
Identification of Genomic Structural Variations in Xinjiang Brown Cattle by Deep Sequencing and Their Association with Body Conformation Traits

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Genome Data Description
2.2. Detection of Genomic Structural Variations
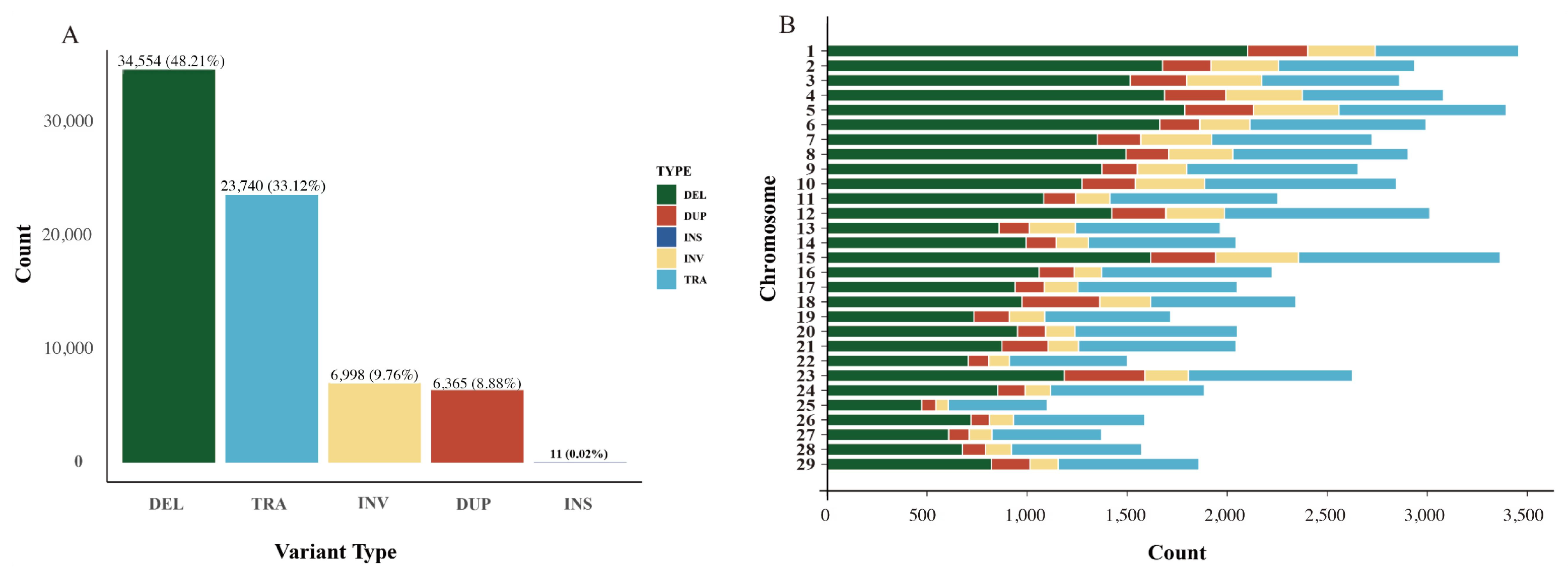
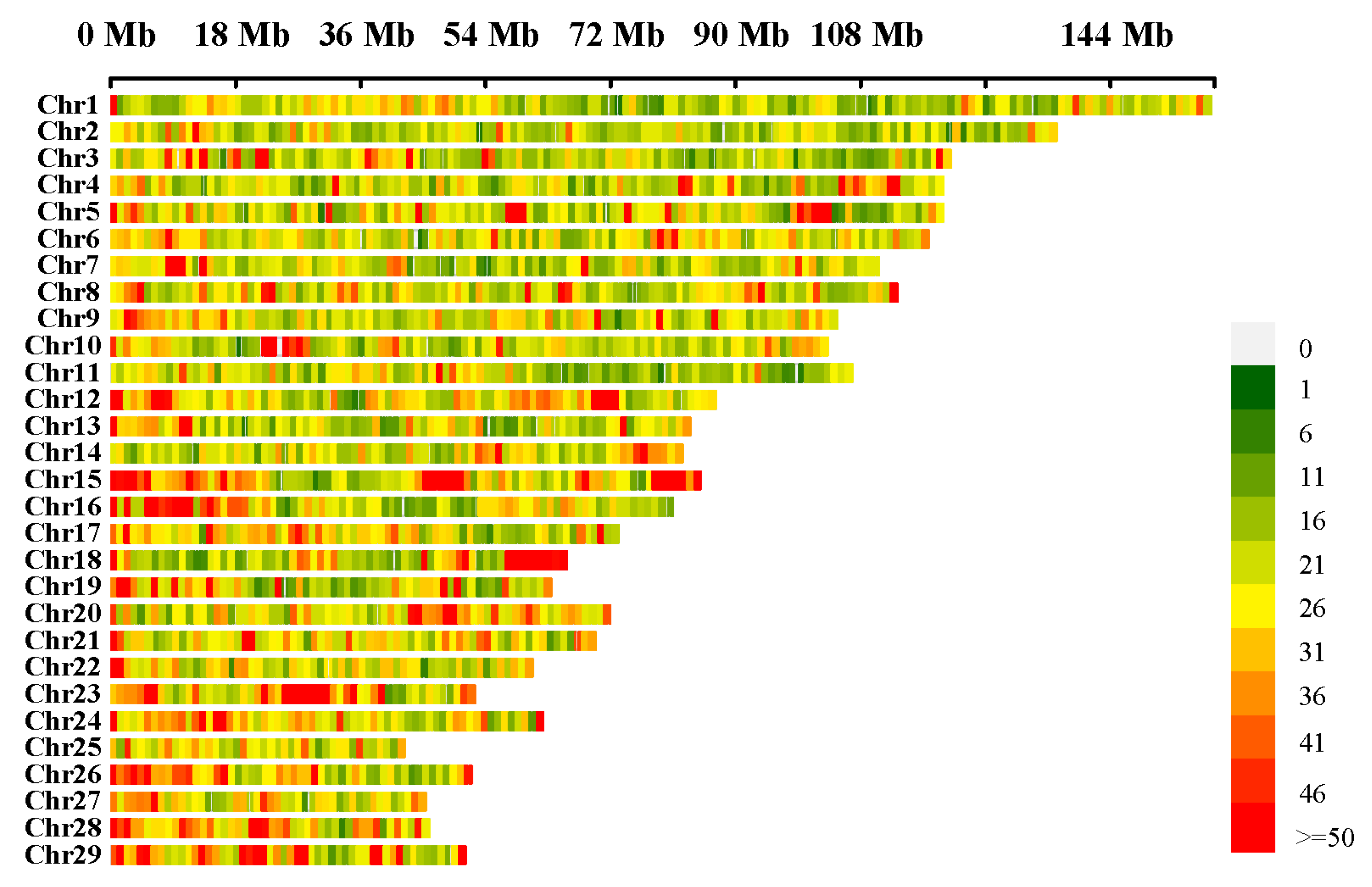
2.3. Characteristics of Structural Variations
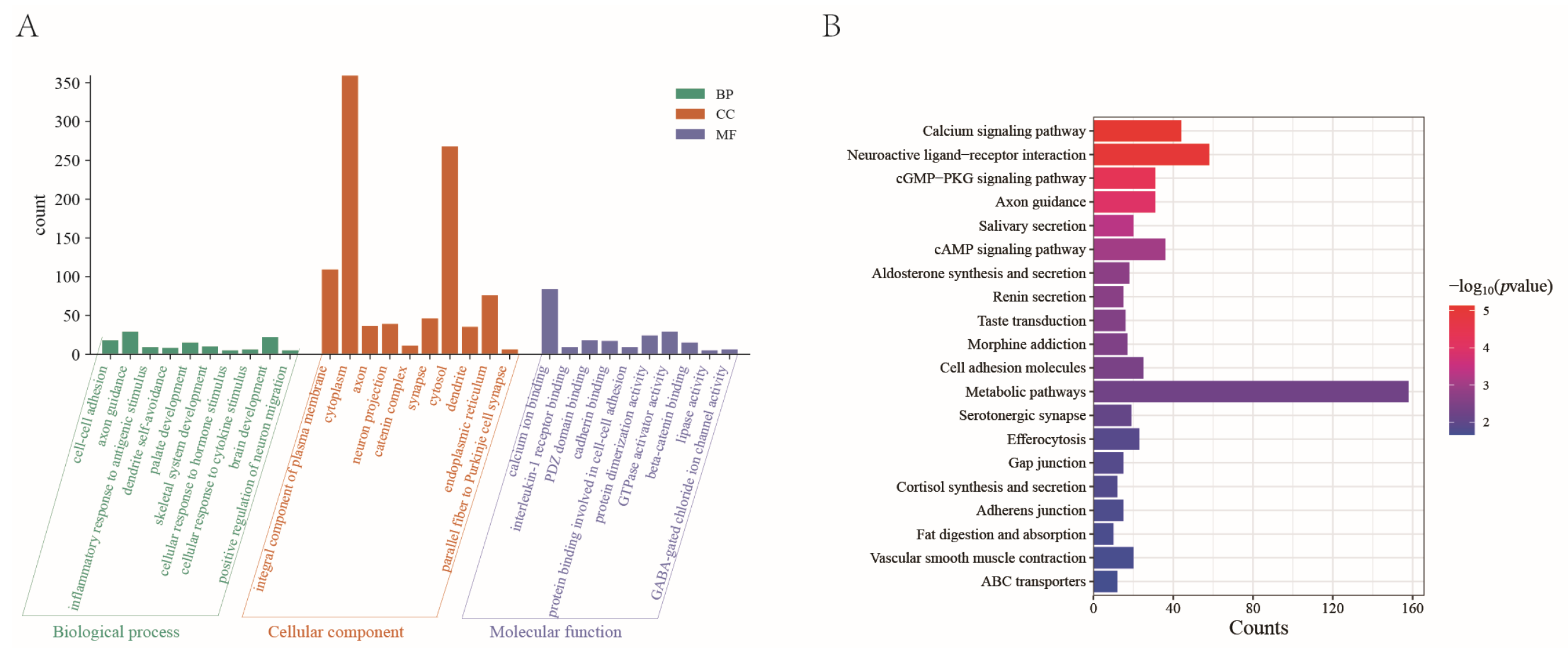
2.4. Annotation and Enrichment Analysis of High-Frequency SVs in Xinjiang Brown Cattle
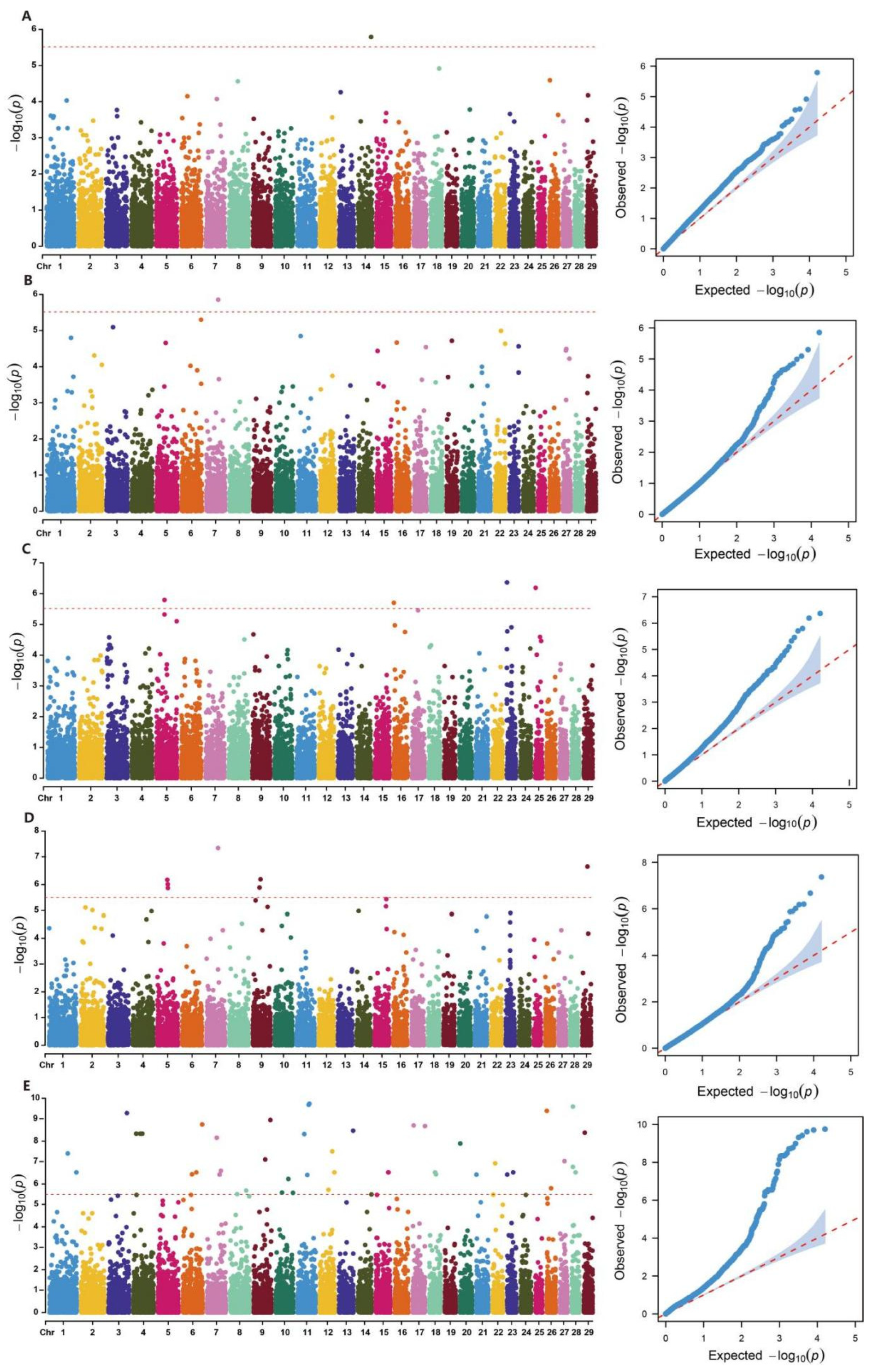
2.5. Genome-Wide Association Analysis of Body Conformation Traits in Xinjiang Brown Cattle
3. Discussion
4. Materials and Methods
4.1. Experimental Materials
4.2. Genomic DNA Extraction
4.3. Genome Sequencing, Quality Control, and Read Alignment
4.4. Structural Variation Detection and Genotyping
4.5. Variant Annotation
4.6. Genome-Wide Association Studies
4.7. Functional Gene Enrichment Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sturtevant, A.H. The Linear Arrangement of Six Sex-Linked Factors in Drosophila, as Shown by Their Mode of Association. J. Exp. Zool. 1913, 14, 43–59. [Google Scholar] [CrossRef]
- Mcclintock, B. The origin and behavior of mutable loci in maize. Proc. Natl. Acad. Sci. USA 1950, 36, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.D.; Celniker, S.E.; Holt, R.A.; Evans, C.A.; Gocayne, J.D.; Amanatides, P.G.; Scherer, S.E.; Li, P.W.; Hoskins, R.A.; Galle, R.F.; et al. The Genome Sequence of Drosophila melanogaster. Science 2000, 287, 2185–2195. [Google Scholar] [CrossRef] [PubMed]
- Feuk, L.; Carson, A.R.; Scherer, S.W. Structural variation in the human genome. Nat. Rev. Genet. 2006, 7, 85–97. [Google Scholar] [CrossRef]
