
Arsenite-Mediated Transcriptional Regulation of Glutathione Synthesis in Mammalian Primary Cortical Astrocytes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
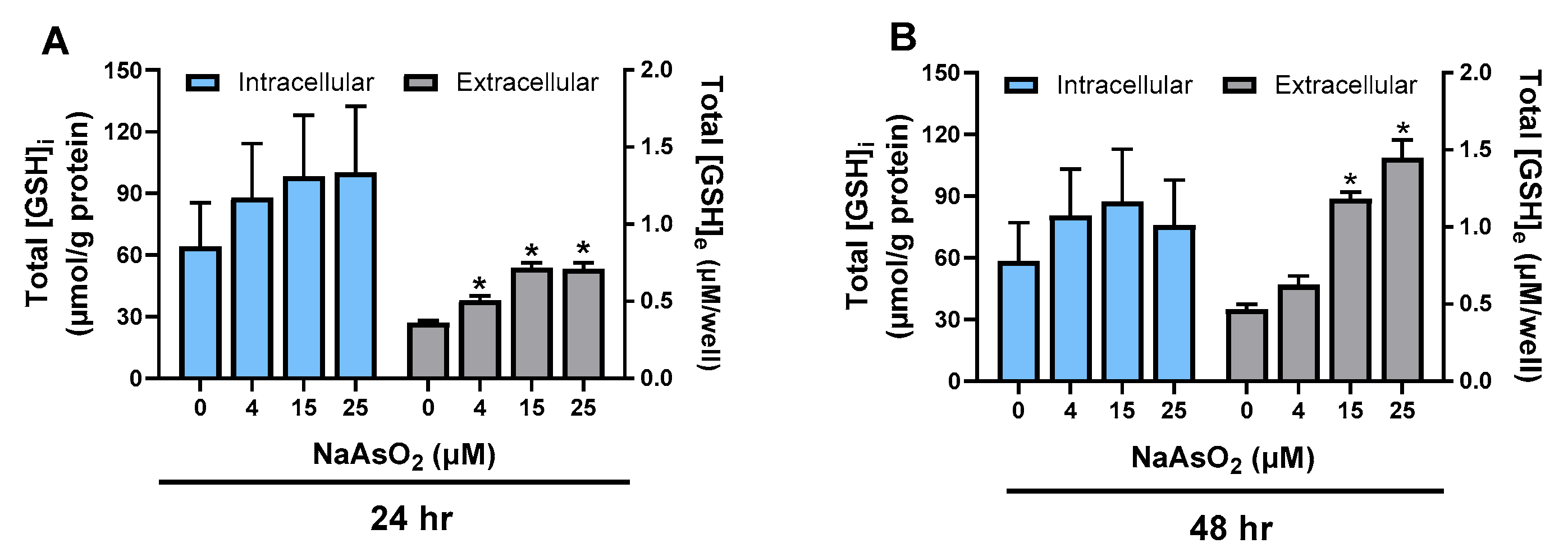
2.1. Effect of Arsenite on Astrocyte GSH Levels
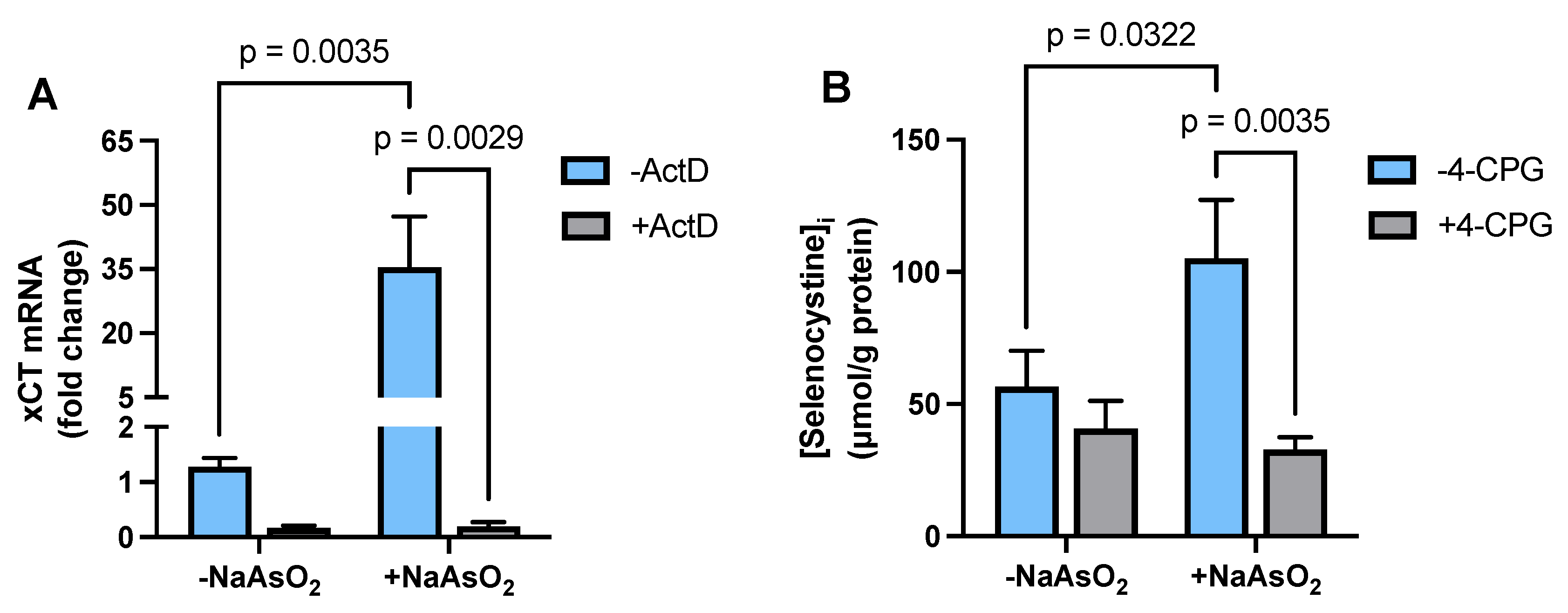
2.2. Effect of Arsenite on Astrocyte xCT mRNA Expression and Transporter Activity
2.3. Effect of Arsenite on Astrocyte GCL mRNA and Protein Expression
2.4. Effect of Arsenite on Astrocyte GS mRNA and Protein Expression
2.5. Transcriptional Regulation of Astrocytic GSH Levels by Arsenite
3. Discussion
4. Materials and Methods
4.1. Experimental Media and Buffers
- Media Stock (MS): Minimum Essential Media with Earle’s salts purchased without L-glutamine (Corning, Corning, NY, USA) supplemented with L-glutamine (L-gln; Gibco, Burlington, ON, Canada), glucose, and sodium bicarbonate to a final concentration of 2.0, 25.7, and 28.2 mM, respectively, and stored at 4 °C. This MS, which contained 100 µM cystine, as provided in the base MEM formulation, served as the foundation for all other cell culture media used in this study.
- Astrocyte Plating Media (APM): MS was supplemented with 10% Calf Serum (CS; Hyclone Laboratories, Marlboro, MA, USA), 10% Fetal Bovine Serum (FBS; Hyclone), 50 IU penicillin, and 50 µg/mL streptomycin (Gibco) and stored at 4 °C. L-gln and epidermal growth factor (EGF; Thermo Fischer Scientific, Waltham, MA, USA) were added to a final concentration of 2 mM and 10 ng/mL, respectively. Medium was warmed to 37 °C immediately prior to use.
- Maintenance Media-1 (MM-1): MS was supplemented with 10% CS, 50 IU penicillin, and 50 µg/mL streptomycin and stored at 4 °C. L-gln was supplemented to a final concentration 2 mM. Medium was warmed to 37 °C just prior to use.
- Maintenance Media-2 (MM-2): MS was supplemented with 3% CS, 50 IU penicillin, and 50 µg/mL streptomycin and stored at 4 °C. L-gln was supplemented to a final concentration 2 mM. Medium was warmed to 37 °C just prior to use.
