
Potassium Iodide Induces Apoptosis in Salivary Gland Cancer Cells

 , , ,
, , ,  , , , ,
, , , , 
Abstract

1. Introduction
2. Results
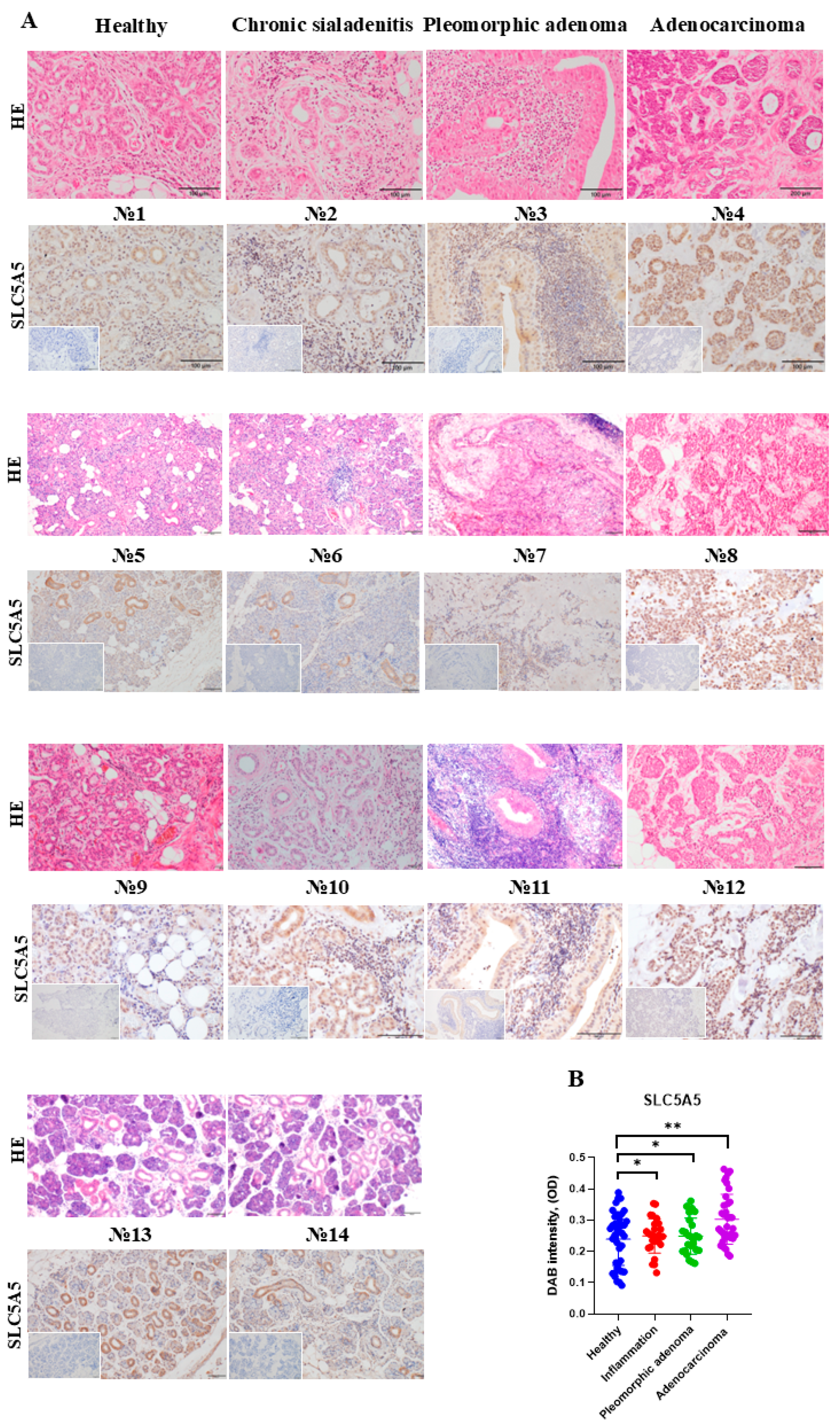
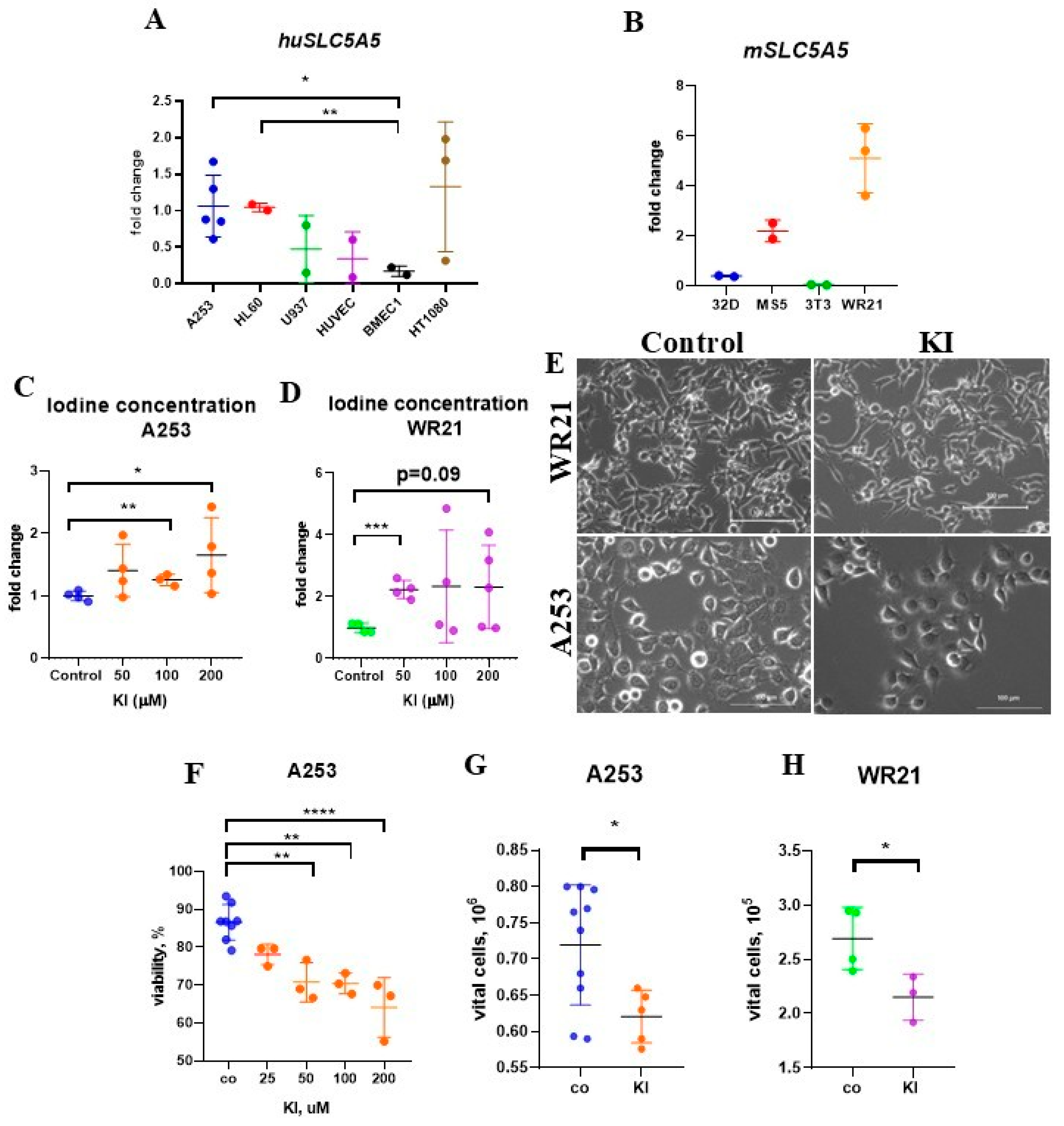
2.1. Enhanced Iodide Uptake in SLC5A5-Expressing SGC Cells
2.2. KI Treatment Does Not Affect the Migration of SGC Cells
2.3. Treatment with KI Impairs the Growth and Viability of SGC Cells
2.4. KI Promotes Apoptosis in SGC Cells
2.5. KI and All-Trans Retinoic Acid Treatment Do Not Alter SLC5A5 Expression
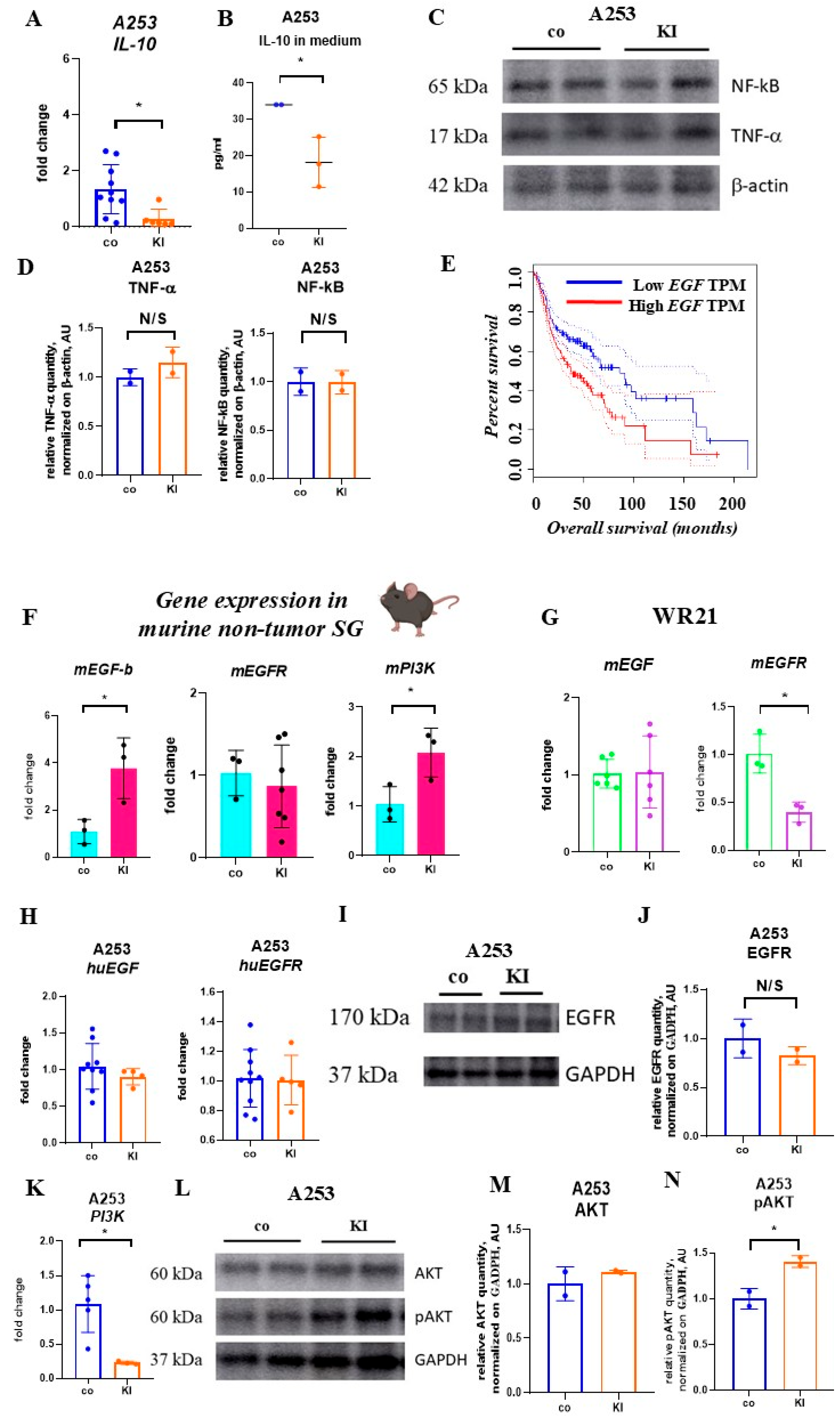
2.6. KI Does Not Alter Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells and Tumor Necrosis Factor-Alpha-Expression
2.7. KI Enhances EGF Expression in Non-Malignant but Not Malignant SG Cells
2.8. KI-Driven Reactive Oxygen Species (ROS) Generation Controls SGC Cell Proliferation
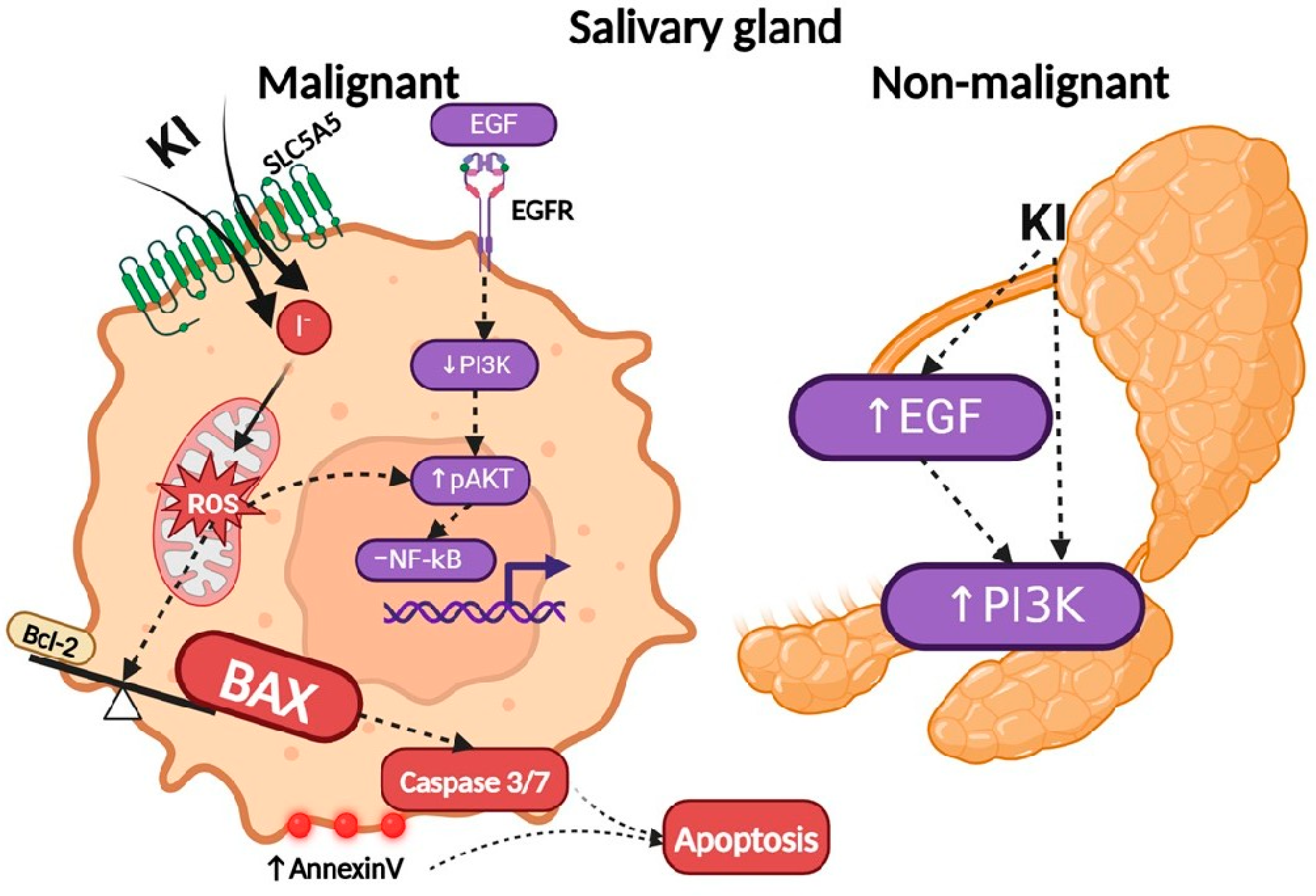
3. Discussion
Limitation of the Study
4. Materials and Methods
4.1. SG Clinical Samples
4.2. HE and SLC5A5 Staining of Human Tissue Sections
4.3. KI and Cytokines
4.4. Cell Lines
4.5. Mice
4.6. Cell Proliferation or Cytotoxicity Assay After Drug Addition
4.7. Cell Cycle
4.8. Proliferation CCK-8 Assay
4.9. Apoptotic Assay
4.10. Caspase 3/7 Assay
4.11. Detection of Total ROS
4.12. Iodine Concentration Detection in Cell Lysate
4.13. Protein Concentration
4.14. Western Blotting
4.15. Migration Assay
4.16. ELISA
4.17. Cytokine Expression in Cell Lines After Drug Addition
4.18. Quantification of mRNA Expression by qPCR
4.19. Bioinformatics Analysis
4.20. Statistical Analysis
4.21. Cultured Cell Image Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.L.; Nishimoto, I.N.; Califano, J.A.; Kowalski, L.P. Trends in incidence and prognosis for head and neck cancer in the United States: A Site-specific Analysis of the SEER Database. Int. J. Cancer 2005, 114, 806–816. [Google Scholar] [CrossRef] [PubMed]
- Steuer, C.E.; Hanna, G.J.; Viswanathan, K.; Bates, J.E.; Kaka, A.S.; Schmitt, N.C.; Ho, A.L.; Saba, N.F. The evolving landscape of salivary gland tumors. CA Cancer J. Clin. 2023, 73, 597–619. [Google Scholar] [CrossRef] [PubMed]
- Geiger, J.L.; Ismaila, N.; Beadle, B.; Caudell, J.J.; Chau, N.; Deschler, D.; Glastonbury, C.; Kaufman, M.; Lamarre, E.; Lau, H.Y.; et al. Management of salivary gland malignancy: ASCO Guideline. J. Clin. Oncol. 2021, 39, 1909–1941. [Google Scholar] [CrossRef]
- Zhang, Q.; Yang, Y.; Yang, P.; Tan, Y.; Liu, X.; Xiong, B.; Qiu, J. Cosmetic approach selection in parotidectomy for benign parotid gland tumour according to its location. J. Plast. Reconstr. Aesthetic Surg. 2020, 73, 921–926. [Google Scholar] [CrossRef]
- Airoldi, M.; Pedani, F.; Succo, G.; Gabriele, A.M.; Ragona, R.; Marchionatti, S.; Bumma, C. Phase II randomized trial comparing vinorelbine versus vinorelbine plus cisplatin in patients with recurrent salivary gland malignancies. Cancer 2001, 91, 541–547. [Google Scholar] [CrossRef]
