
The Anti-Parkinsonian A2A Receptor Antagonist Istradefylline (KW-6002) Attenuates Behavioral Abnormalities, Neuroinflammation, and Neurodegeneration in Cerebral Ischemia: An Adenosinergic Signaling Link Between Stroke and Parkinson’s Disease

 ,
, 
 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
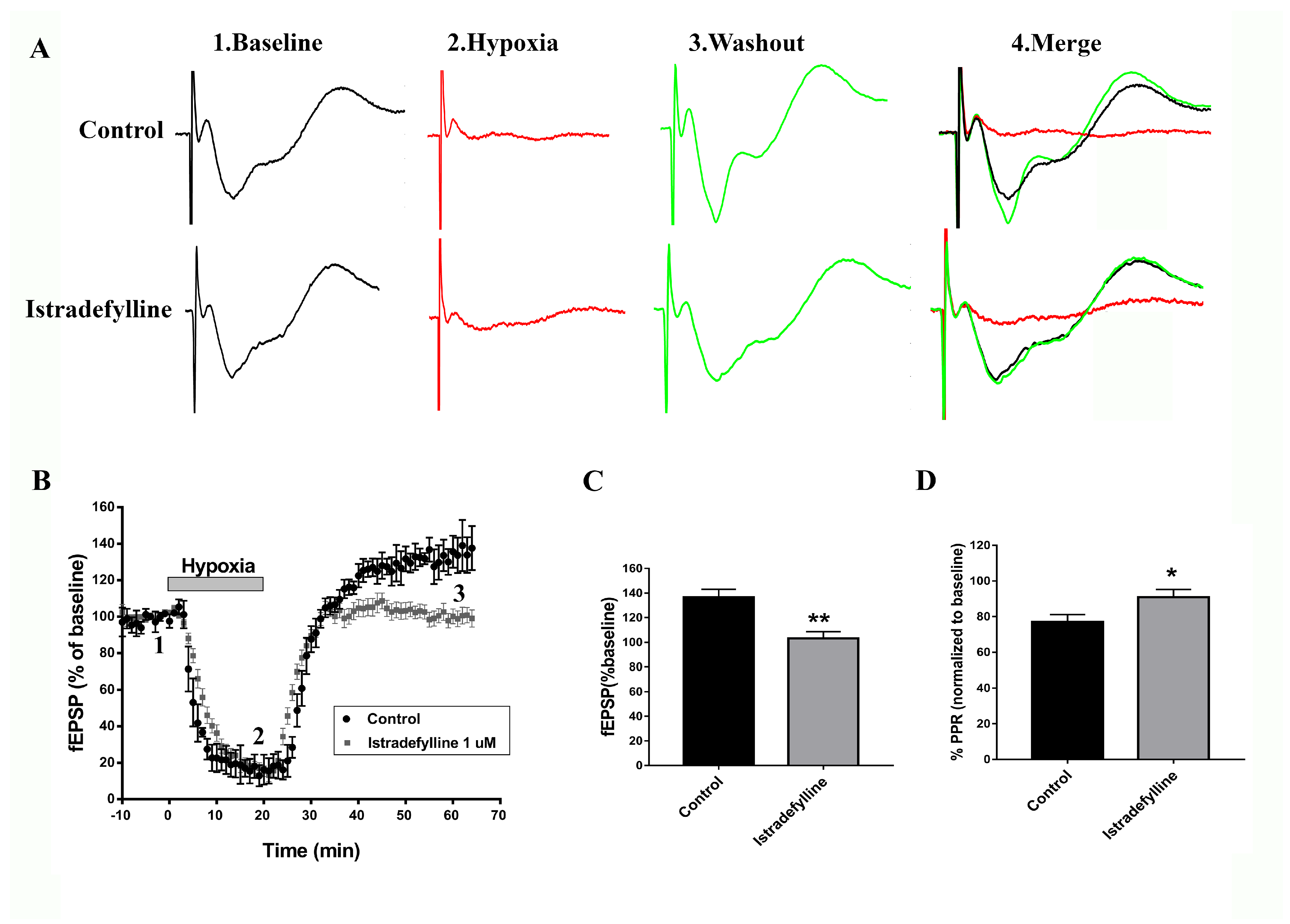
2.1. Istradefylline Shows Neuroprotective Potential in Ex Vivo Ischemic Stroke Model
2.2. Istradefylline Attenuated Hypoxia-Induced Hippocampal Cell Death
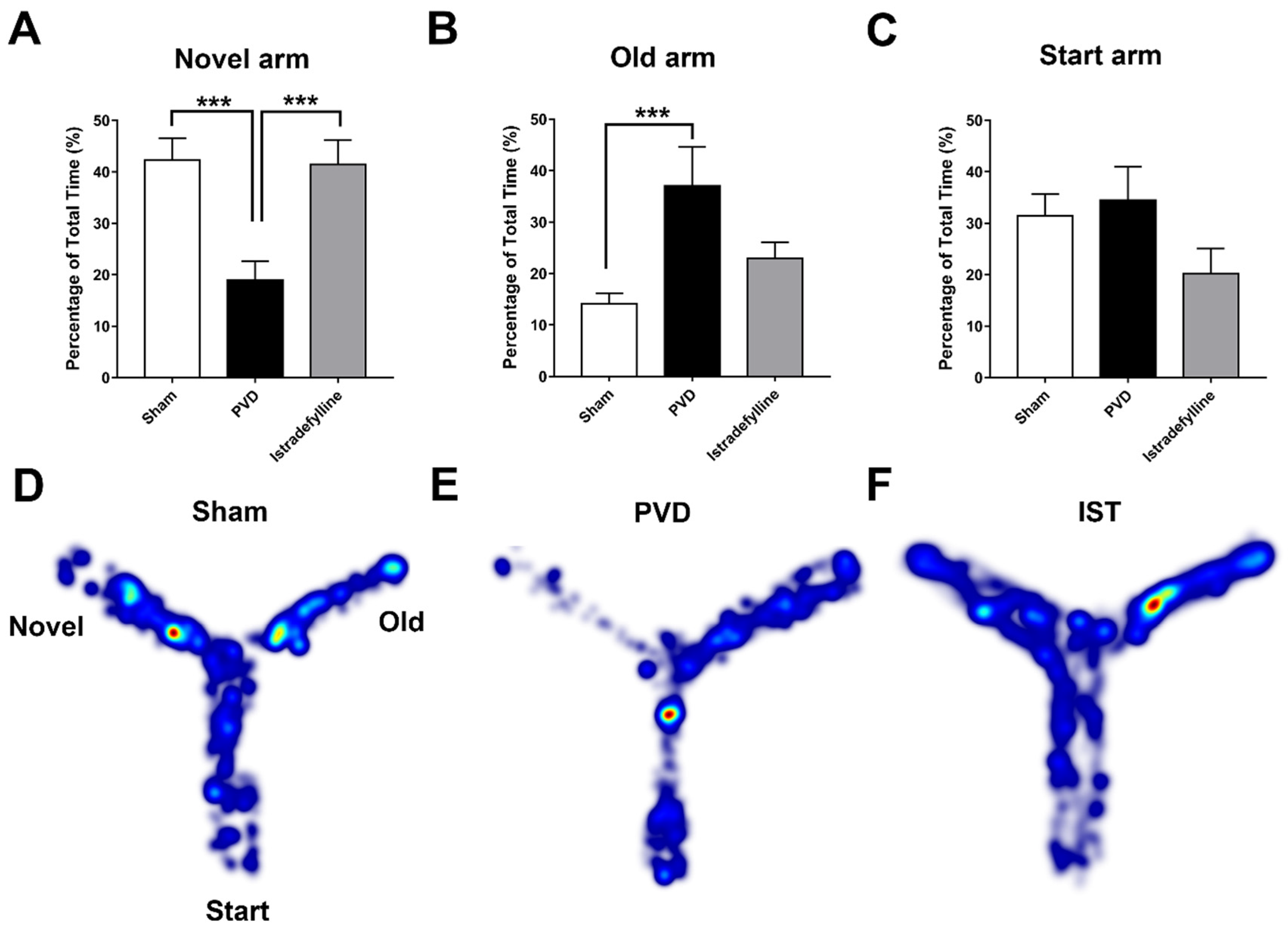
2.3. Istradefylline Prevented PVD-Induced Memory Deficits
2.4. Istradefylline Attenuated PVD-Induced Motor Deficits and Anxiety-like Behavior
2.5. Istradefylline Attenuated Post-Stroke Depression
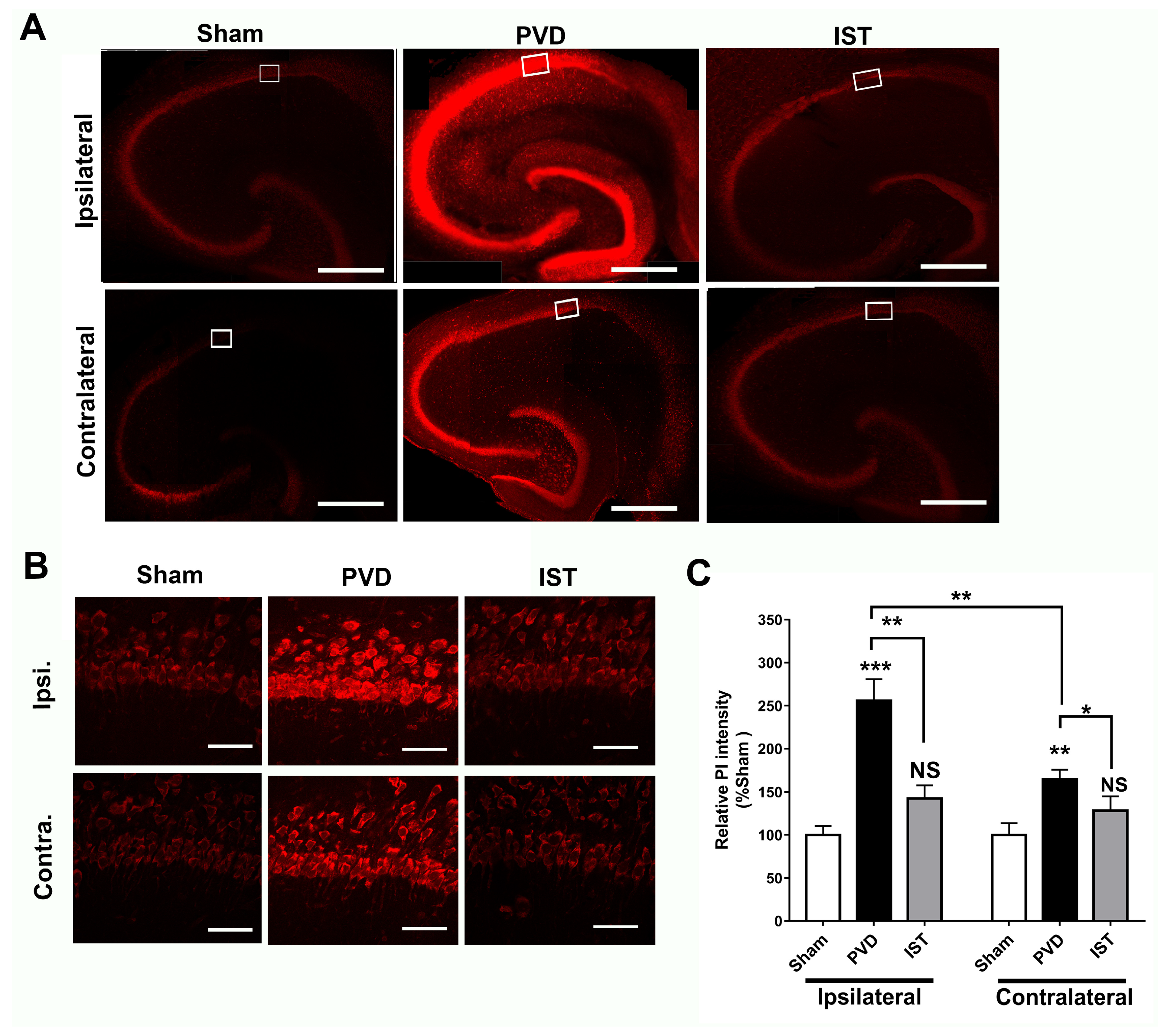
2.6. Istradefylline Inhibited PVD-Induced Hippocampal Cell Death
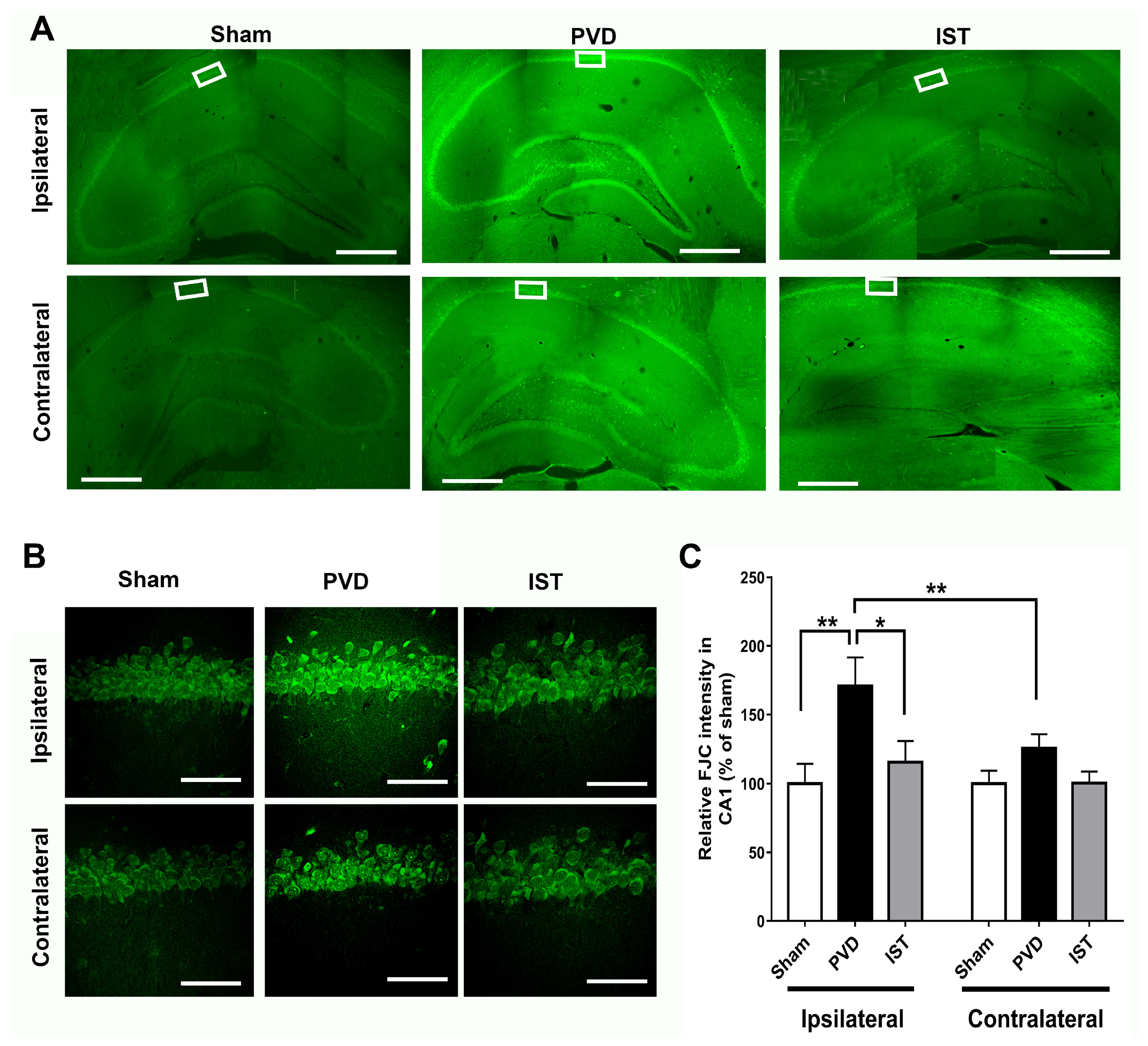
2.7. Istradefylline Decreased Hippocampal Neurodegeneration Caused by Focal Cortical Ischemia
2.8. Istradefylline Inhibited Microglia- and Astrocyte-Induced Neuroinflammation
3. Discussion
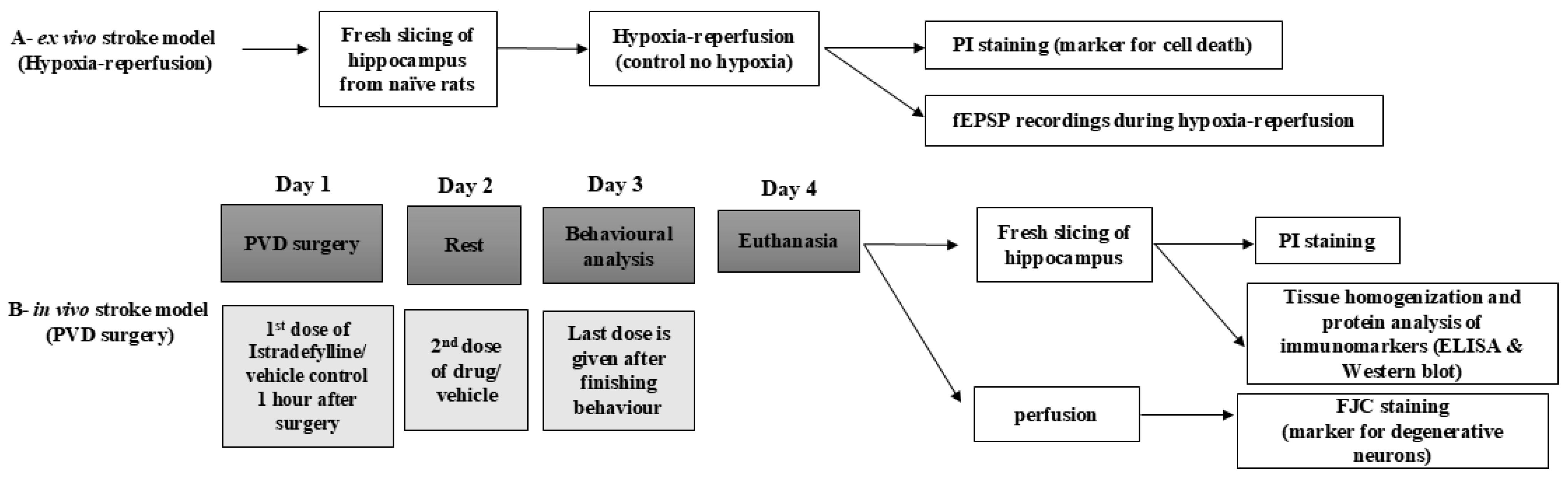
4. Materials and Methods
4.1. Animal Subjects
4.2. Hippocampal Slice Preparation and Electrophysiological Recordings
4.3. Drug Treatments
4.4. Biochemistry and Western Blotting
4.5. Enzyme-Linked Immunosorbent Assay (ELISA)
4.6. Propidium Iodide Staining
4.7. FluoroJade C Staining
4.8. Pial Vessel Disruption as a Model of Small-Vessel Stroke
4.9. Y-Maze
4.10. Open Field Test
4.11. Forced Swim Test (FST)
4.12. Rotarod
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hankey, G.J. Stroke. Lancet 2017, 389, 641–654. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, M.P.; Norrving, B.; Sacco, R.L.; Brainin, M.; Hacke, W.; Martins, S.; Pandian, J.; Feigin, V. World Stroke Organization (WSO): Global Stroke Fact Sheet 2019. Int. J. Stroke Off. J. Int. Stroke Soc. 2019, 14, 806–817. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; de Ferranti, S.D.; et al. Heart Disease and Stroke Statistics—2018 Update: A Report From the American Heart Association. Circulation 2018, 137, e67–e492. [Google Scholar] [CrossRef] [PubMed]
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart disease and stroke statistics—2020 update: A report from the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef]
- Katsanos, A.H.; Palaiodimou, L.; Zand, R.; Yaghi, S.; Kamel, H.; Navi, B.B.; Turc, G.; Romoli, M.; Sharma, V.; Mavridis, D.; et al. Abstract P82: The Impact of SARS-CoV-2 on Stroke Epidemiology and Care: A Meta-Analysis. Stroke 2021, 52 (Suppl. 1), AP82. [Google Scholar] [CrossRef]
- Mizuno, Y.; Kondo, T.; Japanese Istradefylline Study, G. Adenosine A2A receptor antagonist istradefylline reduces daily OFF time in Parkinson’s disease. Mov. Disord. 2013, 28, 1138–1141. [Google Scholar] [CrossRef] [PubMed]
- Kouli, A.; Torsney, K.M.; Kuan, W.L. Parkinson’s Disease: Etiology, Neuropathology, and Pathogenesis. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Brisbane, Australia, 2018. [Google Scholar]
- Lohmann, S.; Grigoletto, J.; Bernis, M.E.; Pesch, V.; Ma, L.; Reithofer, S.; Tamgüney, G. Ischemic stroke causes Parkinson’s disease-like pathology and symptoms in transgenic mice overexpressing alpha-synuclein. Acta Neuropathol. Commun. 2022, 10, 26. [Google Scholar] [CrossRef]
