
Neutralizing Antibodies: Role in Immune Response and Viral Vector Based Gene Therapy
 , ,
, , 
Abstract
1. Introduction
2. Definition of nAbs
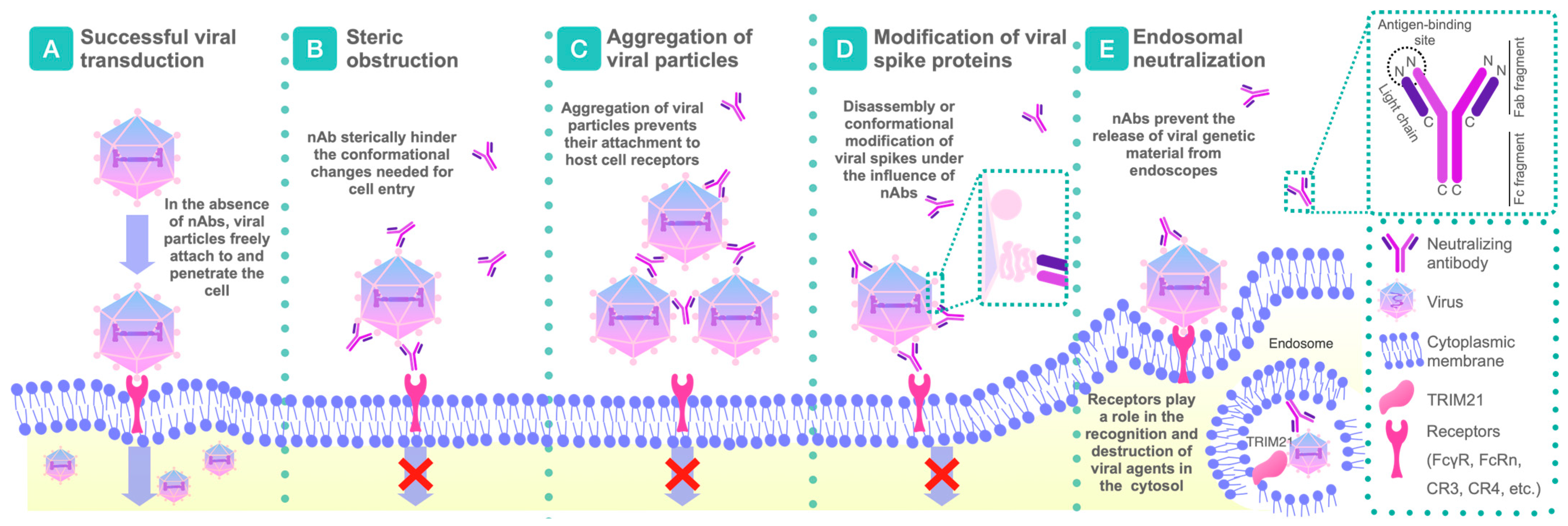
2.1. Mechanisms of Action of nAbs
2.1.1. Disruption or Conformational Modifications of Viral Spikes
2.1.2. Aggregation by nAbs
2.1.3. Steric Obstruction Following Viral Attachment
2.1.4. Intracytoplasmic Neutralization
2.1.5. Steric Blockade of the Receptor-Binding Site Before Virus Attachment
2.1.6. Prevention of Conformational Changes Necessary for the Fusion of Virus and Cell Membrane
2.1.7. Antibody-Dependent Cellular Cytotoxicity
2.1.8. Antibody-Dependent Cellular Phagocytosis
2.1.9. Complement Activation
3. Mechanisms of nAbs’ Formation
3.1. Immune Response to Viral Vectors
3.2. Immune Response to Therapeutic Genes
3.3. Factors Affecting the Formation of nAbs
4. nAbs’ Impact on Gene Therapy Efficacy
4.1. Reduced Efficacy
4.1.1. Neutralization of Viral Vectors Preventing Gene Delivery to Target Cells
4.1.2. Accelerated Clearance of Viral Vectors from the Body
4.1.3. Induction of Cytotoxic Immune Responses
4.2. Risks and Side Effects
Immunopathology and Disease Development

{kind=link}
| Viral Vector Type | Examples of Applications | Potential Role of Neutralizing Antibodies | Side Effects and Limitations | Sources |
|---|---|---|---|---|
| Adeno-Associated Viruses | Gene therapy for inherited diseases (e.g., spinal muscular atrophy, hemophilia), oncology. | nAbs to AAV can reduce therapy effectiveness by preventing vector entry into target cells. Patients with pre-existing antibodies to AAV may require vector serotype switching. | Low immunogenicity, but inflammatory reactions in the liver are possible. Limited genome capacity (~4.9 kb). | [89,119,120,121,122] |
| Lentiviruses | Treatment of hemoglobinopathies (e.g., β-thalassemia), CAR-T therapy. | Lentiviruses are less susceptible to neutralization by antibodies but may elicit an immune response to viral proteins. | Risk of insertional mutagenesis due to integration into the host genome. Oncogene activation is possible. | [123,124,125,126,127,128] |
| Retroviruses | Ex vivo therapy (e.g., treatment of SCID), oncology. | Neutralizing antibodies can limit repeated vector administration. | Infect only dividing cells. High risk of insertional mutagenesis. | [129,130,131,132,133] |
| Adenoviruses | Vaccines (e.g., against COVID-19, HPV), oncology. | High levels of pre-existing antibodies to adenoviruses in the population can reduce effectiveness. | Strong immune response, risk of cytokine storm. Limited use upon re-administration. | [134,135,136,137,138] |
| Herpes Simplex Viruses | Neurodegenerative diseases, oncology. | Antibodies can reduce delivery efficiency. | Ability to infect only non-dividing cells. Neurotoxicity, inflammatory reactions in the CNS are possible. | [139,140,141,142] |
4.3. Complications Associated with Repeated Injections
4.3.1. Reduced Therapeutic Efficacy Due to Elevated nAb Levels
4.3.2. Increased Risk of Adverse Effects
5. Strategies to Overcome Challenges Associated with nAbs
5.1. Surface Modification of Viral Vectors to Reduce Immunogenicity
5.2. Immunomodulation
Immunosuppressive Therapy and Induction of Immune Tolerance
5.3. Alternative Strategies for Preventing nAb Functioning
5.3.1. Vexosomes and Extracellular Vesicle-Associated AAVs
5.3.2. Degradation of nAbs by Pre-Injection of Enzymes That Break Down Ig
5.4. Modification of Delivery Methods
5.5. Selection of Optimal Doses
6. Perspectives and Future Research
6.1. Development of New, Safer Viral Vectors
6.2. Development of New Immunomodulation Methods
6.3. Investigating Individual Differences in Immune Response to Gene Therapy
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Schlieben, L.D.; Prokisch, H.; Yepez, V.A. How Machine Learning and Statistical Models Advance Molecular Diagnostics of Rare Disorders Via Analysis of RNA Sequencing Data. Front. Mol. Biosci. 2021, 8, 647277. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, P.; Pariser, A.R.; Austin, C. From scientific discovery to treatments for rare diseases—The view from the National Center for Advancing Translational Sciences—Office of Rare Diseases Research. Orphanet J. Rare Dis. 2018, 13, 196. [Google Scholar] [CrossRef]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Lefebvre, E.; Laporte, J. Comparative in vivo characterization of newly discovered myotropic adeno-associated vectors. Skelet. Muscle 2024, 14, 9. [Google Scholar] [CrossRef]
- McCarron, A.; Farrow, N.; Cmielewski, P.; Knight, E.; Donnelley, M.; Parsons, D. Breaching the Delivery Barrier: Chemical and Physical Airway Epithelium Disruption Strategies for Enhancing Lentiviral-Mediated Gene Therapy. Front. Pharmacol. 2021, 12, 669635. [Google Scholar] [CrossRef] [PubMed]
- Gross, D.A.; Tedesco, N.; Leborgne, C.; Ronzitti, G. Overcoming the Challenges Imposed by Humoral Immunity to AAV Vectors to Achieve Safe and Efficient Gene Transfer in Seropositive Patients. Front. Immunol. 2022, 13, 857276. [Google Scholar] [CrossRef]
- Li, W.; Feng, S.L.; Herrschaft, L.; Samulski, R.J.; Li, C. Rationally engineered novel AAV capsids for intra-articular gene delivery. Mol. Ther. Methods Clin. Dev. 2024, 32, 101211. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, L.; Van Kaer, L. Role of canonical and noncanonical autophagy pathways in shaping the life journey of B cells. Front. Immunol. 2024, 15, 1426204. [Google Scholar] [CrossRef]
- Daeron, M. The function of antibodies. Immunol. Rev. 2024, 328, 113–125. [Google Scholar] [CrossRef]
- Pantaleo, G.; Correia, B.; Fenwick, C.; Joo, V.S.; Perez, L. Antibodies to combat viral infections: Development strategies and progress. Nat. Rev. Drug Discov. 2022, 21, 676–696. [Google Scholar] [CrossRef]
- Arlotta, K.J.; Owen, S.C. Antibody and antibody derivatives as cancer therapeutics. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2019, 11, e1556. [Google Scholar] [CrossRef] [PubMed]
- Calvert, R.A.; Nyamboya, R.A.; Beavil, A.J.; Sutton, B.J. The evolution of flexibility and function in the Fc domains of IgM, IgY, and IgE. Front. Immunol. 2024, 15, 1389494. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.L.; Suscovich, T.J.; Fortune, S.M.; Alter, G. Beyond binding: Antibody effector functions in infectious diseases. Nat. Rev. Immunol. 2018, 18, 46–61. [Google Scholar] [CrossRef]
