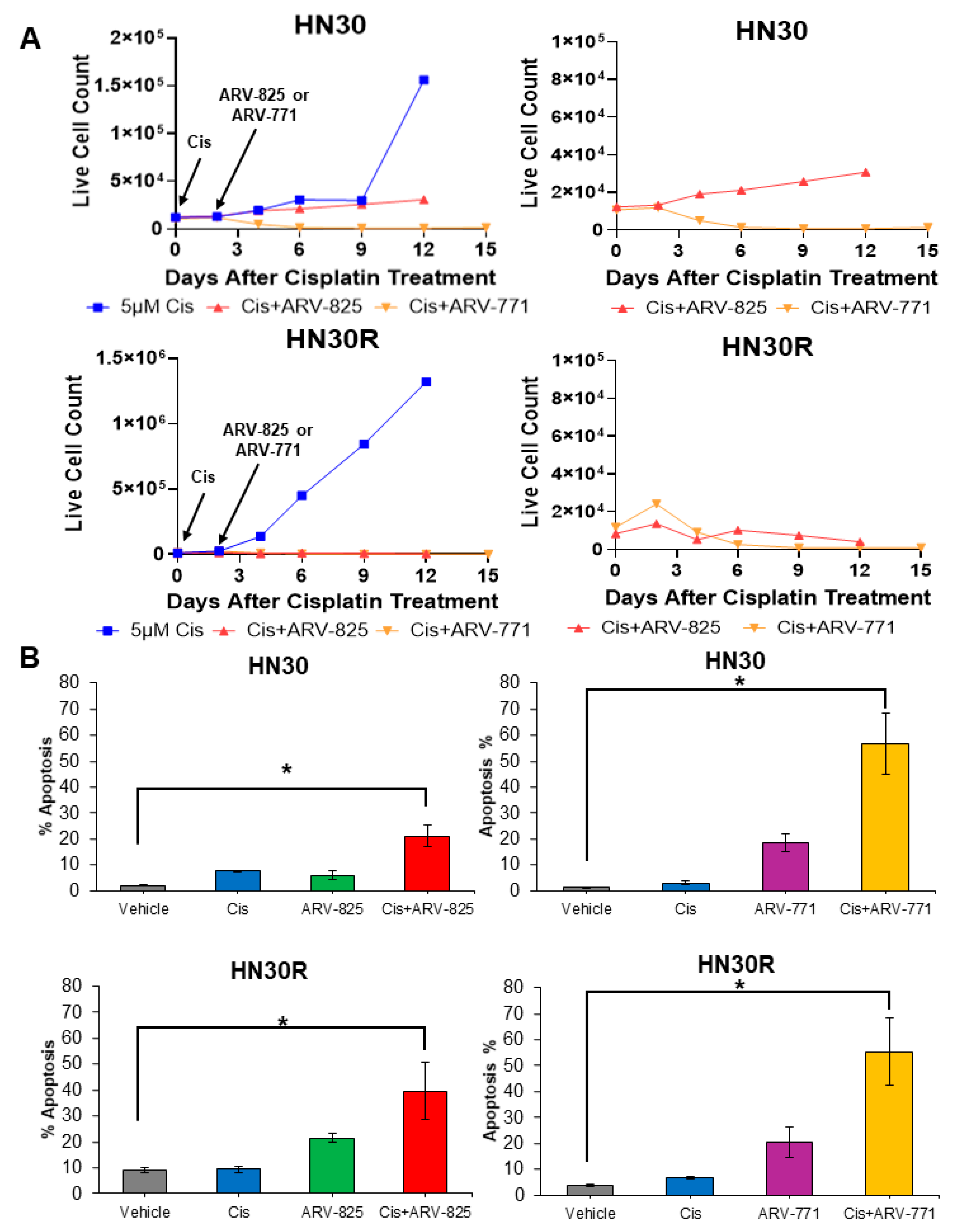
2.5. PROTAC BET Degraders Sensitize Parental and Resistant HNSCC Cells to Cisplatin
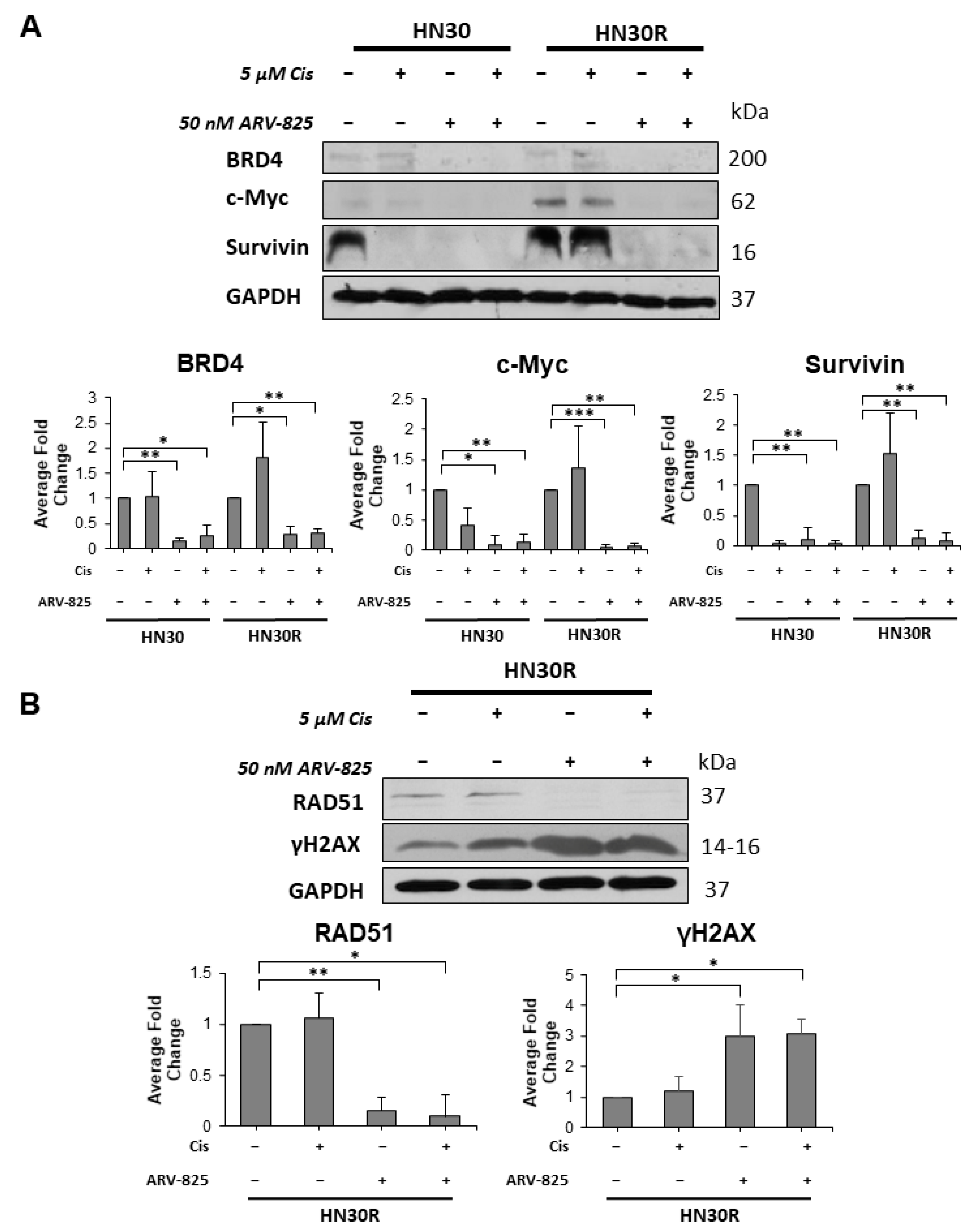
2.6. Suppression of BRD4 Promotes a Reduction in Target Proteins and the Induction of DNA Damage in HN30R Cells Treated with Cisplatin and ARV-825
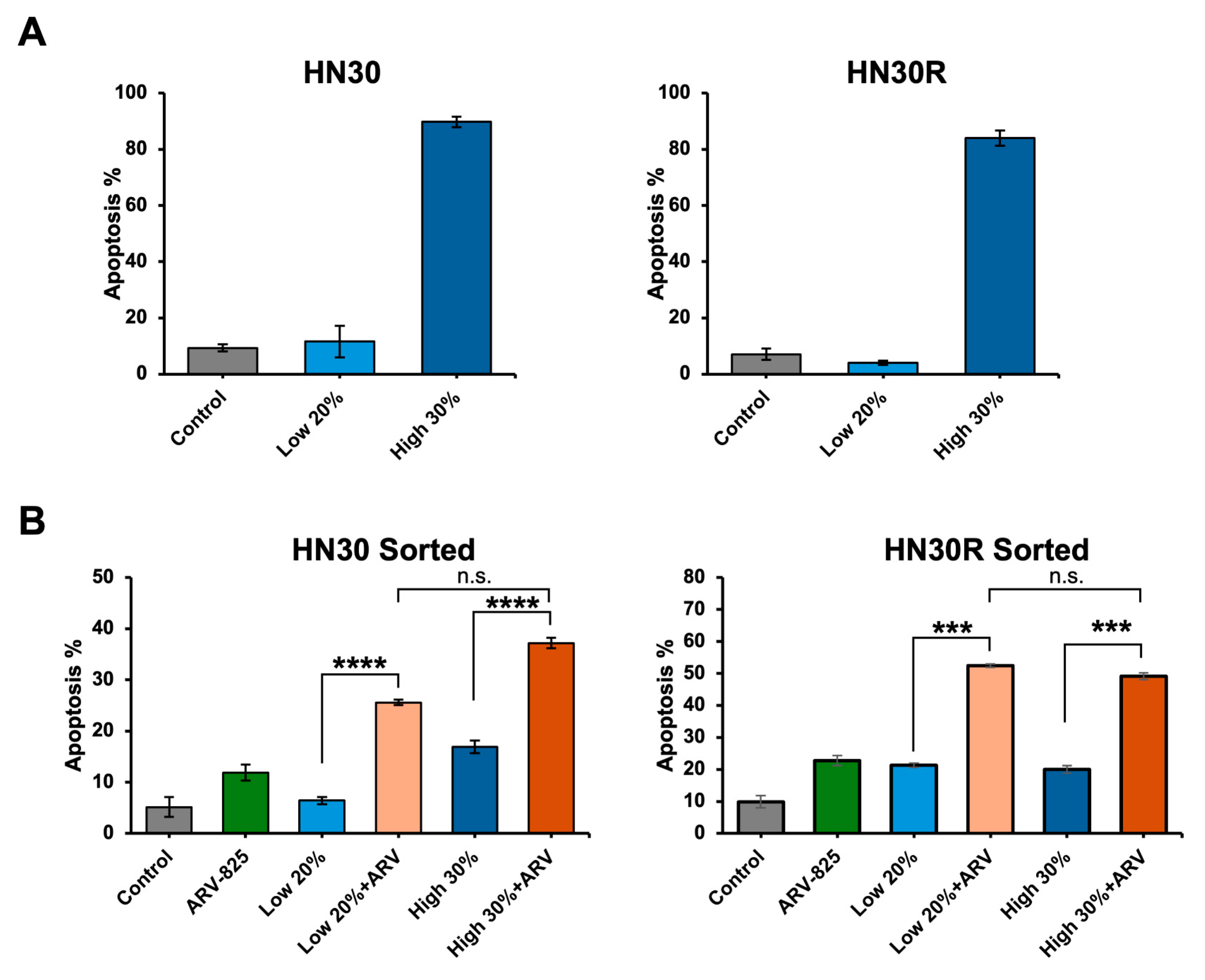
2.7. ARV-825 Does Not Appear to Be Acting as a Senolytic
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Drug Treatment
4.2. Cell Viability Assay
4.3. In Vitro Drug Sensitivity Assay
4.4. SA-β-Galactosidase Staining/Enrichment
4.5. Annexin-V/PI Staining
4.6. Western Blotting
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burtness, B.; Harrington, K.J.; Greil, R.; Soulières, D.; Tahara, M.; de Castro, G.; Psyrri, A.; Basté, N.; Neupane, P.; Bratland, Å.; et al. Pembrolizumab Alone or with Chemotherapy versus Cetuximab with Chemotherapy for Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck (KEYNOTE-048): A Randomised, Open-Label, Phase 3 Study. Lancet 2019, 394, 1915–1928. [Google Scholar] [CrossRef] [PubMed]
- Vermorken, J.; Specenier, P. Cetuximab: Its Unique Place in Head and Neck Cancer Treatment. Biol. Targets Ther. 2013, 7, 77–90. [Google Scholar] [CrossRef]
- Cohen, E.E.W.; Soulières, D.; Le Tourneau, C.; Dinis, J.; Licitra, L.; Ahn, M.-J.; Soria, A.; Machiels, J.-P.; Mach, N.; Mehra, R.; et al. Pembrolizumab versus Methotrexate, Docetaxel, or Cetuximab for Recurrent or Metastatic Head-and-Neck Squamous Cell Carcinoma (KEYNOTE-040): A Randomised, Open-Label, Phase 3 Study. Lancet 2019, 393, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Jou, A.; Hess, J. Epidemiology and Molecular Biology of Head and Neck Cancer. Oncol. Res. Treat. 2017, 40, 328–332. [Google Scholar] [CrossRef]