- 1× BSS10: Cell-culture-grade water (Corning) containing 116.4 mM sodium chloride (NaCl; Thermo Fischer Scientific), 5.4 mM potassium chloride (KCl; Thermo Fischer Scientific), 0.8 mM magnesium sulfate heptahydrate (MgSO4·7H2O; Sigma-Aldrich, St. Louis, MO, USA), 1 mM sodium phosphate monobasic monohydrate (NaH2PO4·H2O; Thermo Fischer Scientific), 10 mM glucose, and 26 mM sodium bicarbonate was used. A 10× stock was generated and filter-sterilized before being stored at room temperature. A 1× BSS10 solution was then generated from the stock solution with sterile cell-culture-grade water in a sterile environment. Just prior to use, 1.8 mM CaCl2 was slowly added for a duration of five minutes while CO2 was bubbled into the solution. The 1× BSS10 was also stored at room temperature.
4.2. Astrocyte Cultures
4.3. Drug Stocks and Exposures
- Arsenite (Na2AsO2): Arsenite stock (25 mM) was prepared by dissolving sodium arsenite (Sigma-Aldrich) in MS and adjusting the pH to 7.0 using 1.65 N HCl. This stock solution was stored at 4 °C. Arsenite sub-stocks were generated by diluting the stock in MS to concentrations of 4, 10, 15, or 25 µM. Micromolar levels of arsenite have been commonly used in vitro to facilitate the identification of potential pathways and targets affected by arsenic, including those involved in the regulation of GSH synthesis [17,37,76]. On the day of experimentation, the astrocyte culture medium was fully aspirated, and cells were washed twice with MS (400 µL or 1.6 mL for 24- or 6-well plates, respectively). Cultures were treated with arsenite by either direct addition of the sub-stocks or by spiking the stock solution into wells to the desired final concentration. The exposure duration lasted 4 to 6 h depending on the specific requirements of the experimental protocol.
- Actinomycin D (ActD): To inhibit transcription, ActD (Cayman, Ann Arbor, MI, USA) was utilized [77]. ActD stock (10 mg/mL) was prepared in DMSO and stored at −20 °C. A sub-stock (10 µg/mL) was made by diluting the stock solution into MS, resulting in a final DMSO concentration of =0.1%. Vehicle controls were prepared by adding DMSO to MS to the same final concentration. Astrocyte cultures were pretreated with either ActD (10 µg/mL) or vehicle (0.1% DMSO) prior to arsenite exposure (15 µM final concentration). The concentration of ActD used in these studies was consistent with prior work by our group and others demonstrating effective transcriptional inhibition while preserving astrocyte viability [78,79,80].