- O’Reilly, S.; Byrne, J.; Feeney, E.R.; Mallon, P.W.G.; Gautier, V. Navigating the Landscape of B Cell Mediated Immunity and Antibody Monitoring in SARS-CoV-2 Vaccine Efficacy: Tools, Strategies and Clinical Trial Insights. Vaccines 2024, 12, 1089. [Google Scholar] [CrossRef]
- Pisil, Y.; Yazici, Z.; Shida, H.; Miura, T. Is SARS-CoV-2 Neutralized More Effectively by IgM and IgA than IgG Having the Same Fab Region? Pathogens 2021, 10, 751. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Gan, H.; Liang, Z.; Liu, L.; Liu, Q.; Mai, Y.; Chen, H.; Lei, B.; Yu, S.; Chen, H.; et al. Review of therapeutic mechanisms and applications based on SARS-CoV-2 neutralizing antibodies. Front. Microbiol. 2023, 14, 1122868. [Google Scholar] [CrossRef]
- Burton, D.R. Antiviral neutralizing antibodies: From in vitro to in vivo activity. Nat. Rev. Immunol. 2023, 23, 720–734. [Google Scholar] [CrossRef]
- Burton, D.R. Antibodies, viruses and vaccines. Nat. Rev. Immunol. 2002, 2, 706–713. [Google Scholar] [CrossRef]
- Burton, D.R.; Poignard, P.; Stanfield, R.L.; Wilson, I.A. Broadly neutralizing antibodies present new prospects to counter highly antigenically diverse viruses. Science 2012, 337, 183–186. [Google Scholar] [CrossRef]
- Adhikari, R.P.; Alem, F.; Kemboi, D.; Kanipakala, T.; Sherchand, S.P.; Kailasan, S.; Purcell, B.K.; Heine, H.S.; Russell-Lodrigue, K.; Etobayeva, I.; et al. Engineered antibodies targeted to bacterial surface integrate effector functions with toxin neutralization to provide superior efficacy against bacterial infections. medRxiv 2024. [Google Scholar] [CrossRef]
- Dispinseri, S.; Secchi, M.; Pirillo, M.F.; Tolazzi, M.; Borghi, M.; Brigatti, C.; De Angelis, M.L.; Baratella, M.; Bazzigaluppi, E.; Venturi, G.; et al. Neutralizing antibody responses to SARS-CoV-2 in symptomatic COVID-19 is persistent and critical for survival. Nat. Commun. 2021, 12, 2670. [Google Scholar] [CrossRef] [PubMed]
- Vandamme, C.; Adjali, O.; Mingozzi, F. Unraveling the Complex Story of Immune Responses to AAV Vectors Trial After Trial. Hum. Gene Ther. 2017, 28, 1061–1074. [Google Scholar] [CrossRef] [PubMed]
- Calcedo, R.; Wilson, J.M. Humoral Immune Response to AAV. Front. Immunol. 2013, 4, 341. [Google Scholar] [CrossRef]
- Kombe Kombe, A.J.; Zahid, A.; Mohammed, A.; Shi, R.; Jin, T. Potent Molecular Feature-based Neutralizing Monoclonal Antibodies as Promising Therapeutics against SARS-CoV-2 Infection. Front. Mol. Biosci. 2021, 8, 670815. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, Q.; Liu, Z.; Wang, Q.; Wu, J.; Hu, Y.; Bai, T.; Xie, T.; Huang, M.; Wu, T.; et al. Spike-specific circulating T follicular helper cell and cross-neutralizing antibody responses in COVID-19-convalescent individuals. Nat. Microbiol. 2021, 6, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Roux, K.H.; Taylor, K.A. AIDS virus envelope spike structure. Curr. Opin. Struct. Biol. 2007, 17, 244–252. [Google Scholar] [CrossRef]
- Murin, C.D.; Wilson, I.A.; Ward, A.B. Antibody responses to viral infections: A structural perspective across three different enveloped viruses. Nat. Microbiol. 2019, 4, 734–747. [Google Scholar] [CrossRef]
- Crispin, M.; Ward, A.B.; Wilson, I.A. Structure and Immune Recognition of the HIV Glycan Shield. Annu. Rev. Biophys. 2018, 47, 499–523. [Google Scholar] [CrossRef]
- Doores, K.J. The HIV glycan shield as a target for broadly neutralizing antibodies. FEBS J. 2015, 282, 4679–4691. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Stieh, D.J.; King, D.F.; Klein, K.; Liu, P.; Shen, X.; Hwang, K.K.; Ferrari, G.; Montefiori, D.C.; Haynes, B.; Pitisuttithum, P.; et al. Aggregate complexes of HIV-1 induced by multimeric antibodies. Retrovirology 2014, 11, 78. [Google Scholar] [CrossRef] [PubMed]
- Klasse, P.J. Neutralization of Virus Infectivity by Antibodies: Old Problems in New Perspectives. Adv. Biol. 2014, 2014, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.F.; Fox, J.M.; Earnest, J.T.; Ng, T.S.; Kim, A.S.; Fibriansah, G.; Kostyuchenko, V.A.; Shi, J.; Shu, B.; Diamond, M.S.; et al. Structural basis of Chikungunya virus inhibition by monoclonal antibodies. Proc. Natl. Acad. Sci. USA 2020, 117, 27637–27645. [Google Scholar] [CrossRef]
- Thompson, B.S.; Moesker, B.; Smit, J.M.; Wilschut, J.; Diamond, M.S.; Fremont, D.H. A therapeutic antibody against west nile virus neutralizes infection by blocking fusion within endosomes. PLoS Pathog. 2009, 5, e1000453. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.; Yan, H.; Lamm, M.E.; Huang, Y.T. Immunoglobulin A antibodies against internal HIV-1 proteins neutralize HIV-1 replication inside epithelial cells. Virology 2006, 356, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Zhang, D.; Qian, P.; Qian, S.; Wu, M.; Chen, H.; Li, X. Swine TRIM21 restricts FMDV infection via an intracellular neutralization mechanism. Antivir. Res. 2016, 127, 32–40. [Google Scholar] [CrossRef]
- Rhodes, D.A.; Isenberg, D.A. TRIM21 and the Function of Antibodies inside Cells. Trends Immunol. 2017, 38, 916–926. [Google Scholar] [CrossRef]
- Tam, E.H.; Peng, Y.; Cheah, M.X.Y.; Yan, C.; Xiao, T. Neutralizing antibodies to block viral entry and for identification of entry inhibitors. Antivir. Res. 2024, 224, 105834. [Google Scholar] [CrossRef]
- Labriola, J.M.; Miersch, S.; Chen, G.; Chen, C.; Pavlenco, A.; Saberianfar, R.; Caccuri, F.; Zani, A.; Sharma, N.; Feng, A.; et al. Peptide-Antibody Fusions Engineered by Phage Display Exhibit an Ultrapotent and Broad Neutralization of SARS-CoV-2 Variants. ACS Chem. Biol. 2022, 17, 1978–1988. [Google Scholar] [CrossRef]
- Hussain, A.; Hasan, A.; Nejadi Babadaei, M.M.; Bloukh, S.H.; Chowdhury, M.E.H.; Sharifi, M.; Haghighat, S.; Falahati, M. Targeting SARS-CoV2 Spike Protein Receptor Binding Domain by Therapeutic Antibodies. Biomed. Pharmacother. 2020, 130, 110559. [Google Scholar] [CrossRef]
- Fedry, J.; Hurdiss, D.L.; Wang, C.; Li, W.; Obal, G.; Drulyte, I.; Du, W.; Howes, S.C.; van Kuppeveld, F.J.M.; Förster, F.; et al. Structural insights into the cross-neutralization of SARS-CoV and SARS-CoV-2 by the human monoclonal antibody 47D11. Sci. Adv. 2021, 7, eabf5632. [Google Scholar] [CrossRef] [PubMed]