- Nakano, K.; Sato, Y.; Sasaki, T.; Shimbashi, W.; Fukushima, H.; Yonekawa, H.; Mitani, H.; Kawabata, K.; Takahashi, S. Combination chemotherapy of carboplatin and paclitaxel for advanced/metastatic salivary gland carcinoma patients: Differences in responses by different pathological diagnoses. Acta Otolaryngol. 2016, 136, 948–951. [Google Scholar] [CrossRef]
- Laurie, S.A.; Siu, L.L.; Winquist, E.; Maksymiuk, A.; Harnett, E.L.; Walsh, W.; Tu, D.; Parulekar, W.R. A Phase 2 study of platinum and gemcitabine in patients with advanced salivary gland cancer. Cancer 2010, 116, 362–368. [Google Scholar] [CrossRef]
- Locati, L.D.; Cavalieri, S.; Bergamini, C.; Resteghini, C.; Alfieri, S.; Calareso, G.; Bossi, P.; Perrone, F.; Tamborini, E.; Quattrone, P.; et al. Phase II trial with axitinib in recurrent and/or metastatic salivary gland cancers of the upper aerodigestive tract. Head Neck 2019, 41, 3670–3676. [Google Scholar] [CrossRef]
- Tchekmedyian, V.; Sherman, E.J.; Dunn, L.; Tran, C.; Baxi, S.; Katabi, N.; Antonescu, C.R.; Ostrovnaya, I.; Haque, S.S.; Pfister, D.G.; et al. Phase II study of lenvatinib in patients with progressive, recurrent or metastatic adenoid cystic carcinoma. J. Clin. Oncol. 2019, 37, 1529–1537. [Google Scholar] [CrossRef]
- La Perle, K.M.D.; Kim, D.C.; Hall, N.C.; Bobbey, A.; Shen, D.H.; Nagy, R.S.; Wakely, P.E.; Lehman, A.; Jarjoura, D.; Jhiang, S.M. Modulation of sodium/iodide symporter expression in the salivary gland. Thyroid 2013, 23, 1029–1036. [Google Scholar] [CrossRef] [PubMed]
- Mandel, S.J.; Mandel, L. Radioactive iodine and the salivary glands. Thyroid 2003, 13, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Aceves, C.; Anguiano, B.; Delgado, G. The extrathyronine actions of iodine as antioxidant, apoptotic, and differentiation factor in various tissues. Thyroid 2013, 23, 938–946. [Google Scholar] [CrossRef] [PubMed]
- Oriel, J.D. The Scars of Venus; Springer: London, UK, 1994; ISBN 978-1-4471-2070-4. [Google Scholar]
- Brouse, S.D.; Johnson, M.L.; Muldrew-Jones, K.M. Iodine-containing compounds. In Comprehensive Handbook of Iodine; Elsevier: Amsterdam, The Netherlands, 2009; pp. 919–925. [Google Scholar]
- Xue, S.; Gu, R.; Wu, T.; Zhang, M.; Wang, X. Oral potassium iodide for the treatment of sporotrichosis. In Cochrane Database of Systematic Reviews; Wu, T., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2006. [Google Scholar]
- Thotan, S.P.; Kumar, V.; Gupta, A.; Mallya, A.; Rao, S. Subcutaneous phycomycosis—Fungal infection mimicking a soft tissue tumor: A case report and review of literature. J. Trop. Pediatr. 2010, 56, 65–66. [Google Scholar] [CrossRef]