- Abecasis, G.R.; Altshuler, D.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Gibbs, R.A.; Hurles, M.E.; Mcvean, G.A. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [CrossRef]
- Letaief, R.; Rebours, E.; Grohs, C.; Meersseman, C.; Fritz, S.; Trouilh, L.; Esquerre, D.; Barbieri, J.; Klopp, C.; Philippe, R.; et al. Identification of copy number variation in French dairy and beef breeds using next-generation sequencing. Genet. Sel. Evol. 2017, 49, 77. [Google Scholar] [CrossRef]
- Roses, A.D.; Akkari, P.A.; Chiba-Falek, O.; Lutz, M.W.; Gottschalk, W.K.; Saunders, A.M.; Saul, B.; Sundseth, S.; Burns, D. Structural variants can be more informative for disease diagnostics, prognostics and translation than current SNP mapping and exon sequencing. Expert Opin. Drug Met. 2016, 12, 135–147. [Google Scholar] [CrossRef]
- Paudel, Y.; Madsen, O.; Megens, H.; Frantz, L.A.F.; Bosse, M.; Crooijmans, R.P.M.A.; Groenen, M.A.M. Copy number variation in the speciation of pigs: A possible prominent role for olfactory receptors. BMC Genom. 2015, 16, 330. [Google Scholar] [CrossRef]
- Gao, Y.; Jiang, J.; Yang, S.; Hou, Y.; Liu, G.E.; Zhang, S.; Zhang, Q.; Sun, D. CNV discovery for milk composition traits in dairy cattle using whole genome resequencing. BMC Genom. 2017, 18, 265. [Google Scholar] [CrossRef]
- Ottaviani, D.; Lecain, M.; Sheer, D. The role of microhomology in genomic structural variation. Trends Genet. 2014, 30, 85–94. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, K.; Wang, L.; Yang, Y.; Ni, Z.; Xie, X.; Shao, X.; Han, J.; Wan, D.; Qiu, Q. Genome-wide patterns of copy number variation in the Chinese yak genome. BMC Genom. 2016, 17, 379. [Google Scholar] [CrossRef] [PubMed]