- 4-carboxyphenylglycine (4-CPG): 4-CPG (Tocris, Avonmouth, Bristol, UK) was used to competitively inhibit Sxc− [34,39,40]. 4-CPG stock (50 mM) was prepared in 1 N NaOH and stored at −20 °C. The stock solution was diluted to a final concentration of 500 µM in 1× BSS10 just prior to addition to cultures.
- Selenocystine (SeCyss): SeCyss (Sigma-Aldrich) was utilized as an analog of cysteine to quantitatively measure Sxc− activity in astrocytes [81]. SeCyss stock (100 mM) was prepared in 1 N NaOH. The solution was stored at −20 °C. On the day of experimentation, a 25 mM sub-stock was generated in 1N NaOH, which was further diluted in 1× BSS10 to a final concentration of 25 µM.
4.4. Glutathione Measurement
4.5. Reverse Transcription–Quantitative Polymerase Chain Reaction (RT-qPCR)
4.6. (Seleno)Cystine Uptake Assay
4.7. Immunoblotting
4.8. Bicinchoninic Acid (BCA) Assay (Thermo Fischer Scientific)
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Duker, A.A.; Carranza, E.J.M.; Hale, M. Arsenic Geochemistry and Health. Environ. Int. 2005, 31, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Ng, J. Arsenic and Arsenic Compounds. In Environmental Health Criteria, 2nd ed.; World Health Organization: Geneva, Switzerland, 2001; ISBN 978-92-4-157224-8. [Google Scholar]
- Frisbie, S.H.; Mitchell, E.J. Arsenic in Drinking Water: An Analysis of Global Drinking Water Regulations and Recommendations for Updates to Protect Public Health. PLoS ONE 2022, 17, e0263505. [Google Scholar] [CrossRef]
- Naujokas, M.F.; Anderson, B.; Ahsan, H.; Aposhian, H.V.; Graziano, J.H.; Thompson, C.; Suk, W.A. The Broad Scope of Health Effects from Chronic Arsenic Exposure: Update on a Worldwide Public Health Problem. Environ. Health Perspect. 2013, 121, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Chávez, L.A.; Rendón-López, C.R.R.; Zepeda, A.; Silva-Adaya, D.; Del Razo, L.M.; Gonsebatt, M.E. Neurological Effects of Inorganic Arsenic Exposure: Altered Cysteine/Glutamate Transport, NMDA Expression and Spatial Memory Impairment. Front. Cell. Neurosci. 2015, 9, 21. [Google Scholar] [CrossRef]
- Martínez-Castillo, M.; García-Montalvo, E.A.; Arellano-Mendoza, M.G.; Sánchez-Peña, L.D.C.; Soria Jasso, L.E.; Izquierdo-Vega, J.A.; Valenzuela, O.L.; Hernández-Zavala, A. Arsenic Exposure and Non-Carcinogenic Health Effects. Hum. Exp. Toxicol. 2021, 40, S826–S850. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, G.; Cai, Y. Thiolated Arsenicals in Arsenic Metabolism: Occurrence, Formation, and Biological Implications. J. Environ. Sci. 2016, 49, 59–73. [Google Scholar] [CrossRef]
- Jiang, X.; McDermott, J.R.; Ajees, A.A.; Rosen, B.P.; Liu, Z. Trivalent Arsenicals and Glucose Use Different Translocation Pathways in Mammalian GLUT1. Metallomics 2010, 2, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Yoshinaga-Sakurai, K.; Shinde, R.; Rodriguez, M.; Rosen, B.P.; El-Hage, N. Comparative Cytotoxicity of Inorganic Arsenite and Methylarsenite in Human Brain Cells. ACS Chem. Neurosci. 2020, 11, 743–751. [Google Scholar] [CrossRef]
- Wang, L.; Xiong, X.; Zhang, L.; Shen, J. Neurovascular Unit: A Critical Role in Ischemic Stroke. CNS Neurosci. Ther. 2021, 27, 7–16. [Google Scholar] [CrossRef]
- Freeman, M.R. Specification and Morphogenesis of Astrocytes. Science 2010, 330, 774–778. [Google Scholar] [CrossRef]
- Liu, L.-R.; Liu, J.-C.; Bao, J.-S.; Bai, Q.-Q.; Wang, G.-Q. Interaction of Microglia and Astrocytes in the Neurovascular Unit. Front. Immunol. 2020, 11, 1024. [Google Scholar] [CrossRef] [PubMed]