- Li, C.J.; Chao, T.L.; Chang, T.Y.; Hsiao, C.C.; Lu, D.C.; Chiang, Y.W.; Lai, G.C.; Tsai, Y.M.; Fang, J.T.; Ieong, S.; et al. Neutralizing Monoclonal Antibodies Inhibit SARS-CoV-2 Infection through Blocking Membrane Fusion. Microbiol. Spectr. 2022, 10, e0181421. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Wang, Z.; Uprety, T.; Sreenivasan, C.C.; Sheng, Z.; Hause, B.M.; Brunick, C.; Wu, H.; Luke, T.; Bausch, C.L.; et al. A fully human monoclonal antibody possesses antibody-dependent cellular cytotoxicity (ADCC) activity against the H1 subtype of influenza A virus by targeting a conserved epitope at the HA1 protomer interface. J. Med. Virol. 2023, 95, e28901. [Google Scholar] [CrossRef]
- Coënon, L.; Villalba, M. From CD16a Biology to Antibody-Dependent Cell-Mediated Cytotoxicity Improvement. Front. Immunol. 2022, 13, 913215. [Google Scholar] [CrossRef]
- Orange, J.S. Formation and function of the lytic NK-cell immunological synapse. Nat. Rev. Immunol. 2008, 8, 713–725. [Google Scholar] [CrossRef]
- Ivanova, M.E.; Lukoyanova, N.; Malhotra, S.; Topf, M.; Trapani, J.A.; Voskoboinik, I.; Saibil, H.R. The pore conformation of lymphocyte perforin. Sci. Adv. 2022, 8, eabk3147. [Google Scholar] [CrossRef] [PubMed]
- Tay, M.Z.; Wiehe, K.; Pollara, J. Antibody-Dependent Cellular Phagocytosis in Antiviral Immune Responses. Front. Immunol. 2019, 10, 332. [Google Scholar] [CrossRef]
- Kamen, L.; Myneni, S.; Langsdorf, C.; Kho, E.; Ordonia, B.; Thakurta, T.; Zheng, K.; Song, A.; Chung, S. A novel method for determining antibody-dependent cellular phagocytosis. J. Immunol. Methods 2019, 468, 55–60. [Google Scholar] [CrossRef]
- Eberhard, S.; Vietzen, H.; Görzer, I.; Jaksch, P.; Puchhammer-Stöckl, E. Analysis and Fine Specificity of the HCMV-Specific Cell-Free and Cell-Associated Antibody-Dependent Cellular Phagocytosis (ADCP) Responses in Lung Transplant Recipients. Int. J. Mol. Sci. 2021, 22, 8206. [Google Scholar] [CrossRef]
- Mellors, J.; Tipton, T.; Fehling, S.K.; Akoi Bore, J.; Koundouno, F.R.; Hall, Y.; Hudson, J.; Alexander, F.; Longet, S.; Taylor, S.; et al. Complement-Mediated Neutralisation Identified in Ebola Virus Disease Survivor Plasma: Implications for Protection and Pathogenesis. Front. Immunol. 2022, 13, 857481. [Google Scholar] [CrossRef]
- Ovcinnikovs, V.; Dijkman, K.; Zom, G.G.; Beurskens, F.J.; Trouw, L.A. Enhancing complement activation by therapeutic anti-tumor antibodies: Mechanisms, strategies, and engineering approaches. Semin. Immunol. 2025, 77, 101922. [Google Scholar] [CrossRef]
- Plasschaert, L.W.; MacDonald, K.D.; Moffit, J.S. Current landscape of cystic fibrosis gene therapy. Front. Pharmacol. 2024, 15, 1476331. [Google Scholar] [CrossRef] [PubMed]
- Salauddin, M.; Saha, S.; Hossain, M.G.; Okuda, K.; Shimada, M. Clinical Application of Adenovirus (AdV): A Comprehensive Review. Viruses 2024, 16, 1094. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.T.; Sugimura, R. Breakthroughs in synthetic controlling strategies for precision in CAR-T therapy. Prog. Mol. Biol. Transl. Sci. 2024, 209, 61–100. [Google Scholar]
- Wang, J.H.; Zhan, W.; Gallagher, T.L.; Gao, G. Recombinant adeno-associated virus as a delivery platform for ocular gene therapy: A comprehensive review. Mol. Ther. 2024, 32, 4185–4207. [Google Scholar] [CrossRef]
- Issa, S.S.; Shaimardanova, A.A.; Solovyeva, V.V.; Rizvanov, A.A. Various AAV Serotypes and Their Applications in Gene Therapy: An Overview. Cells 2023, 12, 785. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Connolly, A.M.; Lehman, K.J.; Griffin, D.A.; Khan, S.Z.; Dharia, S.D.; Quintana-Gallardo, L.; Rodino-Klapac, L.R. Testing preexisting antibodies prior to AAV gene transfer therapy: Rationale, lessons and future considerations. Mol. Ther. Methods Clin. Dev. 2022, 25, 74–83. [Google Scholar] [CrossRef]
- McGinley, L.; McMahon, J.; Strappe, P.; Barry, F.; Murphy, M.; O’Toole, D.; O’Brien, T. Lentiviral vector mediated modification of mesenchymal stem cells & enhanced survival in an in vitro model of ischaemia. Stem Cell Res. Ther. 2011, 2, 12. [Google Scholar]
- Wang, X.; Ma, C.; Rodriguez Labrada, R.; Qin, Z.; Xu, T.; He, Z.; Wei, Y. Recent advances in lentiviral vectors for gene therapy. Sci. China Life Sci. 2021, 64, 1842–1857. [Google Scholar] [CrossRef]
- Matsunaga, W.; Gotoh, A. Adenovirus as a Vector and Oncolytic Virus. Curr. Issues Mol. Biol. 2023, 45, 4826–4840. [Google Scholar] [CrossRef]
- Afzal, S.Y.; MacDougall, M.S.; McGhee, S.A. Immunodeficiency: Gene therapy for primary immune deficiency. Allergy Asthma Proc. 2024, 45, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, M.; Osborne, A.; Yu-Wai-Man, P.; Martin, K. Humoral immune responses to AAV gene therapy in the ocular compartment. Biol. Rev. Camb. Philos. Soc. 2021, 96, 1616–1644. [Google Scholar] [CrossRef]
- Donadeu, L.; Gomez-Olles, S.; Casanova, F.; Torija, A.; Lopez-Meseguer, M.; Boada-Perez, M.; Kervella, D.; Crespo, E.; Carrera-Munoz, C.; Campos-Varela, I.; et al. Role of SARS-CoV-2-specific memory B cells promoting immune protection after booster vaccination in solid organ transplantation. Front. Immunol. 2024, 15, 1463769. [Google Scholar] [CrossRef]
- Hong, Y.; Kwak, K. Both sides now: Evolutionary traits of antigens and B cells in tolerance and activation. Front. Immunol. 2024, 15, 1456220. [Google Scholar] [CrossRef] [PubMed]
- Gruell, H.; Vanshylla, K.; Weber, T.; Barnes, C.O.; Kreer, C.; Klein, F. Antibody-mediated neutralization of SARS-CoV-2. Immunity 2022, 55, 925–944. [Google Scholar] [CrossRef]
- Bouquet, C.; Vignal Clermont, C.; Galy, A.; Fitoussi, S.; Blouin, L.; Munk, M.R.; Valero, S.; Meunier, S.; Katz, B.; Sahel, J.A.; et al. Immune Response and Intraocular Inflammation in Patients With Leber Hereditary Optic Neuropathy Treated With Intravitreal Injection of Recombinant Adeno-Associated Virus 2 Carrying the ND4 Gene: A Secondary Analysis of a Phase 1/2 Clinical Trial. JAMA Ophthalmol. 2019, 137, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Long, B.R.; Robinson, T.M.; Day, J.R.S.; Yu, H.; Lau, K.; Imtiaz, U.; Patton, K.S.; de Hart, G.; Henshaw, J.; Agarwal, S.; et al. Clinical immunogenicity outcomes from GENEr8-1, a phase 3 study of valoctocogene roxaparvovec, an AAV5-vectored gene therapy for hemophilia A. Mol. Ther. 2024, 32, 2052–2063. [Google Scholar] [CrossRef]