- Bickhart, D.M.; Liu, G.E. The challenges and importance of structural variation detection in livestock. Front. Genet. 2014, 5, 37. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, J.M.; Giachetto, P.F.; Da Silva, L.O.; Cintra, L.C.; Paiva, S.R.; Yamagishi, M.E.B.; Caetano, A.R. Genome-wide copy number variation (CNV) detection in Nelore cattle reveals highly frequent variants in genome regions harboring QTLs affecting production traits. BMC Genom. 2016, 17, 454. [Google Scholar] [CrossRef] [PubMed]
- Prinsen, R.T.M.M.; Rossoni, A.; Gredler, B.; Bieber, A.; Bagnato, A.; Strillacci, M.G. A genome wide association study between CNVs and quantitative traits in Brown Swiss cattle. Livest. Sci. 2017, 202, 7–12. [Google Scholar] [CrossRef]
- Xu, L.; Cole, J.B.; Bickhart, D.M.; Hou, Y.; Song, J.; Vanraden, P.M.; Sonstegard, T.S.; Van Tassell, C.P.; Liu, G.E. Genome wide CNV analysis reveals additional variants associated with milk production traits in Holsteins. BMC Genom. 2014, 15, 683. [Google Scholar] [CrossRef]
- Upadhyay, M.; Derks, M.; Andersson, G.; Medugorac, I.; Groenen, M.; Crooijmans, R. Introgression contributes to distribution of structural variations in cattle. Genomics 2021, 113, 3092–3102. [Google Scholar] [CrossRef]
- Bickhart, D.M.; Hou, Y.; Schroeder, S.G.; Alkan, C.; Cardone, M.F.; Matukumalli, L.K.; Song, J.; Schnabel, R.D.; Ventura, M.; Taylor, J.F.; et al. Copy number variation of individual cattle genomes using next-generation sequencing. Genome Res. 2012, 22, 778–790. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, L.; Chen, C.J.; Zhang, M.; Lu, X.; Zhang, Z.; Huang, X.; Shi, Y. Genome-wide association study of milk and reproductive traits in dual-purpose Xinjiang Brown cattle. BMC Genom. 2019, 20, 827. [Google Scholar] [CrossRef]
- Ju, X.; Huang, X.; Zhang, M.; Lan, X.; Wang, D.; Wei, C.; Jiang, H. Effects of eight InDel variants in FHIT on milk traits in Xinjiang brown cattle. Anim. Biotechnol. 2021, 32, 486–494. [Google Scholar] [CrossRef]