- Kummer, B.R.; Diaz, I.; Wu, X.; Aaroe, A.E.; Chen, M.L.; Iadecola, C.; Kamel, H.; Navi, B.B. Associations between cerebrovascular risk factors and parkinson disease. Ann. Neurol. 2019, 86, 572–581. [Google Scholar] [CrossRef]
- Choi, H.L.; Ahn, J.H.; Chang, W.H.; Jung, W.; Kim, B.S.; Han, K.; Youn, J.; Shin, D.W. Risk of Parkinson disease in stroke patients: A nationwide cohort study in South Korea. Eur. J. Neurol. 2024, 31, e16194. [Google Scholar] [CrossRef]
- Melani, A.; Pugliese, A.M.; Pedata, F. Adenosine receptors in cerebral ischemia. Int. Rev. Neurobiol. 2014, 119, 309–348. [Google Scholar] [CrossRef]
- Frenguelli, B.G.; Wigmore, G.; Llaudet, E.; Dale, N. Temporal and mechanistic dissociation of ATP and adenosine release during ischaemia in the mammalian hippocampus1. J. Neurochem. 2007, 101, 1400–1413. [Google Scholar] [CrossRef] [PubMed]
- Medina-Pulido, L.; Molina-Arcas, M.; Justicia, C.; Soriano, E.; Burgaya, F.; Planas, A.M.; Pastor-Anglada, M. Hypoxia and P1 receptor activation regulate the high-affinity concentrative adenosine transporter CNT2 in differentiated neuronal PC12 cells. Biochem. J. 2013, 454, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Andiné, P. Involvement of adenosine in ischemic and postischemic calcium regulation. Mol. Chem. Neuropathol. 1993, 18, 35–49. [Google Scholar] [CrossRef]
- Dunwiddie, T.V.; Masino, S.A. The role and regulation of adenosine in the central nervous system. Annu. Rev. Neurosci. 2001, 24, 31–55. [Google Scholar] [CrossRef] [PubMed]
- Brundege, J.M.; Dunwiddie, T.V. Modulation of excitatory synaptic transmission by adenosine released from single hippocampal pyramidal neurons. J. Neurosci. 1996, 16, 5603–5612. [Google Scholar] [CrossRef]
- Dunwiddie, T.V.; Diao, L. Extracellular adenosine concentrations in hippocampal brain slices and the tonic inhibitory modulation of evoked excitatory responses. J. Pharmacol. Exp. Ther. 1994, 268, 537–545. [Google Scholar] [CrossRef]
- Fredholm, B.B.; Dunwiddie, T.V.; Bergman, B. Levels of adenosine and adenine nucleotides in slices of rat hippocampus. Brain Res. 1984, 295, 127–136. [Google Scholar] [CrossRef]
- Brust, T.B.; Cayabyab, F.S.; MacVicar, B.A. C-Jun N-terminal kinase regulates adenosine A1 receptor-mediated synaptic depression in the rat hippocampus. Neuropharmacology 2007, 53, 906–917. [Google Scholar] [CrossRef]
- Brust, T.B.; Cayabyab, F.S.; Zhou, N.; MacVicar, B.A. p38 mitogen-activated protein kinase contributes to adenosine A1 receptor-mediated synaptic depression in area CA1 of the rat hippocampus. J. Neurosci. 2006, 26, 12427–12438. [Google Scholar] [CrossRef]
- Chen, Z.; Xiong, C.; Pancyr, C.; Stockwell, J.; Walz, W.; Cayabyab, F.S. Prolonged adenosine A1 receptor activation in hypoxia and pial vessel disruption focal cortical ischemia facilitates clathrin-mediated AMPA receptor endocytosis and long-lasting synaptic inhibition in rat hippocampal CA3-CA1 synapses: Differential regulation of GluA2 and GluA1 subunits by p38 MAPK and JNK. J. Neurosci. 2014, 34, 9621–9643. [Google Scholar] [CrossRef]
- Stockwell, J.; Jakova, E.; Cayabyab, F.S. Adenosine A1 and A2A Receptors in the Brain: Current Research and Their Role in Neurodegeneration. Molecules 2017, 22, 676. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, J.; Chen, Z.; Niazi, M.; Nosib, S.; Cayabyab, F.S. Protein phosphatase role in adenosine A1 receptor-induced AMPA receptor trafficking and rat hippocampal neuronal damage in hypoxia/reperfusion injury. Neuropharmacology 2016, 102, 254–265. [Google Scholar] [CrossRef]