- Sakafu, L.L.; Mselle, T.F.; Mwaiselage, J.D.; Maunda, K.K.; Eddin, B.S.; Zafereo, M.E. Thyroid cancer and iodine deficiency status: A 10-year review at a single cancer center in Tanzania. OTO Open 2018, 2, 2473974X18777238. [Google Scholar] [CrossRef]
- Zimmermann, M.B.; Galetti, V. Iodine intake as a risk factor for thyroid cancer: A comprehensive review of animal and human studies. Thyroid Res. 2015, 8, 8. [Google Scholar] [CrossRef]
- Manjer, J.; Sandsveden, M.; Borgquist, S. Serum iodine and breast cancer risk: A prospective nested case–control study stratified for selenium levels. Cancer Epidemiol. Biomark. Prev. 2020, 29, 1335–1340. [Google Scholar] [CrossRef]
- Gulaboglu, M.; Yildiz, L.; Celebi, F.; Gul, M.; Peker, K. Comparison of iodine contents in gastric cancer and surrounding normal tissues. Clin. Chem. Lab. Med. CCLM 2005, 43, 581–584. [Google Scholar] [CrossRef]
- Kwon, Y.-J.; Lee, H.-S.; Kang, S.-W.; Lee, J.-W. Association between consumption of iodine-rich foods and thyroid cancer prevalence: Findings from a large population-based study. Nutrients 2024, 16, 1041. [Google Scholar] [CrossRef]
- Stadel, B. Dietary iodine and risk of breast, endometrial, and ovarian cancer. Lancet 1976, 307, 890–891. [Google Scholar] [CrossRef]
- Cann, S.A.; van Netten, J.P.; van Netten, C. Hypothesis: Iodine, selenium and the development of breast cancer. Cancer Causes Control 2000, 11, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Zouhair, A.; Azria, D.; Ozsahin, M. The epidermal growth factor receptor (egfr) in head and neck cancer: Its role and treatment implications. Radiat. Oncol. 2006, 1, 11. [Google Scholar] [CrossRef]
- Franke, T.F.; Kaplan, D.R.; Cantley, L.C.; Toker, A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science 1997, 275, 665–668. [Google Scholar] [CrossRef]
- Ren, Y.; Hong, Y.; He, W.; Liu, Y.; Chen, W.; Wen, S.; Sun, M. EGF/EGFR promotes salivary adenoid cystic carcinoma cell malignant neural invasion via activation of PI3K/AKT and MEK/ERK signaling. Curr. Cancer Drug Targets 2022, 22, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Ni, W.; Chen, Z.; Zhou, X.; Yang, R.; Yu, M.; Lu, J.; Kaye, F.J.; Wu, L. Targeting Notch and EGFR signaling in human mucoepidermoid carcinoma. Signal Transduct. Target. Ther. 2021, 6, 27. [Google Scholar] [CrossRef]
- Kuroda, M.; Komatsu, N.; Kosai, A.; Hamakubo, T.; Abe, T. Anti-EGFR antibody immunotoxins improve cytotoxic effects in the salivary gland cancer A253 cell Line. J. Oral Maxillofac. Surg. Med. Pathol. 2024, 37, 450–454. [Google Scholar] [CrossRef]
- Huang, Y.; Yu, T.; Fu, X.; Chen, J.; Liu, Y.; Li, C.; Xia, Y.; Zhang, Z.; Li, L. EGFR inhibition prevents in vitro tumor growth of salivary adenoid cystic carcinoma. BMC Cell Biol. 2013, 14, 13. [Google Scholar] [CrossRef] [PubMed]