- Zhang, M.; Luo, H.; Xu, L.; Shi, Y.; Zhou, J.; Wang, D.; Zhang, X.; Huang, X.; Wang, Y. Genomic Selection for Milk Production Traits in Xinjiang Brown Cattle. Animals 2022, 12, 136. [Google Scholar] [CrossRef]
- Layer, R.M.; Chiang, C.; Quinlan, A.R.; Hall, I.M. LUMPY: A probabilistic framework for structural variant discovery. Genome Biol. 2014, 15, R84. [Google Scholar] [CrossRef] [PubMed]
- Rausch, T.; Zichner, T.; Schlattl, A.; Stutz, A.M.; Benes, V.; Korbel, J.O. DELLY: Structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 2012, 28, i333–i339. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Schulz-Trieglaff, O.; Shaw, R.; Barnes, B.; Schlesinger, F.; Kallberg, M.; Cox, A.J.; Kruglyak, S.; Saunders, C.T. Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2016, 32, 1220–1222. [Google Scholar] [CrossRef] [PubMed]
- De Donato, M.; Peters, S.O.; Mitchell, S.E.; Hussain, T.; Imumorin, I.G. Genotyping-by-sequencing (GBS): A novel, efficient and cost-effective genotyping method for cattle using next-generation sequencing. PLoS ONE 2013, 8, e62137. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, L.; Han, X.; Han, J.; Hu, Y.; Li, F.; Xia, H.; Peng, L.; Boschiero, C.; Rosen, B.D.; et al. Assembly of a pangenome for global cattle reveals missing sequences and novel structural variations, providing new insights into their diversity and evolutionary history. Genome Res. 2022, 32, 1585–1601. [Google Scholar] [CrossRef]
- Renzini, A.; Marroncelli, N.; Noviello, C.; Moresi, V.; Adamo, S. HDAC4 Regulates Skeletal Muscle Regeneration via Soluble Factors. Front. Physiol. 2018, 9, 1387. [Google Scholar] [CrossRef]
- Dai, Y.; Wang, Y.M.; Zhang, W.R.; Liu, X.F.; Li, X.; Ding, X.B.; Guo, H. The role of microRNA-1 and microRNA-206 in the proliferation and differentiation of bovine skeletal muscle satellite cells. In Vitro Cell Dev. Biol. Anim. 2016, 52, 27–34. [Google Scholar] [CrossRef]