- Yu, S.; Zhao, Q.; Zhang, C.; Fu, D.; Zhu, X.; Zhou, J.; Ma, W.; Dong, Z.; Zhai, X.; Jiang, L.; et al. Methodological Validation and Inter-Laboratory Comparison of Microneutralization Assay for Detecting Anti-AAV9 Neutralizing Antibody in Human. Viruses 2024, 16, 1512. [Google Scholar] [CrossRef]
- Sayed, N.; Allawadhi, P.; Khurana, A.; Singh, V.; Navik, U.; Pasumarthi, S.K.; Khurana, I.; Banothu, A.K.; Weiskirchen, R.; Bharani, K.K. Gene therapy: Comprehensive overview and therapeutic applications. Life Sci. 2022, 294, 120375. [Google Scholar] [CrossRef]
- Xu, W.; Deng, Z.; Wang, X.; Jiang, H. Network Pharmacology Study of Compound Ligustrazine in Gastric Cancer Therapy. Sichuan Da Xue Xue Bao Yi Xue Ban 2024, 55, 1114–1122. [Google Scholar]
- Shaimardanova, A.A.; Chulpanova, D.S.; Solovyeva, V.V.; Mullagulova, A.I.; Kitaeva, K.V.; Allegrucci, C.; Rizvanov, A.A. Metachromatic Leukodystrophy: Diagnosis, Modeling, and Treatment Approaches. Front. Med. 2020, 7, 576221. [Google Scholar] [CrossRef] [PubMed]
- Biswas, M.; Kumar, S.R.P.; Terhorst, C.; Herzog, R.W. Gene Therapy With Regulatory T Cells: A Beneficial Alliance. Front. Immunol. 2018, 9, 554. [Google Scholar] [CrossRef]
- Patel, S.R.; Lundgren, T.S.; Spencer, H.T.; Doering, C.B. The Immune Response to the fVIII Gene Therapy in Preclinical Models. Front. Immunol. 2020, 11, 494. [Google Scholar] [CrossRef]
- Perna, F.; Parekh, S.; Diorio, C.; Smith, M.; Subklewe, M.; Mehta, R.; Locke, F.L.; Shah, N.N. CAR T-cell toxicities: From bedside to bench, how novel toxicities inform laboratory investigations. Blood Adv. 2024, 8, 4348–4358. [Google Scholar] [CrossRef] [PubMed]
- Ertl, H.C.J. Immunogenicity and toxicity of AAV gene therapy. Front. Immunol. 2022, 13, 975803. [Google Scholar] [CrossRef] [PubMed]
- Sinnett, S.E.; Boyle, E.; Lyons, C.; Gray, S.J. Engineered microRNA-based regulatory element permits safe high-dose miniMECP2 gene therapy in Rett mice. Brain 2021, 144, 3005–3019. [Google Scholar] [CrossRef]
- Endmann, A.; Baden, M.; Weisermann, E.; Kapp, K.; Schroff, M.; Kleuss, C.; Wittig, B.; Juhls, C. Immune response induced by a linear DNA vector: Influence of dose, formulation and route of injection. Vaccine 2010, 28, 3642–3649. [Google Scholar] [CrossRef]
- Mullagulova, A.; Shaimardanova, A.; Solovyeva, V.; Mukhamedshina, Y.; Chulpanova, D.; Kostennikov, A.; Issa, S.; Rizvanov, A. Safety and Efficacy of Intravenous and Intrathecal Delivery of AAV9-Mediated ARSA in Minipigs. Int. J. Mol. Sci. 2023, 24, 9204. [Google Scholar] [CrossRef]
- Shah, V.K.; Firmal, P.; Alam, A.; Ganguly, D.; Chattopadhyay, S. Overview of Immune Response During SARS-CoV-2 Infection: Lessons From the Past. Front. Immunol. 2020, 11, 1949. [Google Scholar] [CrossRef]
- Sbardella, E.; Tomassini, V.; Gasperini, C.; Bellomi, F.; Cefaro, L.A.; Morra, V.B.; Antonelli, G.; Pozzilli, C. Neutralizing antibodies explain the poor clinical response to interferon beta in a small proportion of patients with multiple sclerosis: A retrospective study. BMC Neurol. 2009, 9, 54. [Google Scholar] [CrossRef]
- Gorovits, B.; Azadeh, M.; Buchlis, G.; Fiscella, M.; Harrison, T.; Havert, M.; Janetzki, S.; Jawa, V.; Long, B.; Mahnke, Y.D.; et al. Evaluation of Cellular Immune Response to Adeno-Associated Virus-Based Gene Therapy. AAPS J. 2023, 25, 47. [Google Scholar] [CrossRef]
- Monsey, L.; Best, L.G.; Zhu, J.; DeCroo, S.; Anderson, M.Z. The association of mannose binding lectin genotype and immune response to Chlamydia pneumoniae: The Strong Heart Study. PLoS ONE 2019, 14, e0210640. [Google Scholar] [CrossRef] [PubMed]
- Kubistova, Z.; Mrazek, F.; Petrek, M. Polymorphisms of the immune response genes: Selected biological, methodical and medical aspects. Pap. Med. Fac. Palacky Univ. Olomouc 2009, 153, 93–102. [Google Scholar]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef]
- Arjomandnejad, M.; Dasgupta, I.; Flotte, T.R.; Keeler, A.M. Immunogenicity of Recombinant Adeno-Associated Virus (AAV) Vectors for Gene Transfer. BioDrugs 2023, 37, 311–329. [Google Scholar] [CrossRef] [PubMed]
- Calcedo, R.; Vandenberghe, L.H.; Gao, G.; Lin, J.; Wilson, J.M. Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J. Infect. Dis. 2009, 199, 381–390. [Google Scholar] [CrossRef]
- Greig, J.A.; Calcedo, R.; Grant, R.L.; Peng, H.; Medina-Jaszek, C.A.; Ahonkhai, O.; Qin, Q.; Roy, S.; Tretiakova, A.P.; Wilson, J.M. Intramuscular administration of AAV overcomes pre-existing neutralizing antibodies in rhesus macaques. Vaccine 2016, 34, 6323–6329. [Google Scholar] [CrossRef]
- Boutin, S.; Monteilhet, V.; Veron, P.; Leborgne, C.; Benveniste, O.; Montus, M.F.; Masurier, C. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: Implications for gene therapy using AAV vectors. Hum. Gene Ther. 2010, 21, 704–712. [Google Scholar] [CrossRef]
- Chhabra, A.; Bashirians, G.; Petropoulos, C.J.; Wrin, T.; Paliwal, Y.; Henstock, P.V.; Somanathan, S.; da Fonseca Pereira, C.; Winburn, I.; Rasko, J.E.J. Global seroprevalence of neutralizing antibodies against adeno-associated virus serotypes used for human gene therapies. Mol. Ther. Methods Clin. Dev. 2024, 32, 101273. [Google Scholar] [CrossRef]
- Wang, M.; Crosby, A.; Hastie, E.; Samulski, J.J.; McPhee, S.; Joshua, G.; Samulski, R.J.; Li, C. Prediction of adeno-associated virus neutralizing antibody activity for clinical application. Gene Ther. 2015, 22, 984–992. [Google Scholar] [CrossRef]
- Day, J.W.; Finkel, R.S.; Mercuri, E.; Swoboda, K.J.; Menier, M.; van Olden, R.; Tauscher-Wisniewski, S.; Mendell, J.R. Adeno-associated virus serotype 9 antibodies in patients screened for treatment with onasemnogene abeparvovec. Mol. Ther. Methods Clin. Dev. 2021, 21, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Calcedo, R.; Wang, H.; Bell, P.; Grant, R.; Vandenberghe, L.H.; Sanmiguel, J.; Morizono, H.; Batshaw, M.L.; Wilson, J.M. The pleiotropic effects of natural AAV infections on liver-directed gene transfer in macaques. Mol. Ther. 2010, 18, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, B.; Butler, J.; Felker, G.M.; Ponikowski, P.; Voors, A.A.; Pogoda, J.M.; Provost, R.; Guerrero, J.; Hajjar, R.J.; Zsebo, K.M. Prevalence of AAV1 neutralizing antibodies and consequences for a clinical trial of gene transfer for advanced heart failure. Gene Ther. 2016, 23, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Kruzik, A.; Fetahagic, D.; Hartlieb, B.; Dorn, S.; Koppensteiner, H.; Horling, F.M.; Scheiflinger, F.; Reipert, B.M.; de la Rosa, M. Prevalence of Anti-Adeno-Associated Virus Immune Responses in International Cohorts of Healthy Donors. Mol. Ther. Methods Clin. Dev. 2019, 14, 126–133. [Google Scholar] [CrossRef]