- Arroyo-Helguera, O.; Rojas, E.; Delgado, G.; Aceves, C. Signaling pathways involved in the antiproliferative effect of molecular iodine in normal and tumoral breast cells: Evidence that 6-iodolactone mediates apoptotic effects. Endocr. Relat. Cancer 2008, 15, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Rösner, H.; Torremante, P.; Möller, W.; Gärtner, R. Antiproliferative/cytotoxic activity of molecular iodine and iodolactones in various human carcinoma cell lines. no interfering with EGF-signaling, but evidence for apoptosis. Exp. Clin. Endocrinol. Diabetes 2009, 118, 410–419. [Google Scholar] [CrossRef]
- Bortner, C.D.; Cidlowski, J.A. Uncoupling cell shrinkage from apoptosis reveals that Na+ influx is required for volume loss during programmed cell death. J. Biol. Chem. 2003, 278, 39176–39184. [Google Scholar] [CrossRef]
- Shrivastava, A.; Tiwari, M.; Sinha, R.A.; Kumar, A.; Balapure, A.K.; Bajpai, V.K.; Sharma, R.; Mitra, K.; Tandon, A.; Godbole, M.M. Molecular iodine induces caspase-independent apoptosis in human breast carcinoma cells involving the mitochondria-mediated pathway. J. Biol. Chem. 2006, 281, 19762–19771. [Google Scholar] [CrossRef]
- Dohán, O.; De la Vieja, A.; Carrasco, N. Hydrocortisone and purinergic signaling stimulate sodium/iodide symporter (NIS)-mediated iodide transport in breast cancer cells. Mol. Endocrinol. 2006, 20, 1121–1137. [Google Scholar] [CrossRef] [PubMed]
- Cline, B.L.; Jiang, W.; Lee, C.; Cao, Z.; Yang, X.; Zhan, S.; Chong, H.; Zhang, T.; Han, Z.; Wu, X.; et al. Potassium iodide nanoparticles enhance radiotherapy against breast cancer by exploiting the sodium-iodide symporter. ACS Nano 2021, 15, 17401–17411. [Google Scholar] [CrossRef]
- Skrypnyk, M.; Yatsenko, T.; Riabets, O.; Salama, Y.; Skikevych, M.; Osada, T.; Tobita, M.; Takahashi, S.; Hattori, K.; Heissig, B. Interleukin-10 induces TNF-driven apoptosis and ROS production in salivary gland cancer cells. Heliyon 2024, 10, e31777. [Google Scholar] [CrossRef] [PubMed]
- Guazzo, E.; Cooper, C.; Wilkinson, L.; Feng, S.; King, B.; Simpson, F.; Porceddu, S.; Panizza, B.; Coward, J.I.G. Therapeutic implications of immune-profiling and EGFR expression in salivary gland carcinoma. Head Neck 2021, 43, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Locati, L.D.; Bossi, P.; Perrone, F.; Potepan, P.; Crippa, F.; Mariani, L.; Casieri, P.; Orsenigo, M.; Losa, M.; Bergamini, C.; et al. Cetuximab in recurrent and/or metastatic salivary gland carcinomas: A phase II study. Oral Oncol. 2009, 45, 574–578. [Google Scholar] [CrossRef]
- Tramontano, D.; Veneziani, B.M.; Lombardi, A.; Villone, G.; Ingbar, S.H. Iodine inhibits the proliferation of rat thyroid cells in culture. Endocrinology 1989, 125, 984–992. [Google Scholar] [CrossRef]
- Marquard, F.E.; Jücker, M. PI3K/AKT/MTOR signaling as a molecular target in head and neck cancer. Biochem. Pharmacol. 2020, 172, 113729. [Google Scholar] [CrossRef]