- Florea, A.-M.; Büsselberg, D. Cisplatin as an Anti-Tumor Drug: Cellular Mechanisms of Activity, Drug Resistance and Induced Side Effects. Cancers 2011, 3, 1351–1371. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular Mechanisms of Cisplatin Resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef]
- Schmitt, C.A.; Wang, B.; Demaria, M. Senescence and Cancer—Role and Therapeutic Opportunities. Nat. Rev. Clin. Oncol. 2022, 19, 619–636. [Google Scholar] [CrossRef] [PubMed]
- Song, K.-X.; Wang, J.-X.; Huang, D. Therapy-Induced Senescent Tumor Cells in Cancer Relapse. J. Natl. Cancer Cent. 2023, 3, 273–278. [Google Scholar] [CrossRef]
- Saleh, T.; Greenberg, E.F.; Faber, A.C.; Harada, H.; Gewirtz, D.A. A Critical Appraisal of the Utility of Targeting Therapy-Induced Senescence for Cancer Treatment. Cancer Res. 2025, 85, 1755–1768. [Google Scholar] [CrossRef]
- Ahmadinejad, F.; Bos, T.; Hu, B.; Britt, E.; Koblinski, J.; Souers, A.J.; Leverson, J.D.; Faber, A.C.; Gewirtz, D.A.; Harada, H. Senolytic-Mediated Elimination of Head and Neck Tumor Cells Induced Into Senescence by Cisplatin. Mol. Pharmacol. 2022, 101, 168–180. [Google Scholar] [CrossRef]
- Carpenter, V.J.; Saleh, T.; Gewirtz, D.A. Senolytics for Cancer Therapy: Is All That Glitters Really Gold? Cancers 2021, 13, 723. [Google Scholar] [CrossRef]
- De Paula, B.; Kieran, R.; Koh, S.S.Y.; Crocamo, S.; Abdelhay, E.; Muñoz-Espín, D. Targeting Senescence as a Therapeutic Opportunity for Triple-Negative Breast Cancer. Mol. Cancer Ther. 2023, 22, 583–598. [Google Scholar] [CrossRef]
- Wang, L.; Lankhorst, L.; Bernards, R. Exploiting Senescence for the Treatment of Cancer. Nat. Rev. Cancer 2022, 22, 340–355. [Google Scholar] [CrossRef] [PubMed]
- Rad, A.N.; Grillari, J. Current Senolytics: Mode of Action, Efficacy and Limitations, and Their Future. Mech. Ageing Dev. 2024, 217, 111888. [Google Scholar] [CrossRef]