- Qin, X.; Zaki, M.G.; Chen, Z.; Jakova, E.; Ming, Z.; Cayabyab, F.S. Adenosine Signaling and Clathrin-Mediated Endocytosis of Glutamate AMPA Receptors in Delayed Hypoxic Injury in Rat Hippocampus: Role of Casein Kinase 2. Mol. Neurobiol. 2021, 58, 1932–1951. [Google Scholar] [CrossRef]
- Chen, Z.; Stockwell, J.; Cayabyab, F.S. Adenosine A1 Receptor-Mediated Endocytosis of AMPA Receptors Contributes to Impairments in Long-Term Potentiation (LTP) in the Middle-Aged Rat Hippocampus. Neurochem. Res. 2016, 41, 1085–1097. [Google Scholar] [CrossRef] [PubMed]
- Costenla, A.R.; Diogenes, M.J.; Canas, P.M.; Rodrigues, R.J.; Nogueira, C.; Maroco, J.; Agostinho, P.M.; Ribeiro, J.A.; Cunha, R.A.; de Mendonca, A. Enhanced role of adenosine A(2A) receptors in the modulation of LTP in the rat hippocampus upon ageing. Eur. J. Neurosci. 2011, 34, 12–21. [Google Scholar] [CrossRef] [PubMed]
- White, P.J.; Rose’Meyer, R.B.; Hope, W. Functional characterization of adenosine receptors in the nucleus tractus solitarius mediating hypotensive responses in the rat. Br. J. Pharmacol. 1996, 117, 305–308. [Google Scholar] [CrossRef]
- de Mendonça, A.; Sebastião, A.M.; Ribeiro, J.A. Adenosine: Does it have a neuroprotective role after all? Brain Res. Rev. 2000, 33, 258–274. [Google Scholar] [CrossRef]
- Cunha, R.A. Neuroprotection by adenosine in the brain: From A1 receptor activation to A2A receptor blockade. Purinergic Signal. 2005, 1, 111–134. [Google Scholar] [CrossRef]
- Gao, Y.; Phillis, J.W. CGS 15943, An adenosine A2 receptor antagonist, reduces cerebral ischemic injury in the mongolian gerbil. Life Sci. 1994, 55, PL61–PL65. [Google Scholar] [CrossRef]
- Chen, J.-F.; Huang, Z.; Ma, J.; Zhu, J.; Moratalla, R.; Standaert, D.; Moskowitz, M.A.; Fink, J.S.; Schwarzschild, M.A. A2AAdenosine Receptor Deficiency Attenuates Brain Injury Induced by Transient Focal Ischemia in Mice. J. Neurosci. 1999, 19, 9192–9200. [Google Scholar] [CrossRef]
- Pugliese, A.M.; Traini, C.; Cipriani, S.; Gianfriddo, M.; Mello, T.; Giovannini, M.G.; Galli, A.; Pedata, F. The adenosine A2A receptor antagonist ZM241385 enhances neuronal survival after oxygen-glucose deprivation in rat CA1 hippocampal slices. Br. J. Pharmacol. 2009, 157, 818–830. [Google Scholar] [CrossRef] [PubMed]
- Jakova, E.; Moutaoufik, M.T.; Lee, J.S.; Babu, M.; Cayabyab, F.S. Adenosine A1 receptor ligands bind to α-synuclein: Implications for α-synuclein misfolding and α-synucleinopathy in Parkinson’s disease. Transl. Neurodegener. 2022, 11, 9. [Google Scholar] [CrossRef]
- Lv, Y.-C.; Gao, A.-B.; Yang, J.; Zhong, L.-Y.; Jia, B.; Ouyang, S.-H.; Gui, L.; Peng, T.-H.; Sun, S.; Cayabyab, F.S. Long-term adenosine A1 receptor activation-induced sortilin expression promotes α-synuclein upregulation in dopaminergic neurons. Neural Regen. Res. 2020, 15, 712–723. [Google Scholar] [CrossRef] [PubMed]
- Jakova, E.; Aigbogun, O.P.; Moutaoufik, M.T.; Allen, K.J.H.; Munir, O.; Brown, D.; Taghibiglou, C.; Babu, M.; Phenix, C.P.; Krol, E.S.; et al. The Bifunctional Dimer Caffeine-Indan Attenuates alpha-Synuclein Misfolding, Neurodegeneration and Behavioral Deficits after Chronic Stimulation of Adenosine A1 Receptors. Int. J. Mol. Sci. 2024, 25, 9386. [Google Scholar] [CrossRef]