- Brandebura, A.N.; Paumier, A.; Onur, T.S.; Allen, N.J. Astrocyte Contribution to Dysfunction, Risk and Progression in Neurodegenerative Disorders. Nat. Rev. Neurosci. 2023, 24, 23–39. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-G.; Wheeler, M.A.; Quintana, F.J. Function and Therapeutic Value of Astrocytes in Neurological Diseases. Nat. Rev. Drug Discov. 2022, 21, 339–358. [Google Scholar] [CrossRef]
- Ransom, B.R.; Ransom, C.B. Astrocytes: Multitalented Stars of the Central Nervous System. Methods Mol. Biol. 2012, 814, 3–7. [Google Scholar] [CrossRef]
- Gradisnik, L.; Velnar, T. Astrocytes in the Central Nervous System and Their Functions in Health and Disease: A Review. World J. Clin. Cases 2023, 11, 3385–3394. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R.; Spiller, S.; Neumann, S.; Koehler, Y. Uptake, Metabolic Effects and Toxicity of Arsenate and Arsenite in Astrocytes. Neurochem. Res. 2016, 41, 465–475. [Google Scholar] [CrossRef]
- Dringen, R.; Brandmann, M.; Hohnholt, M.C.; Blumrich, E.-M. Glutathione-Dependent Detoxification Processes in Astrocytes. Neurochem. Res. 2015, 40, 2570–2582. [Google Scholar] [CrossRef]
- Tadepalle, N.; Koehler, Y.; Brandmann, M.; Meyer, N.; Dringen, R. Arsenite Stimulates Glutathione Export and Glycolytic Flux in Viable Primary Rat Brain Astrocytes. Neurochem. Int. 2014, 76, 1–11. [Google Scholar] [CrossRef]
- Aoyama, K. Glutathione in the Brain. Int. J. Mol. Sci. 2021, 22, 5010. [Google Scholar] [CrossRef]
- Dringen, R.; Gutterer, J.M.; Hirrlinger, J. Glutathione Metabolism in Brain Metabolic Interaction between Astrocytes and Neurons in the Defense against Reactive Oxygen Species. Eur. J. Biochem. 2000, 267, 4912–4916. [Google Scholar] [CrossRef]
- Dringen, R.; Kranich, O.; Hamprecht, B. The Gamma-Glutamyl Transpeptidase Inhibitor Acivicin Preserves Glutathione Released by Astroglial Cells in Culture. Neurochem. Res. 1997, 22, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R.; Pfeiffer, B.; Hamprecht, B. Synthesis of the Antioxidant Glutathione in Neurons: Supply by Astrocytes of CysGly as Precursor for Neuronal Glutathione. J. Neurosci. 1999, 19, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.B.; Lindenau, J.; Seyfried, J.; Dichgans, J. Glutathione, Oxidative Stress and Neurodegeneration. Eur. J. Biochem. 2000, 267, 4904–4911. [Google Scholar] [CrossRef]
- Lu, S.C. Glutathione Synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef]
- Lapenna, D. Glutathione and Glutathione-Dependent Enzymes: From Biochemistry to Gerontology and Successful Aging. Ageing Res. Rev. 2023, 92, 102066. [Google Scholar] [CrossRef]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The Cystine/Glutamate Antiporter System Xc− in Health and Disease: From Molecular Mechanisms to Novel Therapeutic Opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef]
- Minich, T.; Riemer, J.; Schulz, J.B.; Wielinga, P.; Wijnholds, J.; Dringen, R. The Multidrug Resistance Protein 1 (Mrp1), but Not Mrp5, Mediates Export of Glutathione and Glutathione Disulfide from Brain Astrocytes. J. Neurochem. 2006, 97, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Hirrlinger, J.; Schulz, J.B.; Dringen, R. Glutathione Release from Cultured Brain Cells: Multidrug Resistance Protein 1 Mediates the Release of GSH from Rat Astroglial Cells. J. Neurosci. Res. 2002, 69, 318–326. [Google Scholar] [CrossRef]