- Saleh, T.; Carpenter, V.J.; Tyutyunyk-Massey, L.; Murray, G.; Leverson, J.D.; Souers, A.J.; Alotaibi, M.R.; Faber, A.C.; Reed, J.; Harada, H.; et al. Clearance of Therapy-induced Senescent Tumor Cells by the Senolytic ABT-263 via Interference with BCL-X L –BAX Interaction. Mol. Oncol. 2020, 14, 2504–2519. [Google Scholar] [CrossRef]
- Softah, A.; Alotaibi, M.R.; Alhoshani, A.R.; Saleh, T.; Alhazzani, K.; Almutairi, M.M.; AlRowis, R.; Alshehri, S.; Albekairy, N.A.; Harada, H.; et al. The Combination of Radiation with PARP Inhibition Enhances Senescence and Sensitivity to the Senolytic, Navitoclax, in Triple Negative Breast Tumor Cells. Biomedicines 2023, 11, 3066. [Google Scholar] [CrossRef]
- Sato, K.; Iwasaki, S.; Yoshino, H. Effects and Related Mechanisms of the Senolytic Agent ABT-263 on the Survival of Irradiated A549 and Ca9-22 Cancer Cells. Int. J. Mol. Sci. 2021, 22, 13233. [Google Scholar] [CrossRef] [PubMed]
- Shahbandi, A.; Rao, S.G.; Anderson, A.Y.; Frey, W.D.; Olayiwola, J.O.; Ungerleider, N.A.; Jackson, J.G. BH3 Mimetics Selectively Eliminate Chemotherapy-Induced Senescent Cells and Improve Response in TP53 Wild-Type Breast Cancer. Cell Death Differ. 2020, 27, 3097–3116. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.K.; Roberts, R.L.; Benson, R.D.; Pierce, J.L.; Yu, K.; Hamrick, M.W.; McGee-Lawrence, M.E. The Senolytic Drug Navitoclax (ABT-263) Causes Trabecular Bone Loss and Impaired Osteoprogenitor Function in Aged Mice. Front. Cell Dev. Biol. 2020, 8, 354. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Dai, H.M.; Ling, Y.Y.; Stout, M.B.; Pirtskhalava, T.; Giorgadze, N.; Johnson, K.O.; Giles, C.B.; et al. Identification of a Novel Senolytic Agent, Navitoclax, Targeting the Bcl-2 Family of Anti-apoptotic Factors. Aging Cell 2016, 15, 428–435. [Google Scholar] [CrossRef]
- Gandhi, L.; Camidge, D.R.; Ribeiro de Oliveira, M.; Bonomi, P.; Gandara, D.; Khaira, D.; Hann, C.L.; McKeegan, E.M.; Litvinovich, E.; Hemken, P.M.; et al. Phase I Study of Navitoclax (ABT-263), a Novel Bcl-2 Family Inhibitor, in Patients With Small-Cell Lung Cancer and Other Solid Tumors. J. Clin. Oncol. 2011, 29, 909–916. [Google Scholar] [CrossRef]