- Arbez-Evangelista, C.; Arroyo-Xochihua, O.; Ortega-Ibarra, I.H.; Ortega-Ibarra, E.; De León-Ramírez, Y.M.; Cuevas-Romero, E.; Arroyo-Helguera, O. Excess iodine consumption induces oxidative stress and pancreatic damage independently of chemical form in male wistar rats: Participation of PPAR-γ and C/EBP-β. Biology 2024, 13, 466. [Google Scholar] [CrossRef]
- De Almeida, A.J.P.O.; de Oliveira, J.C.P.L.; da Silva Pontes, L.V.; de Souza Júnior, J.F.; Gonçalves, T.A.F.; Dantas, S.H.; de Almeida Feitosa, M.S.; Silva, A.O.; de Medeiros, I.A. ROS: Basic concepts, sources, cellular signaling, and its implications in aging pathways. Oxidative Med. Cell. Longev. 2022, 2022, 1225578. [Google Scholar] [CrossRef]
- Murata, H.; Ihara, Y.; Nakamura, H.; Yodoi, J.; Sumikawa, K.; Kondo, T. Glutaredoxin Exerts an antiapoptotic effect by regulating the redox state of Akt. J. Biol. Chem. 2003, 278, 50226–50233. [Google Scholar] [CrossRef] [PubMed]
- Riesco-Eizaguirre, G.; Santisteban, P.; De la Vieja, A. The complex regulation of NIS expression and activity in thyroid and extrathyroidal tissues. Endocr. Relat. Cancer 2021, 28, T141–T165. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.M.; Ahn, B.-C. Molecular mechanisms of radioactive iodine refractoriness in differentiated thyroid cancer: Impaired sodium iodide symporter (NIS) expression owing to altered signaling pathway activity and intracellular localization of NIS. Theranostics 2021, 11, 6251–6277. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, G.; Singh, R.; Sharma, R.; Balapure, A.K.; Godbole, M.M. Differential action of iodine on mitochondria from human tumoral- and extra-tumoral tissue in inducing the release of apoptogenic proteins. Mitochondrion 2002, 2, 199–210. [Google Scholar] [CrossRef]
- Sun, S.-Y. N-Acetylcysteine, reactive oxygen species and beyond. Cancer Biol. Ther. 2010, 9, 109–110. [Google Scholar] [CrossRef]
- Zhang, L.; Sharma, S.; Zhu, L.X.; Kogai, T.; Hershman, J.M.; Brent, G.A.; Dubinett, S.M.; Huang, M. Nonradioactive iodide effectively induces apoptosis in genetically modified lung cancer cells. Cancer Res. 2003, 63, 5065–5072. [Google Scholar]
- Vitale, M.; Di Matola, T.; D’Ascoli, F.; Salzano, S.; Bogazzi, F.; Fenzi, G.; Martino, E.; Rossi, G. Iodide excess induces apoptosis in thyroid cells through a p53-independent mechanism involving oxidative stress. Endocrinology 2000, 141, 598–605. [Google Scholar] [CrossRef]
- Toshiyuki, M.; Reed, J.C. Tumor suppressor p53 is a direct transcriptional activator of the human Bax gene. Cell 1995, 80, 293–299. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-mediated cellular signaling. Oxidative Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef]
- Tang, D.; Okada, H.; Ruland, J.; Liu, L.; Stambolic, V.; Mak, T.W.; Ingram, A.J. Akt is activated in response to an apoptotic signal. J. Biol. Chem. 2001, 276, 30461–30466. [Google Scholar] [CrossRef]