- Chen, F.; Chen, H.; Wang, J.; Niu, H.; Lan, X.; Hua, L.; Li, Z.; Lei, C.; Fang, X. MEF2A Gene Polymorphisms are Associated with GrowthTraits in Chinese Indigenous Cattle Breeds. J. Anim. Vet. Adv. 2010, 4, 814–819. [Google Scholar]
- Zhou, Y.; Liu, Y.; Jiang, X.; Du, H.; Li, X.; Zhu, Q. Polymorphism of chicken myocyte-specific enhancer-binding factor 2A gene and its association with chicken carcass traits. Mol. Biol. Rep. 2010, 37, 587–594. [Google Scholar] [CrossRef]
- Chen, Z.; Zhu, M.; Wu, Q.; Lu, H.; Lei, C.; Ahmed, Z.; Sun, J. Analysis of genetic diversity and selection characteristics using the whole genome sequencing data of five buffaloes, including Xilin buffalo, in Guangxi, China. Front. Genet. 2022, 13, 1084824. [Google Scholar] [CrossRef]
- Zhou, H.; Cheng, L.; Azimu, W.; Hodge, S.; Edwards, G.R.; Hickford, J.G.H. Variation in the bovine FABP4 gene affects milk yield and milk protein content in dairy cows. Sci. Rep. 2015, 5, 10023. [Google Scholar] [CrossRef] [PubMed]
- Mehar, S.K.; Thomson, P.C.; Tammen, I.; Raadsma, H.W. Quantitative trait loci mapping in dairy cattle: Review and meta-analysis. Genet. Sel. Evol. 2004, 36, 163. [Google Scholar] [CrossRef]
- Shi, L.; Liu, L.; Ma, Z.; Lv, X.; Li, C.; Xu, L.; Han, B.; Li, Y.; Zhao, F.; Yang, Y.; et al. Identification of genetic associations of ECHS1 gene with milk fatty acid traits in dairy cattle. Anim. Genet. 2019, 50, 430–438. [Google Scholar] [CrossRef]
- Bakhtiarizadeh, M.R.; Mirzaei, S.; Norouzi, M.; Sheybani, N.; Vafaei, S.M. Identification of Gene Modules and Hub Genes Involved in Mastitis Development Using a Systems Biology Approach. Front. Genet. 2020, 11, 722. [Google Scholar] [CrossRef]