- Stridh, M.H.; Tranberg, M.; Weber, S.G.; Blomstrand, F.; Sandberg, M. Stimulated Efflux of Amino Acids and Glutathione from Cultured Hippocampal Slices by Omission of Extracellular Calcium: Likely Involvement of Connexin Hemichannels. J. Biol. Chem. 2008, 283, 10347–10356. [Google Scholar] [CrossRef]
- Rana, S.; Dringen, R. Gap Junction Hemichannel-Mediated Release of Glutathione from Cultured Rat Astrocytes. Neurosci. Lett. 2007, 415, 45–48. [Google Scholar] [CrossRef]
- Ballatori, N.; Hammond, C.L.; Cunningham, J.B.; Krance, S.M.; Marchan, R. Molecular Mechanisms of Reduced Glutathione Transport: Role of the MRP/CFTR/ABCC and OATP/SLC21A Families of Membrane Proteins. Toxicol. Appl. Pharmacol. 2005, 204, 238–255. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Lee, T.K.; Meier, P.J.; Ballatori, N. Identification of Glutathione as a Driving Force and Leukotriene C4 as a Substrate for Oatp1, the Hepatic Sinusoidal Organic Solute Transporter. J. Biol. Chem. 1998, 273, 16184–16191. [Google Scholar] [CrossRef] [PubMed]
- Fogal, B.; Li, J.; Lobner, D.; McCullough, L.D.; Hewett, S.J. System Xc− Activity and Astrocytes Are Necessary for Interleukin-1 Beta-Mediated Hypoxic Neuronal Injury. J. Neurosci. 2007, 27, 10094–10105. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Jackman, N.A.; Thorn, T.L.; Vought, V.E.; Hewett, S.J. Interleukin-1β Protects Astrocytes against Oxidant-Induced Injury via an NF-κB-Dependent Upregulation of Glutathione Synthesis. Glia 2015, 63, 1568–1580. [Google Scholar] [CrossRef]
- Lee, T.D.; Yang, H.; Whang, J.; Lu, S.C. Cloning and Characterization of the Human Glutathione Synthetase 5′-Flanking Region. Biochem. J. 2005, 390, 521–528. [Google Scholar] [CrossRef]
- Meyer, N.; Koehler, Y.; Tulpule, K.; Dringen, R. Arsenate Accumulation and Arsenate-Induced Glutathione Export in Astrocyte-Rich Primary Cultures. Neurochem. Int. 2013, 62, 1012–1019. [Google Scholar] [CrossRef]
- Lutgen, V.; Qualmann, K.; Resch, J.; Kong, L.; Choi, S.; Baker, D.A. Reduction in Phencyclidine Induced Sensorimotor Gating Deficits in the Rat Following Increased System Xc− Activity in the Medial Prefrontal Cortex. Psychopharmacology 2013, 226, 531–540. [Google Scholar] [CrossRef]
- Gochenauer, G.E.; Robinson, M.B. Dibutyryl-cAMP (dbcAMP) up-Regulates Astrocytic Chloride-Dependent L-[3H]Glutamate Transport and Expression of Both System Xc− Subunits. J. Neurochem. 2001, 78, 276–286. [Google Scholar] [CrossRef]
- Patel, S.A.; Warren, B.A.; Rhoderick, J.F.; Bridges, R.J. Differentiation of Substrate and Non-Substrate Inhibitors of Transport System Xc−: An Obligate Exchanger of L-Glutamate and L-Cystine. Neuropharmacology 2004, 46, 273–284. [Google Scholar] [CrossRef]
- Sosnoski, H.M.; Sears, S.M.S.; He, Y.; Frare, C.; Hewett, S.J. Sexually Dimorphic and Brain Region-Specific Transporter Adaptations in System Xc− Null Mice. Neurochem. Int. 2020, 141, 104888. [Google Scholar] [CrossRef]
- Shanker, G.; Allen, J.W.; Mutkus, L.A.; Aschner, M. The Uptake of Cysteine in Cultured Primary Astrocytes and Neurons. Brain Res. 2001, 902, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Flora, S.J.S.; Bhadauria, S.; Pant, S.C.; Dhaked, R.K. Arsenic Induced Blood and Brain Oxidative Stress and Its Response to Some Thiol Chelators in Rats. Life Sci. 2005, 77, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Sato, H.; Kuriyama-Matsumura, K.; Sato, K.; Maebara, K.; Wang, H.; Tamba, M.; Itoh, K.; Yamamoto, M.; Bannai, S. Electrophile Response Element-Mediated Induction of the Cystine/Glutamate Exchange Transporter Gene Expression. J. Biol. Chem. 2002, 277, 44765–44771. [Google Scholar] [CrossRef] [PubMed]