- Nakatani, K.; Thompson, D.A.; Barthel, A.; Sakaue, H.; Liu, W.; Weigel, R.J.; Roth, R.A. Up-regulation of Akt3 in estrogen receptor-deficient breast cancers and androgen-independent prostate cancer lines. J. Biol. Chem. 1999, 274, 21528–21532. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Huang, J.; Wang, J.; Hu, S.; Li, S.; Xiong, C.; Liu, F.; Yuan, C.; Hu, Y.; Sun, W. Potassium iodide promotes the pyroptosis of thyroid follicular epithelial cells through the PARP1-NF-ΚB-NLRP3 inflammasome. Chin. J. Endocrinol. Metab. 2021, 37, 820–829. [Google Scholar] [CrossRef]
- Faria, M.; Domingues, R.; Paixão, F.; Bugalho, M.J.; Matos, P.; Silva, A.L. TNFα-mediated activation of NF-ΚB downregulates sodium-iodide symporter expression in thyroid cells. PLoS ONE 2020, 15, e0228794. [Google Scholar] [CrossRef] [PubMed]
- Haddad, R.; Colevas, A.D.; Krane, J.F.; Cooper, D.; Glisson, B.; Amrein, P.C.; Weeks, L.; Costello, R.; Posner, M. Herceptin in patients with advanced or metastatic salivary gland carcinomas. A phase II study. Oral Oncol. 2003, 39, 724–727. [Google Scholar] [CrossRef]
- Takahashi, H.; Tada, Y.; Saotome, T.; Akazawa, K.; Ojiri, H.; Fushimi, C.; Masubuchi, T.; Matsuki, T.; Tani, K.; Osamura, R.Y.; et al. Phase II trial of trastuzumab and docetaxel in patients with human epidermal growth factor receptor 2–positive salivary duct carcinoma. J. Clin. Oncol. 2019, 37, 125–134. [Google Scholar] [CrossRef]
- Kurzrock, R.; Bowles, D.W.; Kang, H.; Meric-Bernstam, F.; Hainsworth, J.; Spigel, D.R.; Bose, R.; Burris, H.; Sweeney, C.J.; Beattie, M.S.; et al. Targeted therapy for advanced salivary gland carcinoma based on molecular profiling: Results from MyPathway, a phase iia multiple basket study. Ann. Oncol. 2020, 31, 412–421. [Google Scholar] [CrossRef]
- Dagogo-jack, S. Dietary iodine affects epidermal growth factor levels in mouse thyroid and submaxillary glands. Endocr. Res. 1994, 20, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Vega, A.; Vega-Riveroll, L.; Ayala, T.; Peralta, G.; Torres-Martel, J.M.; Rojas, J.; Mondragón, P.; Domínguez, A.; De Obaldía, R.; Avecilla-Guerrero, C.; et al. Adjuvant Effect of Molecular Iodine in Conventional Chemo-therapy for Breast Cancer. Randomized Pilot Study. Nutrients 2019, 11, 1623. [Google Scholar] [CrossRef]
- Crissman, H.A.; Steinkamp, J.A. Rapid, simultaneous measurement of DNA, protein, and cell volume in single cells from large mammalian cell populations. J. Cell Biol. 1973, 59, 766–771. [Google Scholar] [CrossRef]
- Kim, H.; Xue, X. Detection of total reactive oxygen species in adherent cells by 2’,7’-dichlorodihydrofluorescein diacetate staining. J. Vis. Exp. 2020, 23, e60682. [Google Scholar] [CrossRef]