- Rossi, U.A.; Hasenauer, F.C.; Caffaro, M.E.; Neumann, R.; Salatin, A.; Poli, M.A.; Rossetti, C.A. A haplotype at intron 8 of PTPRT gene is associated with resistance to Brucella infection in Argentinian creole goats. Vet. Microbiol. 2017, 207, 133–137. [Google Scholar] [CrossRef]
- Du, H.; Zheng, X.; Zhao, Q.; Hu, Z.; Wang, H.; Zhou, L.; Liu, J. Analysis of Structural Variants Reveal Novel Selective Regions in the Genome of Meishan Pigs by Whole Genome Sequencing. Front. Genet. 2021, 12, 550676. [Google Scholar] [CrossRef]
- Michael, J.; Griffin, Y.Z.S.K. Early B-cell Factor-1 (EBF1) Is a Key Regulator of Metabolic and Inflammatory Signaling Pathways in Mature Adipocytes. J. Biol. Chem. 2013, 288, 35925–35939. [Google Scholar]
- Caimi, K.; Blanco, F.; Soria, M.; Bigi, F. Transcriptional response of bovine monocyte-derived macrophages after the infection with different Argentinean Mycobacterium bovis isolates. Biomed. Res. Int. 2013, 2013, 458278. [Google Scholar] [CrossRef]
- Phatak, M.; Sonawane, M. Functional characterisation of romeharsha and clint1 reaffirms the link between plasma membrane homeostasis, cell size maintenance and tissue homeostasis in developing zebrafish epidermis. J. Biosci. 2018, 43, 605–619. [Google Scholar] [CrossRef]
- Short, M.K.; Hallett, J.P.; Tar, K.; Dange, T.; Schmidt, M.; Moir, R.; Willis, I.M.; Jubinsky, P.T. The yeast magmas ortholog pam16 has an essential function in fermentative growth that involves sphingolipid metabolism. PLoS ONE 2012, 7, e39428. [Google Scholar] [CrossRef]
- Chen, X.; Ghazanfar, B.; Khan, A.R.; Hayat, S.; Cheng, Z. Comparative analysis of putative orthologues of mitochondrial import motor subunit: Pam18 and Pam16 in plants. PLoS ONE 2013, 8, e78400. [Google Scholar] [CrossRef] [PubMed]
- Iso-Touru, T.; Sahana, G.; Guldbrandtsen, B.; Lund, M.S.; Vilkki, J. Genome-wide association analysis of milk yield traits in Nordic Red Cattle using imputed whole genome sequence variants. BMC Genet. 2016, 17, 55. [Google Scholar] [CrossRef] [PubMed]