- Palma-Lara, I.; Martínez-Castillo, M.; Quintana-Pérez, J.C.; Arellano-Mendoza, M.G.; Tamay-Cach, F.; Valenzuela-Limón, O.L.; García-Montalvo, E.A.; Hernández-Zavala, A. Arsenic Exposure: A Public Health Problem Leading to Several Cancers. Regul. Toxicol. Pharmacol. 2020, 110, 104539. [Google Scholar] [CrossRef]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Kerins, M.J.; Ooi, A. The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid. Redox Signal. 2018, 29, 1756–1773. [Google Scholar] [CrossRef]
- He, Q.; Yang, J.; Pan, Z.; Zhang, G.; Chen, B.; Li, S.; Xiao, J.; Tan, F.; Wang, Z.; Chen, P.; et al. Biochanin A Protects against Iron Overload Associated Knee Osteoarthritis via Regulating Iron Levels and NRF2/System Xc−/GPX4 Axis. Biomed. Pharmacother. 2023, 157, 113915. [Google Scholar] [CrossRef]
- Hu, K.; Li, K.; Lv, J.; Feng, J.; Chen, J.; Wu, H.; Cheng, F.; Jiang, W.; Wang, J.; Pei, H.; et al. Suppression of the SLC7A11/Glutathione Axis Causes Synthetic Lethality in KRAS-Mutant Lung Adenocarcinoma. J. Clin. Investig. 2020, 130, 1752–1766. [Google Scholar] [CrossRef]
- He, F.; Zhang, P.; Liu, J.; Wang, R.; Kaufman, R.J.; Yaden, B.C.; Karin, M. ATF4 Suppresses Hepatocarcinogenesis by Inducing SLC7A11 (xCT) to Block Stress-Related Ferroptosis. J. Hepatol. 2023, 79, 362–377. [Google Scholar] [CrossRef]
- Xu, Y.; Li, Y.; Li, J.; Chen, W. Ethyl Carbamate Triggers Ferroptosis in Liver through Inhibiting GSH Synthesis and Suppressing Nrf2 Activation. Redox Biol. 2022, 53, 102349. [Google Scholar] [CrossRef]
- Shih, A.Y.; Erb, H.; Sun, X.; Toda, S.; Kalivas, P.W.; Murphy, T.H. Cystine/Glutamate Exchange Modulates Glutathione Supply for Neuroprotection from Oxidative Stress and Cell Proliferation. J. Neurosci. 2006, 26, 10514–10523. [Google Scholar] [CrossRef] [PubMed]
- D’Ezio, V.; Colasanti, M.; Persichini, T. Amyloid-β 25-35 Induces Neurotoxicity through the Up-Regulation of Astrocytic System Xc−. Antioxidants 2021, 10, 1685. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Lu, M.; Mei, M.; Wang, H.; Han, Z.; Chen, M.; Yao, H.; Song, N.; Ding, X.; Ding, J.; et al. Pyridoxine Induces Glutathione Synthesis via PKM2-Mediated Nrf2 Transactivation and Confers Neuroprotection. Nat. Commun. 2020, 11, 941. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.-Q.; Xu, T.; Ji, W.; Wang, C.; Ren, Y.; Xiong, X.; Zhou, X.; Lin, S.-H.; Xu, Y.; Qiu, Y. Herpes Simplex Virus 1-Induced Ferroptosis Contributes to Viral Encephalitis. mBio 2023, 14, e0237022. [Google Scholar] [CrossRef]
- Ozturk, M.; Metin, M.; Altay, V.; Bhat, R.A.; Ejaz, M.; Gul, A.; Unal, B.T.; Hasanuzzaman, M.; Nibir, L.; Nahar, K.; et al. Arsenic and Human Health: Genotoxicity, Epigenomic Effects, and Cancer Signaling. Biol. Trace Elem. Res. 2022, 200, 988–1001. [Google Scholar] [CrossRef]
- Litwin, I.; Bocer, T.; Dziadkowiec, D.; Wysocki, R. Oxidative Stress and Replication-Independent DNA Breakage Induced by Arsenic in Saccharomyces Cerevisiae. PLoS Genet. 2013, 9, e1003640. [Google Scholar] [CrossRef]
- Büscher, M.; Rahmsdorf, H.J.; Litfin, M.; Karin, M.; Herrlich, P. Activation of the C-Fos Gene by UV and Phorbol Ester: Different Signal Transduction Pathways Converge to the Same Enhancer Element. Oncogene 1988, 3, 301–311. [Google Scholar]
- Dosch, J.; Kaina, B. Induction of C-Fos, c-Jun, junB and junD mRNA and AP-1 by Alkylating Mutagens in Cells Deficient and Proficient for the DNA Repair Protein O6-Methylguanine-DNA Methyltransferase (MGMT) and Its Relationship to Cell Death, Mutation Induction and Chromosomal Instability. Oncogene 1996, 13, 1927–1935. [Google Scholar]