- Cayabyab, F.S.; Gowribai, K.; Walz, W. Involvement of matrix metalloproteinases-2 and -9 in the formation of a lacuna-like cerebral cavity. J. Neurosci. Res. 2013, 91, 920–933. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef]
- Kaur, D.; Sharma, V.; Deshmukh, R. Activation of microglia and astrocytes: A roadway to neuroinflammation and Alzheimer’s disease. Inflammopharmacology 2019, 27, 663–677. [Google Scholar] [CrossRef]
- Bhusal, A.; Afridi, R.; Lee, W.-H.; Suk, K. Bidirectional communication between microglia and astrocytes in neuroinflammation. Curr. Neuropharmacol. 2023, 21, 2020. [Google Scholar] [CrossRef] [PubMed]
- Uchida, S.; Tashiro, T.; Kawai-Uchida, M.; Mori, A.; Jenner, P.; Kanda, T. Adenosine A₂A-receptor antagonist istradefylline enhances the motor response of L-DOPA without worsening dyskinesia in MPTP-treated common marmosets. J. Pharmacol. Sci. 2014, 124, 480–485. [Google Scholar] [CrossRef]
- Kanda, T.; Jackson, M.J.; Smith, L.A.; Pearce, R.K.; Nakamura, J.; Kase, H.; Kuwana, Y.; Jenner, P. Combined use of the adenosine A(2A) antagonist KW-6002 with L-DOPA or with selective D1 or D2 dopamine agonists increases antiparkinsonian activity but not dyskinesia in MPTP-treated monkeys. Exp. Neurol. 2000, 162, 321–327. [Google Scholar] [CrossRef]
- Dungo, R.; Deeks, E.D. Istradefylline: First Global Approval. Drugs 2013, 73, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Campbell Burton, C.A.; Murray, J.; Holmes, J.; Astin, F.; Greenwood, D.; Knapp, P. Frequency of anxiety after stroke: A systematic review and meta-analysis of observational studies. Int. J. Stroke 2013, 8, 545–559. [Google Scholar] [CrossRef] [PubMed]
- Hatem, S.M.; Saussez, G.; Della Faille, M.; Prist, V.; Zhang, X.; Dispa, D.; Bleyenheuft, Y. Rehabilitation of Motor Function after Stroke: A Multiple Systematic Review Focused on Techniques to Stimulate Upper Extremity Recovery. Front. Hum. Neurosci. 2016, 10, 442. [Google Scholar] [CrossRef]
- Chun, H.-Y.Y.; Whiteley, W.N.; Dennis, M.S.; Mead, G.E.; Carson, A.J. Anxiety After Stroke: The Importance of Subtyping. Stroke 2018, 49, 556–564. [Google Scholar] [CrossRef]
- Towfighi, A.; Ovbiagele, B.; Husseini, N.E.; Hackett, M.L.; Jorge, R.E.; Kissela, B.M.; Mitchell, P.H.; Skolarus, L.E.; Whooley, M.A.; Williams, L.S. Poststroke Depression: A Scientific Statement for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke 2017, 48, e30–e43. [Google Scholar] [CrossRef]
- Honig, L.S.; Tang, M.X.; Albert, S.; Costa, R.; Luchsinger, J.; Manly, J.; Stern, Y.; Mayeux, R. Stroke and the risk of Alzheimer disease. Arch. Neurol. 2003, 60, 1707–1712. [Google Scholar] [CrossRef]
- Shen, H.-Y.; Chen, J.-F. Adenosine A2A receptors in psychopharmacology: Modulators of behavior, mood and cognition. Curr. Neuropharmacol. 2009, 7, 195. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B.; Dunwiddie, T.V. How does adenosine inhibit transmitter release? Trends Pharmacol. Sci. 1988, 9, 130–134. [Google Scholar] [CrossRef]
- Rebholz, H.; Nishi, A.; Liebscher, S.; Nairn, A.C.; Flajolet, M.; Greengard, P. CK2 negatively regulates Gαs signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 14096–14101. [Google Scholar] [CrossRef]
- Blanquet, P.R. Casein kinase 2 as a potentially important enzyme in the nervous system. Prog. Neurobiol. 2000, 60, 211–246. [Google Scholar] [CrossRef]
- Borgo, C.; D’Amore, C.; Sarno, S.; Salvi, M.; Ruzzene, M. Protein kinase CK2: A potential therapeutic target for diverse human diseases. Signal Transduct. Target. Ther. 2021, 6, 183. [Google Scholar] [CrossRef] [PubMed]