- Manno, C.S.; Pierce, G.F.; Arruda, V.R.; Glader, B.; Ragni, M.; Rasko, J.J.; Ozelo, M.C.; Hoots, K.; Blatt, P.; Konkle, B.; et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 2006, 12, 342–347. [Google Scholar] [CrossRef]
- Tse, L.V.; Klinc, K.A.; Madigan, V.J.; Castellanos Rivera, R.M.; Wells, L.F.; Havlik, L.P.; Smith, J.K.; Agbandje-McKenna, M.; Asokan, A. Structure-guided evolution of antigenically distinct adeno-associated virus variants for immune evasion. Proc. Natl. Acad. Sci. USA 2017, 114, E4812–E4821. [Google Scholar] [CrossRef]
- Tse, L.V.; Moller-Tank, S.; Asokan, A. Strategies to circumvent humoral immunity to adeno-associated viral vectors. Expert. Opin. Biol. Ther. 2015, 15, 845–855. [Google Scholar] [CrossRef]
- Tseng, Y.S.; Agbandje-McKenna, M. Mapping the AAV Capsid Host Antibody Response toward the Development of Second Generation Gene Delivery Vectors. Front. Immunol. 2014, 5, 9. [Google Scholar] [CrossRef]
- Scallan, C.D.; Jiang, H.; Liu, T.; Patarroyo-White, S.; Sommer, J.M.; Zhou, S.; Couto, L.B.; Pierce, G.F. Human immunoglobulin inhibits liver transduction by AAV vectors at low AAV2 neutralizing titers in SCID mice. Blood 2006, 107, 1810–1817. [Google Scholar] [CrossRef]
- George, L.A.; Sullivan, S.K.; Giermasz, A.; Rasko, J.E.J.; Samelson-Jones, B.J.; Ducore, J.; Cuker, A.; Sullivan, L.M.; Majumdar, S.; Teitel, J.; et al. Hemophilia B Gene Therapy with a High-Specific-Activity Factor IX Variant. N. Engl. J. Med. 2017, 377, 2215–2227. [Google Scholar] [CrossRef]
- Meadows, A.S.; Pineda, R.J.; Goodchild, L.; Bobo, T.A.; Fu, H. Threshold for Pre-existing Antibody Levels Limiting Transduction Efficiency of Systemic rAAV9 Gene Delivery: Relevance for Translation. Mol. Ther. Methods Clin. Dev. 2019, 13, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Nathwani, A.C.; Tuddenham, E.G.; Rangarajan, S.; Rosales, C.; McIntosh, J.; Linch, D.C.; Chowdary, P.; Riddell, A.; Pie, A.J.; Harrington, C.; et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N. Engl. J. Med. 2011, 365, 2357–2365. [Google Scholar] [CrossRef]
- Zaiss, A.K.; Cotter, M.J.; White, L.R.; Clark, S.A.; Wong, N.C.; Holers, V.M.; Bartlett, J.S.; Muruve, D.A. Complement is an essential component of the immune response to adeno-associated virus vectors. J. Virol. 2008, 82, 2727–2740. [Google Scholar] [CrossRef]
- Muhuri, M.; Maeda, Y.; Ma, H.; Ram, S.; Fitzgerald, K.A.; Tai, P.W.; Gao, G. Overcoming innate immune barriers that impede AAV gene therapy vectors. J. Clin. Investig. 2021, 131, e143780. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, Z.; Leborgne, C.; Barbon, E.; Masat, E.; Ronzitti, G.; van Wittenberghe, L.; Vignaud, A.; Collaud, F.; Charles, S.; Simon Sola, M.; et al. Influence of Pre-existing Anti-capsid Neutralizing and Binding Antibodies on AAV Vector Transduction. Mol. Ther. Methods Clin. Dev. 2018, 9, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Palaschak, B.; Marsic, D.; Herzog, R.W.; Zolotukhin, S.; Markusic, D.M. An Immune-Competent Murine Model to Study Elimination of AAV-Transduced Hepatocytes by Capsid-Specific CD8+ T Cells. Mol. Ther. Methods Clin. Dev. 2017, 5, 142–152. [Google Scholar] [CrossRef]
- Hosel, M.; Broxtermann, M.; Janicki, H.; Esser, K.; Arzberger, S.; Hartmann, P.; Gillen, S.; Kleeff, J.; Stabenow, D.; Odenthal, M.; et al. Toll-like receptor 2-mediated innate immune response in human nonparenchymal liver cells toward adeno-associated viral vectors. Hepatology 2012, 55, 287–297. [Google Scholar] [CrossRef]
- Martino, A.T.; Suzuki, M.; Markusic, D.M.; Zolotukhin, I.; Ryals, R.C.; Moghimi, B.; Ertl, H.C.; Muruve, D.A.; Lee, B.; Herzog, R.W. The genome of self-complementary adeno-associated viral vectors increases Toll-like receptor 9-dependent innate immune responses in the liver. Blood 2011, 117, 6459–6468. [Google Scholar] [CrossRef]
- Pipe, S.; Leebeek, F.W.G.; Ferreira, V.; Sawyer, E.K.; Pasi, J. Clinical Considerations for Capsid Choice in the Development of Liver-Targeted AAV-Based Gene Transfer. Mol. Ther. Methods Clin. Dev. 2019, 15, 170–178. [Google Scholar] [CrossRef]
- Costa Verdera, H.; Kuranda, K.; Mingozzi, F. AAV Vector Immunogenicity in Humans: A Long Journey to Successful Gene Transfer. Mol. Ther. 2020, 28, 723–746. [Google Scholar] [CrossRef]
- Rogers, G.L.; Martino, A.T.; Aslanidi, G.V.; Jayandharan, G.R.; Srivastava, A.; Herzog, R.W. Innate Immune Responses to AAV Vectors. Front. Microbiol. 2011, 2, 194. [Google Scholar] [CrossRef]
- Dhungel, B.P.; Winburn, I.; Pereira, C.D.F.; Huang, K.; Chhabra, A.; Rasko, J.E.J. Understanding AAV vector immunogenicity: From particle to patient. Theranostics 2024, 14, 1260–1288. [Google Scholar] [CrossRef] [PubMed]
- Zaiss, A.K.; Liu, Q.; Bowen, G.P.; Wong, N.C.; Bartlett, J.S.; Muruve, D.A. Differential activation of innate immune responses by adenovirus and adeno-associated virus vectors. J. Virol. 2002, 76, 4580–4590. [Google Scholar] [CrossRef]
- Kotterman, M.A.; Yin, L.; Strazzeri, J.M.; Flannery, J.G.; Merigan, W.H.; Schaffer, D.V. Antibody neutralization poses a barrier to intravitreal adeno-associated viral vector gene delivery to non-human primates. Gene Ther. 2015, 22, 116–126. [Google Scholar] [CrossRef]
- Mullard, A. Gene therapy community grapples with toxicity issues, as pipeline matures. Nat. Rev. Drug Discov. 2021, 20, 804–805. [Google Scholar] [CrossRef] [PubMed]
- Day, J.W.; Mendell, J.R.; Mercuri, E.; Finkel, R.S.; Strauss, K.A.; Kleyn, A.; Tauscher-Wisniewski, S.; Tukov, F.F.; Reyna, S.P.; Chand, D.H. Clinical Trial and Postmarketing Safety of Onasemnogene Abeparvovec Therapy. Drug Saf. 2021, 44, 1109–1119. [Google Scholar] [CrossRef] [PubMed]