- Leverson, J.D.; Phillips, D.C.; Mitten, M.J.; Boghaert, E.R.; Diaz, D.; Tahir, S.K.; Belmont, L.D.; Nimmer, P.; Xiao, Y.; Ma, X.M.; et al. Exploiting Selective BCL-2 Family Inhibitors to Dissect Cell Survival Dependencies and Define Improved Strategies for Cancer Therapy. Sci. Transl. Med. 2015, 7, 279ra40. [Google Scholar] [CrossRef]
- Tan, X.; Tong, J.; Wang, Y.-J.; Fletcher, R.; Schoen, R.E.; Yu, J.; Shen, L.; Zhang, L. BET Inhibitors Potentiate Chemotherapy and Killing of SPOP-Mutant Colon Cancer Cells via Induction of DR5. Cancer Res. 2019, 79, 1191–1203. [Google Scholar] [CrossRef] [PubMed]
- Shorstova, T.; Foulkes, W.D.; Witcher, M. Achieving Clinical Success with BET Inhibitors as Anti-Cancer Agents. Br. J. Cancer 2021, 124, 1478–1490. [Google Scholar] [CrossRef]
- Leonard, B.; Brand, T.M.; O’Keefe, R.A.; Lee, E.D.; Zeng, Y.; Kemmer, J.D.; Li, H.; Grandis, J.R.; Bhola, N.E. BET Inhibition Overcomes Receptor Tyrosine Kinase-Mediated Cetuximab Resistance in HNSCC. Cancer Res. 2018, 78, 4331–4343. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, Y.; Diao, P.; Zhang, W.; Li, J.; Ge, H.; Song, Y.; Li, Z.; Wang, D.; Liu, L.; et al. Therapeutic Targeting of BRD4 in Head Neck Squamous Cell Carcinoma. Theranostics 2019, 9, 1777–1793. [Google Scholar] [CrossRef] [PubMed]
- Girardini, M.; Maniaci, C.; Hughes, S.J.; Testa, A.; Ciulli, A. Cereblon versus VHL: Hijacking E3 Ligases against Each Other Using PROTACs. Bioorg Med. Chem. 2019, 27, 2466–2479. [Google Scholar] [CrossRef]
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K.; et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22, 755–763. [Google Scholar] [CrossRef]