- Brach, M.A.; Hass, R.; Sherman, M.L.; Gunji, H.; Weichselbaum, R.; Kufe, D. Ionizing Radiation Induces Expression and Binding Activity of the Nuclear Factor Kappa B. J. Clin. Investig. 1991, 88, 691–695. [Google Scholar] [CrossRef]
- Mohan, N.; Meltz, M.L. Induction of Nuclear Factor Kappa B after Low-Dose Ionizing Radiation Involves a Reactive Oxygen Intermediate Signaling Pathway. Radiat. Res. 1994, 140, 97–104. [Google Scholar] [CrossRef]
- Lastro, M.; Kourtidis, A.; Farley, K.; Conklin, D.S. xCT Expression Reduces the Early Cell Cycle Requirement for Calcium Signaling. Cell. Signal. 2008, 20, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Kumanova, M.; Hart, L.S.; Sloane, K.; Zhang, H.; De Panis, D.N.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Ron, D.; Koumenis, C. The GCN2-ATF4 Pathway Is Critical for Tumour Cell Survival and Proliferation in Response to Nutrient Deprivation. EMBO J. 2010, 29, 2082–2096. [Google Scholar] [CrossRef] [PubMed]
- Griffith, O.W.; Mulcahy, R.T. The Enzymes of Glutathione Synthesis: Gamma-Glutamylcysteine Synthetase. Adv. Enzymol. Relat. Areas Mol. Biol. 1999, 73, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.A.; White, C.C.; Cox, D.P.; Chan, J.Y.; Kavanagh, T.J.; Fausto, N.; Franklin, C.C. Distinct Nrf1/2-Independent Mechanisms Mediate As3+-Induced Glutamate-Cysteine Ligase Subunit Gene Expression in Murine Hepatocytes. Free Radic. Biol. Med. 2009, 46, 1614–1625. [Google Scholar] [CrossRef]
- Zipper, L.M.; Mulcahy, R.T. Inhibition of ERK and P38 MAP Kinases Inhibits Binding of Nrf2 and Induction of GCS Genes. Biochem. Biophys. Res. Commun. 2000, 278, 484–492. [Google Scholar] [CrossRef]
- Liu, X.; Shah, A.; Gangwani, M.R.; Silverstein, P.S.; Fu, M.; Kumar, A. HIV-1 Nef Induces CCL5 Production in Astrocytes through P38-MAPK and PI3K/Akt Pathway and Utilizes NF-kB, CEBP and AP-1 Transcription Factors. Sci. Rep. 2014, 4, 4450. [Google Scholar] [CrossRef]
- Nookala, A.R.; Shah, A.; Noel, R.J.; Kumar, A. HIV-1 Tat-Mediated Induction of CCL5 in Astrocytes Involves NF-κB, AP-1, C/EBPα and C/EBPγ Transcription Factors and JAK, PI3K/Akt and P38 MAPK Signaling Pathways. PLoS ONE 2013, 8, e78855. [Google Scholar] [CrossRef]
- Kirsch, K.; Zeke, A.; Tőke, O.; Sok, P.; Sethi, A.; Sebő, A.; Kumar, G.S.; Egri, P.; Póti, Á.L.; Gooley, P.; et al. Co-Regulation of the Transcription Controlling ATF2 Phosphoswitch by JNK and P38. Nat. Commun. 2020, 11, 5769. [Google Scholar] [CrossRef]
- Richman, P.G.; Meister, A. Regulation of Gamma-Glutamyl-Cysteine Synthetase by Nonallosteric Feedback Inhibition by Glutathione. J. Biol. Chem. 1975, 250, 1422–1426. [Google Scholar] [CrossRef]
- Liu, Y.; Hyde, A.S.; Simpson, M.A.; Barycki, J.J. Emerging Regulatory Paradigms in Glutathione Metabolism. Adv. Cancer Res. 2014, 122, 69–101. [Google Scholar] [CrossRef]
- Backos, D.S.; Fritz, K.S.; Roede, J.R.; Petersen, D.R.; Franklin, C.C. Posttranslational Modification and Regulation of Glutamate-Cysteine Ligase by the α,β-Unsaturated Aldehyde 4-Hydroxy-2-Nonenal. Free Radic. Biol. Med. 2011, 50, 14–26. [Google Scholar] [CrossRef]
- Yang, H.; Magilnick, N.; Xia, M.; Lu, S.C. Effects of Hepatocyte Growth Factor on Glutathione Synthesis, Growth, and Apoptosis Is Cell Density-Dependent. Exp. Cell Res. 2008, 314, 398–412. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.Z.; Chen, C.; Zeng, Z.; Yang, H.; Oh, J.; Chen, L.; Lu, S.C. Mechanism and Significance of Increased Glutathione Level in Human Hepatocellular Carcinoma and Liver Regeneration. FASEB J. 2001, 15, 19–21. [Google Scholar] [CrossRef] [PubMed]