- Leal-Gutierrez, J.D.; Elzo, M.A.; Johnson, D.D.; Hamblen, H.; Mateescu, R.G. Genome wide association and gene enrichment analysis reveal membrane anchoring and structural proteins associated with meat quality in beef. BMC Genom. 2019, 20, 151. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; He, X.Y.; Jiang, Y.T.; Lan, R.; Li, M.; Li, Z.M.; Yang, W.F.; Hong, Q.H.; Chu, M.X. Combined approaches to reveal genes associated with litter size in Yunshang black goats. Anim. Genet. 2020, 51, 924–934. [Google Scholar] [CrossRef]
- Li, G.; Liu, W.; Zhang, F.; Zhu, F.; Yang, F.; Hao, J.; Hou, Z. Genome-wide association study of bone quality and feed efficiency-related traits in Pekin ducks. Genomics 2020, 112, 5021–5028. [Google Scholar] [CrossRef]
- Xu, L.; Wu, J.; Yu, Y.; Li, H.; Sun, S.; Zhang, T.; Wang, M. Dok5 regulates proliferation and differentiation of osteoblast via canonical Wnt/beta-catenin signaling. J. Musculoskelet. Neuronal Interact. 2022, 22, 113–122. [Google Scholar]
- Ahn, H.; Roh, J.S.; Lee, S.; Beon, J.; Lee, B.; Sohn, D.H.; Kim, S. Myeloid IPMK promotes the resolution of serum transfer-induced arthritis in mice. Anim. Cells Syst. 2021, 25, 219–226. [Google Scholar] [CrossRef]
- Wijayanti, D.; Erdenee, S.; Akhatayeva, Z.; Li, H.; Li, J.; Cai, Y.; Jiang, F.; Xu, H.; Lan, X. Genetic polymorphisms within the ETAA1 gene associated with growth traits in Chinese sheep breeds. Anim. Genet. 2022, 53, 460–465. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, Y.; Chen, Q.; Wang, D.; Zhang, X.; Huang, X.; Xu, L. Genome-Wide Association Study on Body Conformation Traits in Xinjiang Brown Cattle. Int. J. Mol. Sci. 2024, 25, 10557. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Heng, L. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Okonechnikov, K.; Conesa, A.; Garcia-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef] [PubMed]