- Canas, P.M.; Porciúncula, L.O.; Simões, A.P.; Augusto, E.; Silva, H.B.; Machado, N.J.; Gonçalves, N.; Alfaro, T.M.; Gonçalves, F.Q.; Araújo, I.M.; et al. Neuronal Adenosine A2A Receptors Are Critical Mediators of Neurodegeneration Triggered by Convulsions. eneuro 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Orr, A.G.; Lo, I.; Schumacher, H.; Ho, K.; Gill, M.; Guo, W.; Kim, D.H.; Knox, A.; Saito, T.; Saido, T.C.; et al. Istradefylline reduces memory deficits in aging mice with amyloid pathology. Neurobiol. Dis. 2018, 110, 29–36. [Google Scholar] [CrossRef]
- Temido-Ferreira, M.; Ferreira, D.G.; Batalha, V.L.; Marques-Morgado, I.; Coelho, J.E.; Pereira, P.; Gomes, R.; Pinto, A.; Carvalho, S.; Canas, P.M.; et al. Age-related shift in LTD is dependent on neuronal adenosine A2A receptors interplay with mGluR5 and NMDA receptors. Mol. Psychiatry 2018, 25, 1876–1900. [Google Scholar] [CrossRef]
- Batalha, V.L.; Pego, J.M.; Fontinha, B.M.; Costenla, A.R.; Valadas, J.S.; Baqi, Y.; Radjainia, H.; Muller, C.E.; Sebastiao, A.M.; Lopes, L.V. Adenosine A2A receptor blockade reverts hippocampal stress-induced deficits and restores corticosterone circadian oscillation. Mol. Psychiatry 2013, 18, 320–331. [Google Scholar] [CrossRef]
- Horita, T.K.; Kobayashi, M.; Mori, A.; Jenner, P.; Kanda, T. Effects of the adenosine A 2A antagonist istradefylline on cognitive performance in rats with a 6-OHDA lesion in prefrontal cortex. Psychopharmacology 2013, 230, 345–352. [Google Scholar] [CrossRef]
- Kaster, M.P.; Machado, N.J.; Silva, H.B.; Nunes, A.; Ardais, A.P.; Santana, M.; Baqi, Y.; Müller, C.E.; Rodrigues, A.L.S.; Porciúncula, L.O.; et al. Caffeine acts through neuronal adenosine A2A receptors to prevent mood and memory dysfunction triggered by chronic stress. Proc. Natl. Acad. Sci. USA 2015, 112, 7833–7838. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.C.; Lemos, C.; Gonçalves, F.Q.; Pliássova, A.V.; Machado, N.J.; Silva, H.B.; Canas, P.M.; Cunha, R.A.; Lopes, J.P.; Agostinho, P. Blockade of adenosine A2A receptors recovers early deficits of memory and plasticity in the triple transgenic mouse model of Alzheimer’s disease. Neurobiol. Dis. 2018, 117, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, F.Q.; Lopes, J.P.; Silva, H.B.; Lemos, C.; Silva, A.C.; Gonçalves, N.; Tomé, Â.R.; Ferreira, S.G.; Canas, P.M.; Rial, D. Synaptic and memory dysfunction in a β-amyloid model of early Alzheimer’s disease depends on increased formation of ATP-derived extracellular adenosine. Neurobiol. Dis. 2019, 132, 104570. [Google Scholar] [CrossRef]
- Cunha, G.M.A.; Canas, P.M.; Oliveira, C.R.; Cunha, R.A. Increased density and synapto-protective effect of adenosine A2A receptors upon sub-chronic restraint stress. Neuroscience 2006, 141, 1775–1781. [Google Scholar] [CrossRef]
- Machado, N.J.; Simões, A.P.; Silva, H.B.; Ardais, A.P.; Kaster, M.P.; Garção, P.; Rodrigues, D.I.; Pochmann, D.; Santos, A.I.; Araújo, I.M.; et al. Caffeine Reverts Memory But Not Mood Impairment in a Depression-Prone Mouse Strain with Up-Regulated Adenosine A2A Receptor in Hippocampal Glutamate Synapses. Mol. Neurobiol. 2017, 54, 1552–1563. [Google Scholar] [CrossRef]
- Chen, J.-F.; Cunha, R.A. The belated US FDA approval of the adenosine A(2A) receptor antagonist istradefylline for treatment of Parkinson’s disease. Purinergic Signal. 2020, 16, 167–174. [Google Scholar] [CrossRef] [PubMed]