- Donati, B.; Lorenzini, E.; Ciarrocchi, A. BRD4 and Cancer: Going beyond Transcriptional Regulation. Mol. Cancer 2018, 17, 164. [Google Scholar] [CrossRef]
- Kotekar, A.; Singh, A.K.; Devaiah, B.N. BRD4 and MYC: Power Couple in Transcription and Disease. FEBS J. 2023, 290, 4820–4842. [Google Scholar] [CrossRef]
- Wakita, M.; Takahashi, A.; Sano, O.; Loo, T.M.; Imai, Y.; Narukawa, M.; Iwata, H.; Matsudaira, T.; Kawamoto, S.; Ohtani, N.; et al. A BET Family Protein Degrader Provokes Senolysis by Targeting NHEJ and Autophagy in Senescent Cells. Nat. Commun. 2020, 11, 1935. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Lim, S.L.; Tao, Y.; Li, X.; Xie, Y.; Yang, C.; Zhang, Z.; Jiang, Y.; Zhang, X.; Cao, X.; et al. PROTAC Bromodomain Inhibitor ARV-825 Displays Anti-Tumor Activity in Neuroblastoma by Repressing Expression of MYCN or c-Myc. Front. Oncol. 2020, 10, 574525. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Qian, X.; Zhang, Z.; Tao, Y.; Li, Z.; Zhang, Q.; Liang, H.; Li, X.; Xie, Y.; Zhuo, R.; et al. ARV-825 Demonstrates Antitumor Activity in Gastric Cancer via MYC-Targets and G2M-Checkpoint Signaling Pathways. Front. Oncol. 2021, 11, 753119. [Google Scholar] [CrossRef] [PubMed]
- Elshazly, A.M.; Sinanian, M.M.; Neely, V.; Chakraborty, E.; Alshehri, M.A.; McGrath, M.K.; Harada, H.; Schoenlein, P.V.; Gewirtz, D.A. BRD4 Inhibition as a Strategy to Prolong the Response to Standard of Care in Estrogen Receptor-Positive Breast Cancer. Cancers 2023, 15, 4066. [Google Scholar] [CrossRef] [PubMed]
- Finnegan, R.M.; Elshazly, A.M.; Patel, N.H.; Tyutyunyk-Massey, L.; Tran, T.H.; Kumarasamy, V.; Knudsen, E.S.; Gewirtz, D.A. The BET Inhibitor/Degrader ARV-825 Prolongs the Growth Arrest Response to Fulvestrant + Palbociclib and Suppresses Proliferative Recovery in ER-Positive Breast Cancer. Front. Oncol. 2022, 12, 966441. [Google Scholar] [CrossRef]