- Trackey, J.L.; Uliasz, T.F.; Hewett, S.J. SIN-1-Induced Cytotoxicity in Mixed Cortical Cell Culture: Peroxynitrite-Dependent and -Independent Induction of Excitotoxic Cell Death. J. Neurochem. 2001, 79, 445–455. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, F.; Liao, Y.; Jin, Y.; Sun, G. Effects of Arsenite in Astrocytes on Neuronal Signaling Transduction. Toxicology 2013, 303, 43–53. [Google Scholar] [CrossRef]
- Lai, W.S.; Arvola, R.M.; Goldstrohm, A.C.; Blackshear, P.J. Inhibiting Transcription in Cultured Metazoan Cells with Actinomycin D to Monitor mRNA Turnover. Methods 2019, 155, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Pshenichkin, S.P.; Szekely, A.M.; Wise, B.C. Transcriptional and Posttranscriptional Mechanisms Involved in the Interleukin-1, Steroid, and Protein Kinase C Regulation of Nerve Growth Factor in Cortical Astrocytes. J. Neurochem. 1994, 63, 419–428. [Google Scholar] [CrossRef]
- Jackman, N.A.; Uliasz, T.F.; Hewett, J.A.; Hewett, S.J. Regulation of System Xc− Activity and Expression in Astrocytes by Interleukin-1β: Implications for Hypoxic Neuronal Injury. Glia 2010, 58, 1806–1815. [Google Scholar] [CrossRef]
- Yu, A.C.H.; Yung, H.W.; Hui, M.H.K.; Lau, L.T.; Chen, X.Q.; Collins, R.A. Cycloheximide and Actinomycin D Delay Death and Affect Bcl-2, Bax, and Ice Gene Expression in Astrocytes under in Vitro Ischemia. J. Neurosci. Res. 2003, 74, 318–325. [Google Scholar] [CrossRef]
- Shimomura, T.; Hirakawa, N.; Ohuchi, Y.; Ishiyama, M.; Shiga, M.; Ueno, Y. Simple Fluorescence Assay for Cystine Uptake via the xCT in Cells Using Selenocystine and a Fluorescent Probe. ACS Sens. 2021, 6, 2125–2128. [Google Scholar] [CrossRef]
- Hewett, J.A.; Hewett, S.J.; Winkler, S.; Pfeiffer, S.E. Inducible Nitric Oxide Synthase Expression in Cultures Enriched for Mature Oligodendrocytes Is Due to Microglia. J. Neurosci. Res. 1999, 56, 189–198. [Google Scholar] [CrossRef]
- Uliasz, T.F.; Hewett, S.J. A Microtiter Trypan Blue Absorbance Assay for the Quantitative Determination of Excitotoxic Neuronal Injury in Cell Culture. J. Neurosci. Methods 2000, 100, 157–163. [Google Scholar] [CrossRef] [PubMed]


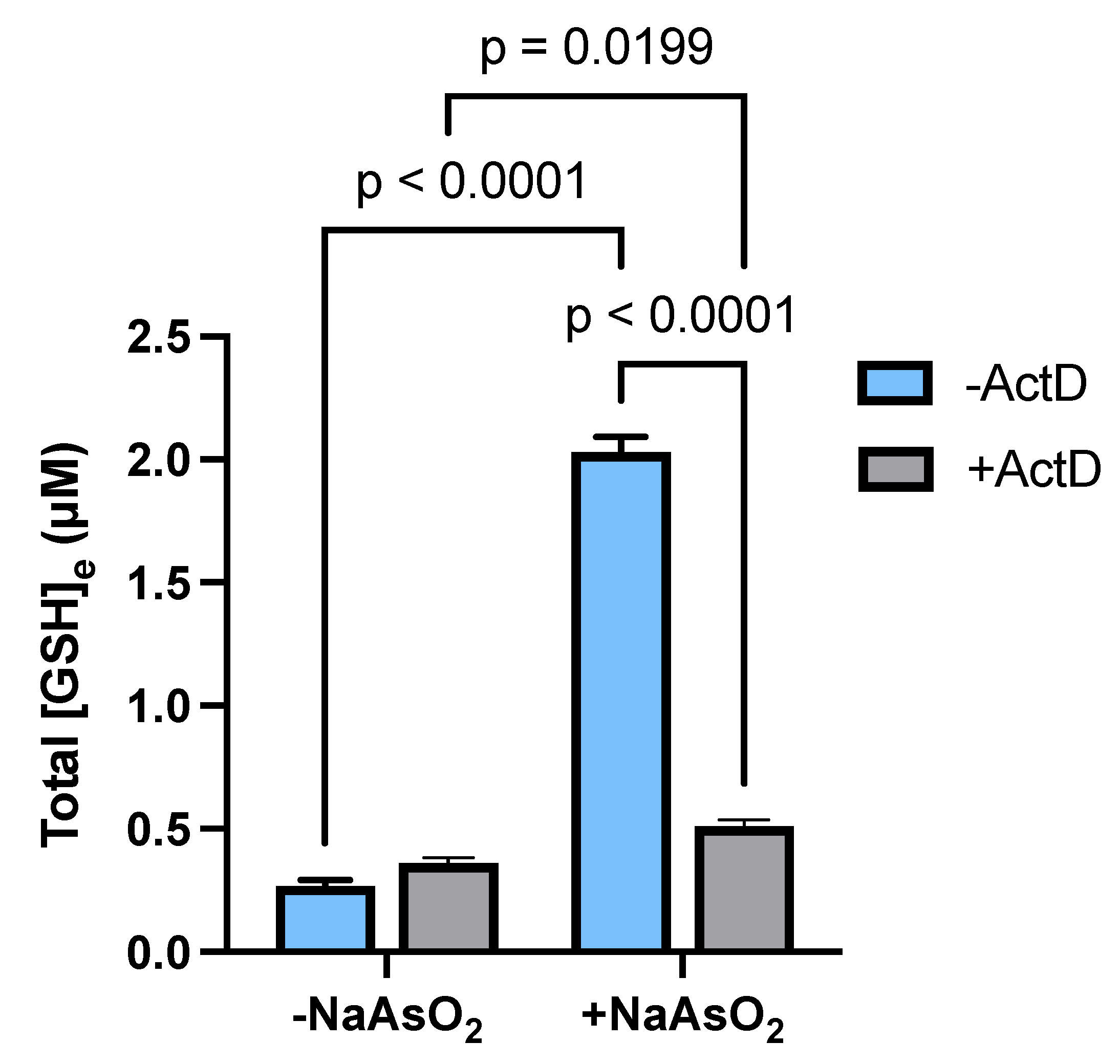
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leisawitz, J.P.; He, J.; Baggeroer, C.; Hewett, S.J. Arsenite-Mediated Transcriptional Regulation of Glutathione Synthesis in Mammalian Primary Cortical Astrocytes. Int. J. Mol. Sci. 2025, 26, 5375. https://doi.org/10.3390/ijms26115375
Leisawitz JP, He J, Baggeroer C, Hewett SJ. Arsenite-Mediated Transcriptional Regulation of Glutathione Synthesis in Mammalian Primary Cortical Astrocytes. International Journal of Molecular Sciences. 2025; 26(11):5375. https://doi.org/10.3390/ijms26115375
Chicago/Turabian StyleLeisawitz, Jacob P., Jiali He, Caroline Baggeroer, and Sandra J. Hewett. 2025. "Arsenite-Mediated Transcriptional Regulation of Glutathione Synthesis in Mammalian Primary Cortical Astrocytes" International Journal of Molecular Sciences 26, no. 11: 5375. https://doi.org/10.3390/ijms26115375
APA StyleLeisawitz, J. P., He, J., Baggeroer, C., & Hewett, S. J. (2025). Arsenite-Mediated Transcriptional Regulation of Glutathione Synthesis in Mammalian Primary Cortical Astrocytes. International Journal of Molecular Sciences, 26(11), 5375. https://doi.org/10.3390/ijms26115375