- LeWitt, P.A.; Guttman, M.; Tetrud, J.W.; Tuite, P.J.; Mori, A.; Chaikin, P.; Sussman, N.M.; Group, U.S. Adenosine A2A receptor antagonist istradefylline (KW-6002) reduces “off” time in Parkinson’s disease: A double-blind, randomized, multicenter clinical trial (6002-US-005). Ann. Neurol. 2008, 63, 295–302. [Google Scholar] [CrossRef]
- Kalda, A.; Yu, L.; Oztas, E.; Chen, J.-F. Novel neuroprotection by caffeine and adenosine A2A receptor antagonists in animal models of Parkinson’s disease. J. Neurol. Sci. 2006, 248, 9–15. [Google Scholar] [CrossRef]
- Yu, L.; Shen, H.Y.; Coelho, J.E.; Araújo, I.M.; Huang, Q.Y.; Day, Y.J.; Rebola, N.; Canas, P.M.; Rapp, E.K.; Ferrara, J. Adenosine A2A receptor antagonists exert motor and neuroprotective effects by distinct cellular mechanisms. Ann. Neurol. 2008, 63, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Oztas, E.; Kalda, A.; Xu, K.; Irrizary, M.; Schwarzschild, M.; Chen, J. Caffeine attenuates MPTP-induced loss of dopaminergic neurons in substantia nigra in mice. In Proceedings of the Annual meeting of Society for Neuroscience, Orlando, FL, USA, 2–7 November 2002. [Google Scholar]
- Ascherio, A.; Zhang, S.M.; Hernán, M.A.; Kawachi, I.; Colditz, G.A.; Speizer, F.E.; Willett, W.C. Prospective study of caffeine consumption and risk of Parkinson’s disease in men and women. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2001, 50, 56–63. [Google Scholar] [CrossRef]
- Real, J.I.; Simões, A.P.; Cunha, R.A.; Ferreira, S.G.; Rial, D. Adenosine A2A receptors modulate the dopamine D2 receptor-mediated inhibition of synaptic transmission in the mouse prefrontal cortex. Eur. J. Neurosci. 2018, 47, 1127–1134. [Google Scholar] [CrossRef] [PubMed]
- Dorice, M.H.C.; Khurana, N.; Sharma, N.; Khatik, G.L. Identification of Possible Molecular Targets of Potential Anti-Parkinson Drugs by Predicting Their Binding Affinities Using Molecular Docking. Asian J. Pharm. Clin. Res. 2018, 11, 28–32. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, H.; Wang, H.; Zheng, Z.; Meng, Z.; Xu, Z.; Li, J.; Xue, M. Design, synthesis, and biological activity studies of istradefylline derivatives based on adenine as A2A receptor antagonists. ACS Omega 2021, 6, 4386–4394. [Google Scholar] [CrossRef]
- El Yacoubi, M.; Costentin, J.; Vaugeois, J.-M. Adenosine A2A receptors and depression. Neurology 2003, 61, S82–S87. [Google Scholar] [CrossRef]
- Yacoubi, M.E.; Ledent, C.; Parmentier, M.; Bertorelli, R.; Ongini, E.; Costentin, J.; Vaugeois, J.M. Adenosine A2A receptor antagonists are potential antidepressants: Evidence based on pharmacology and A2A receptor knockout mice. Br. J. Pharmacol. 2001, 134, 68–77. [Google Scholar] [CrossRef]
- Yamada, K.; Kobayashi, M.; Mori, A.; Jenner, P.; Kanda, T. Antidepressant-like activity of the adenosine A2A receptor antagonist, istradefylline (KW-6002), in the forced swim test and the tail suspension test in rodents. Pharmacol. Biochem. Behav. 2013, 114, 23–30. [Google Scholar] [CrossRef]
- Orr, A.G.; Orr, A.L.; Li, X.-J.; Gross, R.E.; Traynelis, S.F. Adenosine A2A receptor mediates microglial process retraction. Nat. Neurosci. 2009, 12, 872–878. [Google Scholar] [CrossRef]
- Madeira, M.H.; Boia, R.; Elvas, F.; Martins, T.; Cunha, R.A.; Ambrósio, A.F.; Santiago, A.R. Selective A2A receptor antagonist prevents microglia-mediated neuroinflammation and protects retinal ganglion cells from high intraocular pressure–induced transient ischemic injury. Transl. Res. 2016, 169, 112–128. [Google Scholar] [CrossRef] [PubMed]