- Ohashi, T.; Yamaki, M.; Pandav, C.S.; Karmarkar, M.G.; Irie, M. Simple microplate method for determination of urinary iodine. Clin. Chem. 2000, 46, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Number | Diagnosis, Histological Findings | Treatment | Age | Gender |
|---|---|---|---|---|
| 1 | Calculous sialadenitis of the right submandibular SG. Pathological biopsy included adjacent tissues; morphologically healthy tissue in the biopsy material. | The entire submandibular SG was extracted. | 48 | Male |
| 2 | Chronic sialadenitis caused by multiple concrements in the parenchyma of the submandibular SGs. Chronic inflammatory changes in glandular tissue. | The entire glands were extracted. | 60 | Female |
| 3 | Pleomorphic adenoma of the right parotid SG. Benign mixed tumor characteristics in the glandular tissue. | The tumor was surgically excised along with the adjacent parotid SG. | 34 | Female |
| 4 | Adenoid cystic carcinoma of the left parotid SG. Malignant tumor with perineural invasion and a characteristic cribriform pattern (Tissue block 4). | The tumor, along with the entire SG, was surgically excised. | 45 | Female |
| 5 | Calculous sialadenitis of the right submandibular SG. Morphologically healthy SG tissue in the biopsy material. | The entire SG was surgically excised. | 40 | Male |
| 6 | Calculous sialadenitis of the right submandibular SG. Inflammatory changes in the tissue biopsy material. | The entire SG was surgically excised. | 40 | Male |
| 7 | Pleomorphic adenoma of the left parotid SG. Benign mixed tumor characteristic of pleomorphic adenoma. | The tumor was surgically excised along with the adjacent parotid SG. | 82 | Female |
| 8 | Adenoid cystic carcinoma of the left parotid SG. Malignant tumor with perineural invasion and a characteristic cribriform pattern (Tissue block 5). | The entire SG was surgically extracted. | 45 | Female |
| 9 | Calculous sialadenitis of the left submandibular SG. Morphologically intact and healthy biopsy material. | The entire SG was surgically extracted. | 31 | Male |
| 10 | Exacerbation of chronic submandibular sialadenitis. Inflammatory changes in the submandibular SG. | The entire SG was surgically extracted. | 42 | Male |
| 11 | Pleomorphic adenoma of the right submandibular SG. Presence of a benign pleomorphic adenoma. | The tumor was surgically excised along with the adjacent parotid SG. | 51 | Male |
| 12 | Adenoid cystic carcinoma of the left parotid SG. Malignant tumor with perineural invasion and a characteristic cribriform pattern. The entire gland was surgically excised (Tissue block 2). | The tumor, along with the entire SG, was surgically excised. | 45 | Female |
| 13 | Calculous sialadenitis of the left submandibular SG. Morphologically healthy tissue in the biopsy material. | Extirpation of the affected SG. | 46 | Female |