- Deng, Y.; Yu, C.; Chen, L.; Zhang, X.; Lei, Q.; Liu, Q.; Cai, G.; Liu, F. ARV-771 Acts as an Inducer of Cell Cycle Arrest and Apoptosis to Suppress Hepatocellular Carcinoma Progression. Front. Pharmacol. 2022, 13, 858901. [Google Scholar] [CrossRef]
- Raina, K.; Lu, J.; Qian, Y.; Altieri, M.; Gordon, D.; Rossi, A.M.K.; Wang, J.; Chen, X.; Dong, H.; Siu, K.; et al. PROTAC-Induced BET Protein Degradation as a Therapy for Castration-Resistant Prostate Cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 7124–7129. [Google Scholar] [CrossRef]
- Adelstein, D.J.; Li, Y.; Adams, G.L.; Wagner, H.; Kish, J.A.; Ensley, J.F.; Schuller, D.E.; Forastiere, A.A. An Intergroup Phase III Comparison of Standard Radiation Therapy and Two Schedules of Concurrent Chemoradiotherapy in Patients with Unresectable Squamous Cell Head and Neck Cancer. J. Clin. Oncol. 2003, 21, 92–98. [Google Scholar] [CrossRef]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and Neck Squamous Cell Carcinoma. Nat. Rev. Dis. Primers 2020, 6, 92. [Google Scholar] [CrossRef]
- Shoemaker, A.R.; Mitten, M.J.; Adickes, J.; Oleksijew, A.; Zhang, H.; Bauch, J.; Marsh, K.; Frost, D.J.; Madar, D.; Tse, C.; et al. The Bcl-2 Family Inhibitor ABT-263 Shows Significant but Reversible Thrombocytopenia in Mice. Blood 2006, 108, 1107. [Google Scholar] [CrossRef]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A Potent and Orally Bioavailable Bcl-2 Family Inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef] [PubMed]
- Shahar, N.; Larisch, S. Inhibiting the Inhibitors: Targeting Anti-Apoptotic Proteins in Cancer and Therapy Resistance. Drug Resist. Updates 2020, 52, 100712. [Google Scholar] [CrossRef]
- Maji, S.; Panda, S.; Samal, S.K.; Shriwas, O.; Rath, R.; Pellecchia, M.; Emdad, L.; Das, S.K.; Fisher, P.B.; Dash, R. Bcl-2 Antiapoptotic Family Proteins and Chemoresistance in Cancer. In Advances in Cancer Research; Elsevier: Amsterdam, The Netherlands, 2018; Volume 137, pp. 37–75. ISBN 978-0-12-815123-5. [Google Scholar]