- Guillou, J.; de Pellegars, A.; Porcheret, F.; Fremeaux-Bacchi, V.; Allain-Launay, E.; Debord, C.; Denis, M.; Pereon, Y.; Barnerias, C.; Desguerre, I.; et al. Fatal thrombotic microangiopathy case following adeno-associated viral SMN gene therapy. Blood Adv. 2022, 6, 4266–4270. [Google Scholar] [CrossRef]
- Wilson, J.M.; Flotte, T.R. Moving Forward After Two Deaths in a Gene Therapy Trial of Myotubular Myopathy. Hum. Gene Ther. 2020, 31, 695–696. [Google Scholar] [CrossRef]
- Kashiwakura, Y.; Baatartsogt, N.; Yamazaki, S.; Nagao, A.; Amano, K.; Suzuki, N.; Matsushita, T.; Sawada, A.; Higasa, S.; Yamasaki, N.; et al. The seroprevalence of neutralizing antibodies against the adeno-associated virus capsids in Japanese hemophiliacs. Mol. Ther. Methods Clin. Dev. 2022, 27, 404–414. [Google Scholar] [CrossRef]
- Pichard, V.; Guilbaud, M.; Devaux, M.; Jaulin, N.; Journou, M.; Cospolite, M.; Garcia, A.; Ferry, N.; Michalak-provost, S.; Gernoux, G.; et al. Incomplete elimination of viral genomes is associated with chronic inflammation in nonhuman primate livers after AAV-mediated gene transfer. Gene Ther. 2025, 32, 287–298. [Google Scholar] [CrossRef]
- Maturana, C.J.; Verpeut, J.L.; Kooshkbaghi, M.; Engel, E.A. Novel tool to quantify with single-cell resolution the number of incoming AAV genomes co-expressed in the mouse nervous system. Gene Ther. 2023, 30, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Bower, J.J.; Song, L.; Bastola, P.; Hirsch, M.L. Harnessing the Natural Biology of Adeno-Associated Virus to Enhance the Efficacy of Cancer Gene Therapy. Viruses 2021, 13, 1205. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ling, S.; Wang, D.; Wang, X.; Hao, F.; Yin, L.; Yuan, Z.; Liu, L.; Zhang, L.; Li, Y.; et al. Modified lentiviral globin gene therapy for pediatric β0/β0 transfusion-dependent β-thalassemia: A single-center, single-arm pilot trial. Cell Stem Cell 2024, 31, 961–973.e8. [Google Scholar] [CrossRef]
- Kumaresan, P.R.; Wurster, S.; Bavisi, K.; da Silva, T.A.; Hauser, P.; Kinnitt, J.; Albert, N.D.; Bharadwaj, U.; Neelapu, S.; Kontoyiannis, D.P. A novel lentiviral vector-based approach to generate chimeric antigen receptor T cells targeting Aspergillus fumigatus. mBio 2024, 15, e0341323. [Google Scholar] [CrossRef]
- Tada, T.; Zhou, H.; Dcosta, B.M.; Samanovic, M.I.; Chivukula, V.; Herati, R.S.; Hubbard, S.R.; Mulligan, M.J.; Landau, N.R. Increased resistance of SARS-CoV-2 Omicron variant to neutralization by vaccine-elicited and therapeutic antibodies. EBioMedicine 2022, 78, 103944. [Google Scholar] [CrossRef]
- Toon, K.; Bentley, E.M.; Mattiuzzo, G. More Than Just Gene Therapy Vectors: Lentiviral Vector Pseudotypes for Serological Investigation. Viruses 2021, 13, 217. [Google Scholar] [CrossRef] [PubMed]
- Suleman, S.; Payne, A.; Bowden, J.; Haque, S.A.; Zahn, M.; Fawaz, S.; Khalifa, M.S.; Jobling, S.; Hay, D.; Franco, M.; et al. HIV-1 lentivirus tethering to the genome is associated with transcription factor binding sites found in genes that favour virus survival. Gene Ther. 2022, 29, 720–729. [Google Scholar] [CrossRef]
- Ahmad, F.; Hyvärinen, A.; Pirinen, A.; Olsson, V.; Rummukainen, J.; Immonen, A.; Närväinen, J.; Tuunanen, P.; Liimatainen, T.; Kärkkäinen, V.; et al. Lentivirus vector-mediated genetic manipulation of oncogenic pathways induces tumor formation in rabbit brain. Mol. Med. Rep. 2021, 23, 422. [Google Scholar] [CrossRef]
- Migliavacca, M.; Barzaghi, F.; Fossati, C.; Rancoita, P.M.V.; Gabaldo, M.; Dionisio, F.; Giannelli, S.; Salerio, F.A.; Ferrua, F.; Tucci, F.; et al. Long-term and real-world safety and efficacy of retroviral gene therapy for adenosine deaminase deficiency. Nat. Med. 2024, 30, 488–497. [Google Scholar] [CrossRef]
- Lu, X.; Vano, Y.A.; Su, X.; Verkarre, V.; Sun, C.M.; Cheng, W.; Xu, L.; Yan, F.; Kotti, S.; Fridman, W.H.; et al. Stratification system with dual human endogenous retroviruses for predicting immunotherapy efficacy in metastatic clear-cell renal cell carcinoma. J. Immunother. Cancer 2025, 13, e010386. [Google Scholar] [CrossRef]
- Uchiyama, T.; Kawai, T.; Nakabayashi, K.; Nakazawa, Y.; Goto, F.; Okamura, K.; Nishimura, T.; Kato, K.; Watanabe, N.; Miura, A.; et al. Myelodysplasia after clonal hematopoiesis with APOBEC3-mediated CYBB inactivation in retroviral gene therapy for X-CGD. Mol. Ther. J. Am. Soc. Gene Ther. 2023, 31, 3424–3440. [Google Scholar] [CrossRef]
- Osega, C.E.; Bustos, F.J.; Arriagada, G. From Entry to the Nucleus: How Retroviruses Commute. Annu. Rev. Virol. 2024, 11, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Jarosz, A.S.; Halo, J.V. Transcription of Endogenous Retroviruses: Broad and Precise Mechanisms of Control. Viruses 2024, 16, 1312. [Google Scholar] [CrossRef] [PubMed]
- Rappaport, A.R.; Kyi, C.; Lane, M.; Hart, M.G.; Johnson, M.L.; Henick, B.S.; Liao, C.Y.; Mahipal, A.; Shergill, A.; Spira, A.I.; et al. A shared neoantigen vaccine combined with immune checkpoint blockade for advanced metastatic solid tumors: Phase 1 trial interim results. Nat. Med. 2024, 30, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Sheetikov, S.A.; Khmelevskaya, A.A.; Zornikova, K.V.; Zvyagin, I.V.; Shomuradova, A.S.; Serdyuk, Y.V.; Shakirova, N.T.; Peshkova, I.O.; Titov, A.; Romaniuk, D.S.; et al. Clonal structure and the specificity of vaccine-induced T cell response to SARS-CoV-2 Spike protein. Front. Immunol. 2024, 15, 1369436. [Google Scholar] [CrossRef]
- Wan, B.; Qin, L.; Ma, W.; Wang, H. Construction and immune effect of an HPV16/18/58 trivalent therapeutic adenovirus vector vaccine. Infect. Agents Cancer 2022, 17, 5. [Google Scholar] [CrossRef]
- Ono, R.; Nishimae, F.; Wakida, T.; Sakurai, F.; Mizuguchi, H. Effects of pre-existing anti-adenovirus antibodies on transgene expression levels and therapeutic efficacies of arming oncolytic adenovirus. Sci. Rep. 2022, 12, 21560. [Google Scholar] [CrossRef]
- Park, J.; Lindesmith, L.C.; Olia, A.S.; Costantini, V.P.; Brewer-Jensen, P.D.; Mallory, M.L.; Kelley, C.E.; Satterwhite, E.; Longo, V.; Tsybovsky, Y.; et al. Broadly neutralizing antibodies targeting pandemic GII.4 variants or seven GII genotypes of human norovirus. Sci. Transl. Med. 2025, 17, eads8214. [Google Scholar] [CrossRef]