| 14 | Calculous sialadenitis of the left submandibular SG. Inflammatory changes consistent with chronic sialadenitis. | Extirpation of the affected SG. | 46 | Female |
| Target | Forward | Reverse |
|---|---|---|
| Mouse | ||
| BETA-ACTIN | GGCTGTATTCCCCTCCATCG | CCAGTTTGGTAACAATGCCATGT |
| PI3K | CCTGTAGCTGTTTACAATGAGA | GGCAATCTAATGAGCTTCTTCA |
| P21 | TCGCTGTCTTGCACTCTGGTGT | CCAATCTGCGCTTGGAGTGATAG |
| P27 | AGCAGTGTCCAGGGATGAGGAA | TTCTTGGGCGTCTGCTCCACAG |
| SLC5A5 | GCTAATTCGCTGCTCACAG | GCTGAAGCGCAGTTCTAGGT |
| CYCLIND1 | GCAGAAGGAGATTGTGCCATCC | AGGAAGCGGTCCAGGTAGTTCA |
| EGF | ACTGGTGTGACACCAAGAGGTC | CCACAGGTGATCCTCAAACACG |
| EGFR | GGACTGTGTCTCCTGCCAGAAT | GGCAGACATTCTGGATGGCACT |
| MMP9 | AGACGACATAGACGGCATCC | TCGGCTGTGGTTCAGTTGT |
| MMP2 | GGCAGTGCAATACCTGAACA | GCCCAAAGAACTTCTGCATC |
| TIMP2 | AGCCAAAGCAGTGAGCGAGAAG | GCCGTGTAGATAAACTCGATGTC |
| Human | ||
| SLC5A5 | CCAGGGAGAGGGACAGACAG | GGCTCCCTGGGTTAGGAATC |
| P21 | AGGTGGACCTGGAGACTCTCAG | TCCTCTTGGAGAAGATCAGCCG |
| P27 | CTGCAACCGACGATTCTTCT | GCATTTGGGGAACCGTCTGA |
| CYCLIND1 | CCGTCCATGCGGAAGATC | GAAGACCTCCTCCTCGCACT |
| BAX | CATGGAGCTGCAGAGGATGA | CCAGTTGAAGTTGCCGTCAG |
| BCL-2 | GGATAACGGAGGCTGGGATG | GGGCCAAACTGAGCAGAGTC |
| IL-10 | GGCACCCAGTCTGAGAACAG | ACTCTGCTGAAGGCATCTCG |
| BETA-ACTIN | CCAACCGCGAGAAGATGA | CCAGAGGCGTACAGGGATAG |
| EGF | ACGCCCTAAGTCGAGACCGGA | TGGCTGCATTCAGACACATTGCG |
| EGFR | AACACCCTGGTCTGGAAGTACG | TCGTTGGACAGCCTTCAAGACC |
| PI3K | GAAGCACCTGAATAGGCAAGTCG | GAGCATCCATGAAATCTGGTCGC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skrypnyk, M.; Yatsenko, T.; Riabets, O.; Zuieva, O.; Rodionova, I.; Skikevych, M.; Salama, Y.; Osada, T.; Tobita, M.; Takahashi, S.; et al. Potassium Iodide Induces Apoptosis in Salivary Gland Cancer Cells. Int. J. Mol. Sci. 2025, 26, 5199. https://doi.org/10.3390/ijms26115199
Skrypnyk M, Yatsenko T, Riabets O, Zuieva O, Rodionova I, Skikevych M, Salama Y, Osada T, Tobita M, Takahashi S, et al. Potassium Iodide Induces Apoptosis in Salivary Gland Cancer Cells. International Journal of Molecular Sciences. 2025; 26(11):5199. https://doi.org/10.3390/ijms26115199
Chicago/Turabian StyleSkrypnyk, Maksym, Tetiana Yatsenko, Oleksandra Riabets, Olga Zuieva, Iryna Rodionova, Margarita Skikevych, Yousef Salama, Taro Osada, Morikuni Tobita, Satoshi Takahashi, and et al. 2025. "Potassium Iodide Induces Apoptosis in Salivary Gland Cancer Cells" International Journal of Molecular Sciences 26, no. 11: 5199. https://doi.org/10.3390/ijms26115199
APA StyleSkrypnyk, M., Yatsenko, T., Riabets, O., Zuieva, O., Rodionova, I., Skikevych, M., Salama, Y., Osada, T., Tobita, M., Takahashi, S., Hattori, N., Takahashi, K., Hattori, K., & Heissig, B. (2025). Potassium Iodide Induces Apoptosis in Salivary Gland Cancer Cells. International Journal of Molecular Sciences, 26(11), 5199. https://doi.org/10.3390/ijms26115199