- Sarnik, J.; Popławski, T.; Tokarz, P. BET Proteins as Attractive Targets for Cancer Therapeutics. Int. J. Mol. Sci. 2021, 22, 11102. [Google Scholar] [CrossRef] [PubMed]
- Barrows, J.K.; Lin, B.; Quaas, C.E.; Fullbright, G.; Wallace, E.N.; Long, D.T. BRD4 Promotes Resection and Homology-Directed Repair of DNA Double-Strand Breaks. Nat. Commun. 2022, 13, 3016. [Google Scholar] [CrossRef] [PubMed]
- Zanca, C.; Villa, G.R.; Benitez, J.A.; Thorne, A.H.; Koga, T.; D’Antonio, M.; Ikegami, S.; Ma, J.; Boyer, A.D.; Banisadr, A.; et al. Glioblastoma Cellular Cross-Talk Converges on NF-κB to Attenuate EGFR Inhibitor Sensitivity. Genes Dev. 2017, 31, 1212–1227. [Google Scholar] [CrossRef]
- Li, X.; Baek, G.; Ramanand, S.G.; Sharp, A.; Gao, Y.; Yuan, W.; Welti, J.; Rodrigues, D.N.; Dolling, D.; Figueiredo, I.; et al. BRD4 Promotes DNA Repair and Mediates the Formation of TMPRSS2-ERG Gene Rearrangements in Prostate Cancer. Cell Rep. 2018, 22, 796–808. [Google Scholar] [CrossRef]
- Noblejas-López, M.D.M.; Nieto-Jimenez, C.; Burgos, M.; Gómez-Juárez, M.; Montero, J.C.; Esparís-Ogando, A.; Pandiella, A.; Galán-Moya, E.M.; Ocaña, A. Activity of BET-Proteolysis Targeting Chimeric (PROTAC) Compounds in Triple Negative Breast Cancer. J. Exp. Clin. Cancer Res. 2019, 38, 383. [Google Scholar] [CrossRef]
- Lam, F.C.; Kong, Y.W.; Huang, Q.; Vu Han, T.-L.; Maffa, A.D.; Kasper, E.M.; Yaffe, M.B. BRD4 Prevents the Accumulation of R-Loops and Protects against Transcription–Replication Collision Events and DNA Damage. Nat. Commun. 2020, 11, 4083. [Google Scholar] [CrossRef]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocols to Detect Senescence-Associated Beta-Galactosidase (SA-Βgal) Activity, a Biomarker of Senescent Cells in Culture and in Vivo. Nat. Protoc. 2009, 4, 1798–1806. [Google Scholar] [CrossRef]