- Su, D.; Han, L.; Shi, C.; Li, Y.; Qian, S.; Feng, Z.; Yu, L. An updated review of HSV-1 infection-associated diseases and treatment, vaccine development, and vector therapy application. Virulence 2024, 15, 2425744. [Google Scholar] [CrossRef]
- Silke Heilingloh, C.; Lull, C.; Kleiser, E.; Alt, M.; Schipper, L.; Witzke, O.; Trilling, M.; Eis-Hübinger, A.M.; Dittmer, U.; Krawczyk, A. Herpes Simplex Virus Type 2 Is More Difficult to Neutralize by Antibodies Than Herpes Simplex Virus Type 1. Vaccines 2020, 8, 478. [Google Scholar] [CrossRef]
- Kamel, M.S.; Munds, R.A.; Verma, M.S. The Quest for Immunity: Exploring Human Herpesviruses as Vaccine Vectors. Int. J. Mol. Sci. 2023, 24, 16112. [Google Scholar] [CrossRef]
- Epstein, A.L.; Rabkin, S.D. Safety of non-replicative and oncolytic replication-selective HSV vectors. Trends Mol. Med. 2024, 30, 781–794. [Google Scholar] [CrossRef] [PubMed]
- Elmore, Z.C.; Oh, D.K.; Simon, K.E.; Fanous, M.M.; Asokan, A. Rescuing AAV gene transfer from neutralizing antibodies with an IgG-degrading enzyme. JCI Insight 2020, 5, e139881. [Google Scholar] [CrossRef] [PubMed]
- Mingozzi, F.; High, K.A. Overcoming the Host Immune Response to Adeno-Associated Virus Gene Delivery Vectors: The Race Between Clearance, Tolerance, Neutralization, and Escape. Annu. Rev. Virol. 2017, 4, 511–534. [Google Scholar] [CrossRef]
- Earley, J.; Piletska, E.; Ronzitti, G.; Piletsky, S. Evading and overcoming AAV neutralization in gene therapy. Trends Biotechnol. 2023, 41, 836–845. [Google Scholar] [CrossRef]
- Flotte, T.R.E.-i.-C. Revisiting the “New” Inflammatory Toxicities of Adeno-Associated Virus Vectors. Hum. Gene Ther. 2020, 31, 398–399. [Google Scholar] [CrossRef] [PubMed]
- Hinderer, C.; Katz, N.; Buza, E.L.; Dyer, C.; Goode, T.; Bell, P.; Richman, L.K.; Wilson, J.M. Severe Toxicity in Nonhuman Primates and Piglets Following High-Dose Intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Hum. Gene Ther. 2018, 29, 285–298. [Google Scholar] [CrossRef]
- Makela, A.R.; Ernst, W.; Grabherr, R.; Oker-Blom, C. Baculovirus-based display and gene delivery systems. Cold Spring Harb. Protoc. 2010, 2010, pdb.top72. [Google Scholar] [CrossRef]
- Shin, H.Y.; Choi, H.; Kim, N.; Park, N.; Kim, H.; Kim, J.; Kim, Y.B. Unraveling the Genome-Wide Impact of Recombinant Baculovirus Infection in Mammalian Cells for Gene Delivery. Genes 2020, 11, 1306. [Google Scholar] [CrossRef]
- Zheng, H.; Pan, Y.; Wang, X.; Tian, W.; Yao, L.; Sun, J. Impact of Molecular Modification on the Efficiency of Recombinant Baculovirus Vector Invasion to Mammalian Cells and Its Immunogenicity in Mice. Viruses 2022, 14, 140. [Google Scholar] [CrossRef]
- Honda, Y.; Nagao, S.; Kinoh, H.; Liu, X.; Matsudaira, N.; Dirisala, A.; Nitta-Matsutomo, S.; Nomoto, T.; Hayashita-Kinoh, H.; Miura, Y.; et al. Adeno-Associated Virus Self-Assembled with Tannic Acid and Phenylboronic Acid Polymers to Evade Neutralizing Antibodies and Reduce Adverse Events. ACS Nano 2025, 19, 7690–7706. [Google Scholar] [CrossRef]
- Lisowski, L.; Dane, A.P.; Chu, K.; Zhang, Y.; Cunningham, S.C.; Wilson, E.M.; Nygaard, S.; Grompe, M.; Alexander, I.E.; Kay, M.A. Selection and evaluation of clinically relevant AAV variants in a xenograft liver model. Nature 2014, 506, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Vrellaku, B.; Sethw Hassan, I.; Howitt, R.; Webster, C.P.; Harriss, E.; McBlane, F.; Betts, C.; Schettini, J.; Lion, M.; Mindur, J.E.; et al. A systematic review of immunosuppressive protocols used in AAV gene therapy for monogenic disorders. Mol. Ther. J. Am. Soc. Gene Ther. 2024, 32, 3220–3259. [Google Scholar] [CrossRef] [PubMed]
- Flotte, T.R.; Cataltepe, O.; Puri, A.; Batista, A.R.; Moser, R.; McKenna-Yasek, D.; Douthwright, C.; Gernoux, G.; Blackwood, M.; Mueller, C.; et al. AAV gene therapy for Tay-Sachs disease. Nat. Med. 2022, 28, 251–259. [Google Scholar] [CrossRef]
- Kenison, J.E.; Stevens, N.A.; Quintana, F.J. Therapeutic induction of antigen-specific immune tolerance. Nat. Rev. Immunol. 2024, 24, 338–357. [Google Scholar] [CrossRef] [PubMed]
- Sennikov, S.; Kulikova, E.; Knauer, N.Y.; Khantakova, Y.N.J.M.I. Molecular and cellular mechanisms mediated by dendritic cells involved in the induction of tolerance. Med. Immunol. 2017, 19, 359–374. [Google Scholar] [CrossRef]
- Servellita, V.; Sotomayor Gonzalez, A.; Lamson, D.M.; Foresythe, A.; Huh, H.J.; Bazinet, A.L.; Bergman, N.H.; Bull, R.L.; Garcia, K.Y.; Goodrich, J.S.; et al. Adeno-associated virus type 2 in US children with acute severe hepatitis. Nature 2023, 617, 574–580. [Google Scholar] [CrossRef]
- Khan, N.; Maurya, S.; Bammidi, S.; Jayandharan, G.R. AAV6 Vexosomes Mediate Robust Suicide Gene Delivery in a Murine Model of Hepatocellular Carcinoma. Mol. Ther. Methods Clin. Dev. 2020, 17, 497–504. [Google Scholar] [CrossRef]
- Liu, B.; Li, Z.; Huang, S.; Yan, B.; He, S.; Chen, F.; Liang, Y. AAV-Containing Exosomes as a Novel Vector for Improved Gene Delivery to Lung Cancer Cells. Front. Cell Dev. Biol. 2021, 9, 707607. [Google Scholar] [CrossRef]
- György, B.; Fitzpatrick, Z.; Crommentuijn, M.H.; Mu, D.; Maguire, C.A. Naturally enveloped AAV vectors for shielding neutralizing antibodies and robust gene delivery in vivo. Biomaterials 2014, 35, 7598–7609. [Google Scholar] [CrossRef]
- Meliani, A.; Boisgerault, F.; Fitzpatrick, Z.; Marmier, S.; Leborgne, C.; Collaud, F.; Simon Sola, M.; Charles, S.; Ronzitti, G.; Vignaud, A.; et al. Enhanced liver gene transfer and evasion of preexisting humoral immunity with exosome-enveloped AAV vectors. Blood Adv. 2017, 1, 2019–2031. [Google Scholar] [CrossRef] [PubMed]
- Adamiak, M.; Liang, Y.; Sherman, C.; Lodha, S.; Kohlbrenner, E.; Jeong, D.; Ceholski, D.K.; Dogra, N.; Dubois, N.; Hajjar, R.J.; et al. Abstract MP165: Exosome-mediated Encapsulation Alters AAV Antigenicity and Infectivity: Implications for Gene Delivery in the Heart. Circ. Res. 2020, 127 (Suppl. S1), AMP165. [Google Scholar] [CrossRef]
- Bobo, T.A.; Samowitz, P.N.; Robinson, M.I.; Montes, L.I.; Forsberg, L.J.; Feng, R.; Nicely, N.I.; Fu, H. IgG-cleavage protein allows therapeutic AAV gene delivery in passively immunized MPS IIIA mice. Gene Ther. 2023, 30, 377–385. [Google Scholar] [CrossRef]
- Jefferis, R. Isotype and glycoform selection for antibody therapeutics. Arch. Biochem. Biophys. 2012, 526, 159–166. [Google Scholar] [CrossRef]