- Boia, R.; Elvas, F.; Madeira, M.H.; Aires, I.D.; Rodrigues-Neves, A.C.; Tralhão, P.; Szabó, E.C.; Baqi, Y.; Müller, C.E.; Tomé, Â.R. Treatment with A2A receptor antagonist KW6002 and caffeine intake regulate microglia reactivity and protect retina against transient ischemic damage. Cell Death Dis. 2017, 8, e3065. [Google Scholar] [CrossRef] [PubMed]
- Martí Navia, A.; Dal Ben, D.; Lambertucci, C.; Spinaci, A.; Volpini, R.; Marques-Morgado, I.; Coelho, J.E.; Lopes, L.V.; Marucci, G.; Buccioni, M. Adenosine receptors as neuroinflammation modulators: Role of A1 agonists and A2A antagonists. Cells 2020, 9, 1739. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.D.; Al-Khoury, L.; Zivin, J.A. Neuroprotection for Ischemic Stroke: Two Decades of Success and Failure. NeuroRx 2004, 1, 36–45. [Google Scholar] [CrossRef]
- Lees, K.R. Cerestat and other NMDA antagonists in ischemic stroke. Neurology 1997, 49 (Suppl. 4), S66–S69. [Google Scholar] [CrossRef]
- Roundtable, S.T.A.I. Recommendations for standards regarding preclinical neuroprotective and restorative drug development. Stroke 1999, 30, 2752–2758. [Google Scholar]
- Lapchak, P.A.; Zhang, J.H.; Noble-Haeusslein, L.J. RIGOR guidelines: Escalating STAIR and STEPS for effective translational research. Transl. Stroke Res. 2013, 4, 279–285. [Google Scholar] [CrossRef]
- Baron, J.C.; von Kummer, R.; del Zoppo, G.J. Treatment of acute ischemic stroke. Challenging the concept of a rigid and universal time window. Stroke 1995, 26, 2219–2221. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.; Feuerstein, G.; Howells, D.W.; Hurn, P.D.; Kent, T.A.; Savitz, S.I.; Lo, E.H.; Group, S. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke 2009, 40, 2244–2250. [Google Scholar] [CrossRef] [PubMed]
- Meirhaeghe, A.; Cottel, D.; Cousin, B.; Dumont, M.-P.; Marécaux, N.; Amouyel, P.; Dallongeville, J. Sex Differences in Stroke Attack, Incidence, and Mortality Rates in Northern France. J. Stroke Cerebrovasc. Dis. 2018, 27, 1368–1374. [Google Scholar] [CrossRef]
- Suzuki, S.; Brown, C.M.; Wise, P.M. Neuroprotective effects of estrogens following ischemic stroke. Front. Neuroendocrinol. 2009, 30, 201–211. [Google Scholar] [CrossRef]
- Yang, S.-H.; Shi, J.; Day Arthur, L.; Simpkins James, W. Estradiol Exerts Neuroprotective Effects When Administered After Ischemic Insult. Stroke 2000, 31, 745–750. [Google Scholar] [CrossRef]
- Dubal, D.B.; Kashon, M.L.; Pettigrew, L.C.; Ren, J.M.; Finklestein, S.P.; Rau, S.W.; Wise, P.M. Estradiol protects against ischemic injury. J. Cereb. Blood Flow Metab. 1998, 18, 1253–1258. [Google Scholar] [CrossRef]
- Luo, T.; Liu, H.; Kim, J.K. Estrogen protects the female heart from ischemia/reperfusion injury through manganese superoxide dismutase phosphorylation by mitochondrial P38β at threonine 79 and serine 106. PLoS ONE 2016, 11, e0167761. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, M.; Brown, V.J. The effect of the adenosine A 2A antagonist KW-6002 on motor and motivational processes in the rat. Psychopharmacology 2006, 184, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Walz, W. Unusual topographical pattern of proximal astrogliosis around a cortical devascularizing lesion. J. Neurosci. Res. 2003, 73, 497–506. [Google Scholar] [CrossRef]