- Short, S.; Fielder, E.; Miwa, S.; von Zglinicki, T. Senolytics and Senostatics as Adjuvant Tumour Therapy. EBioMedicine 2019, 41, 683–692. [Google Scholar] [CrossRef]
- Segu, V.B.; Li, G.; Metz, S.A. Use of a Soluble Tetrazolium Compound to Assay Metabolic Activation of Intact Beta Cells. Metabolism 1998, 47, 824–830. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A Biomarker That Identifies Senescent Human Cells in Culture and in Aging Skin in Vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Flor, A.; Pagacz, J.; Thompson, D.; Kron, S. Far-Red Fluorescent Senescence-Associated β-Galactosidase Probe for Identification and Enrichment of Senescent Tumor Cells by Flow Cytometry. J. Vis. Exp. 2022, 187, e64176. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luffman, N.; Ahmadinejad, F.; Finnegan, R.M.; Raymond, M.; Gewirtz, D.A.; Harada, H. Effectiveness of PROTAC BET Degraders in Combating Cisplatin Resistance in Head and Neck Cancer Cells. Int. J. Mol. Sci. 2025, 26, 6185. https://doi.org/10.3390/ijms26136185
Luffman N, Ahmadinejad F, Finnegan RM, Raymond M, Gewirtz DA, Harada H. Effectiveness of PROTAC BET Degraders in Combating Cisplatin Resistance in Head and Neck Cancer Cells. International Journal of Molecular Sciences. 2025; 26(13):6185. https://doi.org/10.3390/ijms26136185
Chicago/Turabian StyleLuffman, Natalie, Fereshteh Ahmadinejad, Ryan M. Finnegan, Marissa Raymond, David A. Gewirtz, and Hisashi Harada. 2025. "Effectiveness of PROTAC BET Degraders in Combating Cisplatin Resistance in Head and Neck Cancer Cells" International Journal of Molecular Sciences 26, no. 13: 6185. https://doi.org/10.3390/ijms26136185
APA StyleLuffman, N., Ahmadinejad, F., Finnegan, R. M., Raymond, M., Gewirtz, D. A., & Harada, H. (2025). Effectiveness of PROTAC BET Degraders in Combating Cisplatin Resistance in Head and Neck Cancer Cells. International Journal of Molecular Sciences, 26(13), 6185. https://doi.org/10.3390/ijms26136185