- Jeffares, D.C.; Jolly, C.; Hoti, M.; Speed, D.; Shaw, L.; Rallis, C.; Balloux, F.; Dessimoz, C.; Bähler, J.; Sedlazeck, F.J. Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat. Commun. 2017, 8, 14011–14061. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, D.; Zhang, T.; Zhang, M.; Chen, Q.; Yan, M.; Ma, S.; Wang, J.; Zhang, X.; Ma, K.; Xu, L.; et al. Identification of Genomic Structural Variations in Xinjiang Brown Cattle by Deep Sequencing and Their Association with Body Conformation Traits. Int. J. Mol. Sci. 2025, 26, 5234. https://doi.org/10.3390/ijms26115234
Wang D, Zhang T, Zhang M, Chen Q, Yan M, Ma S, Wang J, Zhang X, Ma K, Xu L, et al. Identification of Genomic Structural Variations in Xinjiang Brown Cattle by Deep Sequencing and Their Association with Body Conformation Traits. International Journal of Molecular Sciences. 2025; 26(11):5234. https://doi.org/10.3390/ijms26115234
Chicago/Turabian StyleWang, Dan, Tao Zhang, Menghua Zhang, Qiuming Chen, Mengjie Yan, Shengchao Ma, Jiangkun Wang, Xiaoxue Zhang, Kailun Ma, Lei Xu, and et al. 2025. "Identification of Genomic Structural Variations in Xinjiang Brown Cattle by Deep Sequencing and Their Association with Body Conformation Traits" International Journal of Molecular Sciences 26, no. 11: 5234. https://doi.org/10.3390/ijms26115234
APA StyleWang, D., Zhang, T., Zhang, M., Chen, Q., Yan, M., Ma, S., Wang, J., Zhang, X., Ma, K., Xu, L., & Huang, X. (2025). Identification of Genomic Structural Variations in Xinjiang Brown Cattle by Deep Sequencing and Their Association with Body Conformation Traits. International Journal of Molecular Sciences, 26(11), 5234. https://doi.org/10.3390/ijms26115234