- Frick, I.M.; Happonen, L.; Wrighton, S.; Nordenfelt, P.; Björck, L. IdeS, a secreted proteinase of Streptococcus pyogenes, is bound to a nuclease at the bacterial surface where it inactivates opsonizing IgG antibodies. J. Biol. Chem. 2023, 299, 105345. [Google Scholar] [CrossRef]
- Winstedt, L.; Järnum, S.; Nordahl, E.A.; Olsson, A.; Runström, A.; Bockermann, R.; Karlsson, C.; Malmström, J.; Palmgren, G.S.; Malmqvist, U.; et al. Complete Removal of Extracellular IgG Antibodies in a Randomized Dose-Escalation Phase I Study with the Bacterial Enzyme IdeS--A Novel Therapeutic Opportunity. PLoS ONE 2015, 10, e0132011. [Google Scholar] [CrossRef]
- Smith, T.J.; Elmore, Z.C.; Fusco, R.M.; Hull, J.A.; Rosales, A.; Martinez, M.; Tarantal, A.F.; Asokan, A. Engineered IgM and IgG cleaving enzymes for mitigating antibody neutralization and complement activation in AAV gene transfer. Mol. Ther. J. Am. Soc. Gene Ther. 2024, 32, 2080–2093. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H. Host and microbial factors in regulation of T cells in the intestine. Front. Immunol. 2013, 4, 141. [Google Scholar] [CrossRef]
- Davis-Gardner, M.E.; Weber, J.A.; Xie, J.; Pekrun, K.; Alexander, E.A.; Weisgrau, K.L.; Furlott, J.R.; Rakasz, E.G.; Kay, M.A.; Gao, G.; et al. A strategy for high antibody expression with low anti-drug antibodies using AAV9 vectors. Front. Immunol. 2023, 14, 1105617. [Google Scholar] [CrossRef]
- Martinez-Alcantar, L.; Talavera-Carrillo, D.K.; Pineda-Salazar, J.U.; Avalos-Viveros, M.; Gutierrez-Ospina, G.; Phillips-Farfan, B.V.; Fuentes-Farias, A.L.; Melendez-Herrera, E. Anterior chamber associated immune deviation to cytosolic neural antigens avoids self-reactivity after optic nerve injury and polarizes the retinal environment to an anti-inflammatory profile. J. Neuroimmunol. 2019, 333, 476964. [Google Scholar] [CrossRef]
- Kharisova, C.B.; Kitaeva, K.V.; Solovyeva, V.V.; Sufianov, A.A.; Sufianova, G.Z.; Akhmetshin, R.F.; Bulgar, S.N.; Rizvanov, A.A. Looking to the Future of Viral Vectors in Ocular Gene Therapy: Clinical Review. Biomedicines 2025, 13, 365. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Fisson, S.; Dalkara, D.; Ail, D. Immune Responses to Gene Editing by Viral and Non-Viral Delivery Vectors Used in Retinal Gene Therapy. Pharmaceutics 2022, 14, 1973. [Google Scholar] [CrossRef] [PubMed]
- Baatartsogt, N.; Kashiwakura, Y.; Hayakawa, M.; Kamoshita, N.; Hiramoto, T.; Mizukami, H.; Ohmori, T. A sensitive and reproducible cell-based assay via secNanoLuc to detect neutralizing antibody against adeno-associated virus vector capsid. Mol. Ther. Methods Clin. Dev. 2021, 22, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Ertl, H.C.J. Mitigating Serious Adverse Events in Gene Therapy with AAV Vectors: Vector Dose and Immunosuppression. Drugs 2023, 83, 287–298. [Google Scholar] [CrossRef]
- Henderson, M.L.; Zieba, J.K.; Li, X.; Campbell, D.B.; Williams, M.R.; Vogt, D.L.; Bupp, C.P.; Edgerly, Y.M.; Rajasekaran, S.; Hartog, N.L.; et al. Gene Therapy for Genetic Syndromes: Understanding the Current State to Guide Future Care. BioTech 2024, 13, 1. [Google Scholar] [CrossRef]
- Au, H.K.E.; Isalan, M.; Mielcarek, M. Gene Therapy Advances: A Meta-Analysis of AAV Usage in Clinical Settings. Front. Med. 2021, 8, 809118. [Google Scholar] [CrossRef]
- George, L.A.; Ragni, M.V.; Rasko, J.E.J.; Raffini, L.J.; Samelson-Jones, B.J.; Ozelo, M.; Hazbon, M.; Runowski, A.R.; Wellman, J.A.; Wachtel, K.; et al. Long-Term Follow-Up of the First in Human Intravascular Delivery of AAV for Gene Transfer: AAV2-hFIX16 for Severe Hemophilia B. Mol. Ther. 2020, 28, 2073–2082. [Google Scholar] [CrossRef]
- Charlesworth, C.T.; Deshpande, P.S.; Dever, D.P.; Camarena, J.; Lemgart, V.T.; Cromer, M.K.; Vakulskas, C.A.; Collingwood, M.A.; Zhang, L.; Bode, N.M.; et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med. 2019, 25, 249–254. [Google Scholar] [CrossRef]
- Kalesnykas, G.; Kokki, E.; Alasaarela, L.; Lesch, H.P.; Tuulos, T.; Kinnunen, K.; Uusitalo, H.; Airenne, K.; Yla-Herttuala, S. Comparative Study of Adeno-associated Virus, Adenovirus, Bacu lovirus and Lentivirus Vectors for Gene Therapy of the Eyes. Curr. Gene Ther. 2017, 17, 235–247. [Google Scholar] [CrossRef]
| Serotype | Global Seroprevalence (%) | Notable Trends/Populations | Geographic Variability |
|---|---|---|---|
| AAV2 | 58–97% (72% average) | Highest overall, increases with age | High across all regions |
| AAV1 | 67% | 2nd highest, co-prevalent with AAV6 | Variable by country |
| AAV5 | 7–35% | Lowest, increases with age | 5.9% (UK) to 51.8% (S. Africa) |
| AAV6 | 20–49% | Intermediate, co-prevalent with AAV1 | Variable |
| AAV8 | 7–46% | Lower in children | Variable |
| AAV9 | 1–56% | Lower in children, higher in adults | Variable |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsaregorodtseva, T.S.; Gubaidullina, A.A.; Kayumova, B.R.; Shaimardanova, A.A.; Issa, S.S.; Solovyeva, V.V.; Sufianov, A.A.; Sufianova, G.Z.; Rizvanov, A.A. Neutralizing Antibodies: Role in Immune Response and Viral Vector Based Gene Therapy. Int. J. Mol. Sci. 2025, 26, 5224. https://doi.org/10.3390/ijms26115224
Tsaregorodtseva TS, Gubaidullina AA, Kayumova BR, Shaimardanova AA, Issa SS, Solovyeva VV, Sufianov AA, Sufianova GZ, Rizvanov AA. Neutralizing Antibodies: Role in Immune Response and Viral Vector Based Gene Therapy. International Journal of Molecular Sciences. 2025; 26(11):5224. https://doi.org/10.3390/ijms26115224
Chicago/Turabian StyleTsaregorodtseva, Tatiana S., Aigul A. Gubaidullina, Beata R. Kayumova, Alisa A. Shaimardanova, Shaza S. Issa, Valeriya V. Solovyeva, Albert A. Sufianov, Galina Z. Sufianova, and Albert A. Rizvanov. 2025. "Neutralizing Antibodies: Role in Immune Response and Viral Vector Based Gene Therapy" International Journal of Molecular Sciences 26, no. 11: 5224. https://doi.org/10.3390/ijms26115224
APA StyleTsaregorodtseva, T. S., Gubaidullina, A. A., Kayumova, B. R., Shaimardanova, A. A., Issa, S. S., Solovyeva, V. V., Sufianov, A. A., Sufianova, G. Z., & Rizvanov, A. A. (2025). Neutralizing Antibodies: Role in Immune Response and Viral Vector Based Gene Therapy. International Journal of Molecular Sciences, 26(11), 5224. https://doi.org/10.3390/ijms26115224