- Wang, K.; Bekar, L.K.; Furber, K.; Walz, W. Vimentin-expressing proximal reactive astrocytes correlate with migration rather than proliferation following focal brain injury. Brain Res. 2004, 1024, 193–202. [Google Scholar] [CrossRef]
- Hua, R.; Walz, W. Minocycline treatment prevents cavitation in rats after a cortical devascularizing lesion. Brain Res. 2006, 1090, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini-Giampietro, D.E.; Zukin, R.S.; Bennett, M.V.; Cho, S.; Pulsinelli, W.A. Switch in glutamate receptor subunit gene expression in CA1 subfield of hippocampus following global ischemia in rats. Proc. Natl. Acad. Sci. USA 1992, 89, 10499–10503. [Google Scholar] [CrossRef] [PubMed]
- Prosser-Loose, E.J.; Verge, V.M.; Cayabyab, F.S.; Paterson, P.G. Protein-energy malnutrition alters hippocampal plasticity-associated protein expression following global ischemia in the gerbil. Curr. Neurovasc. Res. 2010, 7, 341–360. [Google Scholar] [CrossRef] [PubMed]
- Simon, P.; Dupuis, R.; Costentin, J. Thigmotaxis as an index of anxiety in mice. Influence of dopaminergic transmissions. Behav. Brain Res. 1994, 61, 59–64. [Google Scholar] [CrossRef]
- Seibenhener, M.L.; Wooten, M.C. Use of the Open Field Maze to measure locomotor and anxiety-like behavior in mice. J. Vis. Exp. 2015, 96, e52434. [Google Scholar] [CrossRef]
- Yankelevitch-Yahav, R.; Franko, M.; Huly, A.; Doron, R. The forced swim test as a model of depressive-like behavior. J. Vis. Exp. 2015, e52587. [Google Scholar] [CrossRef]
- Deacon, R.M. Measuring motor coordination in mice. J. Vis. Exp. 2013, e2609. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaki, M.G.; Jakova, E.; Pordeli, M.; Setork, E.; Taghibiglou, C.; Cayabyab, F.S. The Anti-Parkinsonian A2A Receptor Antagonist Istradefylline (KW-6002) Attenuates Behavioral Abnormalities, Neuroinflammation, and Neurodegeneration in Cerebral Ischemia: An Adenosinergic Signaling Link Between Stroke and Parkinson’s Disease. Int. J. Mol. Sci. 2025, 26, 5680. https://doi.org/10.3390/ijms26125680
Zaki MG, Jakova E, Pordeli M, Setork E, Taghibiglou C, Cayabyab FS. The Anti-Parkinsonian A2A Receptor Antagonist Istradefylline (KW-6002) Attenuates Behavioral Abnormalities, Neuroinflammation, and Neurodegeneration in Cerebral Ischemia: An Adenosinergic Signaling Link Between Stroke and Parkinson’s Disease. International Journal of Molecular Sciences. 2025; 26(12):5680. https://doi.org/10.3390/ijms26125680
Chicago/Turabian StyleZaki, Michael G., Elisabet Jakova, Mahboubeh Pordeli, Elina Setork, Changiz Taghibiglou, and Francisco S. Cayabyab. 2025. "The Anti-Parkinsonian A2A Receptor Antagonist Istradefylline (KW-6002) Attenuates Behavioral Abnormalities, Neuroinflammation, and Neurodegeneration in Cerebral Ischemia: An Adenosinergic Signaling Link Between Stroke and Parkinson’s Disease" International Journal of Molecular Sciences 26, no. 12: 5680. https://doi.org/10.3390/ijms26125680
APA StyleZaki, M. G., Jakova, E., Pordeli, M., Setork, E., Taghibiglou, C., & Cayabyab, F. S. (2025). The Anti-Parkinsonian A2A Receptor Antagonist Istradefylline (KW-6002) Attenuates Behavioral Abnormalities, Neuroinflammation, and Neurodegeneration in Cerebral Ischemia: An Adenosinergic Signaling Link Between Stroke and Parkinson’s Disease. International Journal of Molecular Sciences, 26(12), 5680. https://doi.org/10.3390/ijms26125680