
Transcriptome-Wide Analysis of Brain Cancer Initiated by Polarity Disruption in Drosophila Type II Neuroblasts

 ,
,  ,
,  ,
,  ,
, 
Abstract
1. Introduction
2. Results
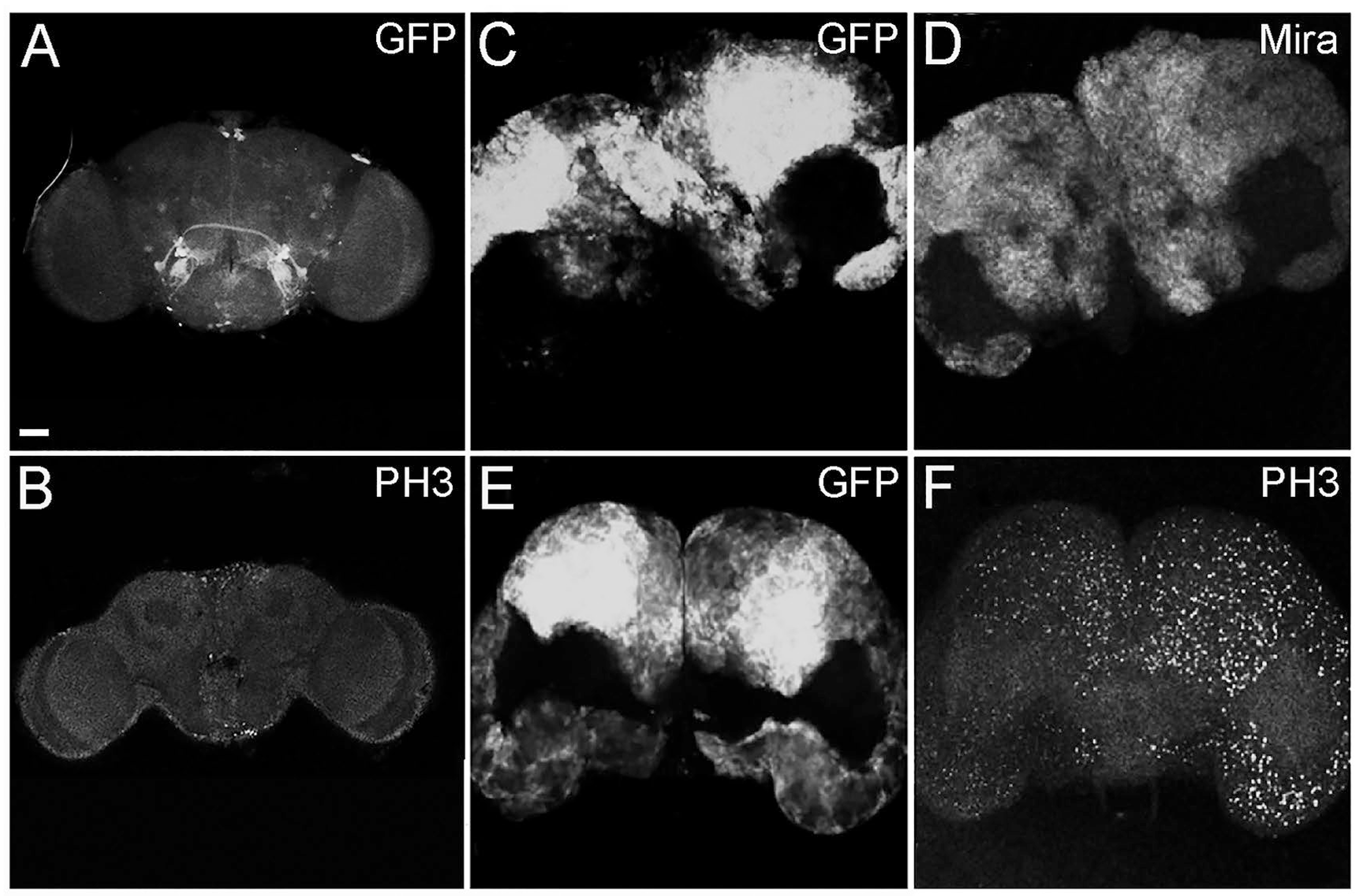
2.1. The Alteration in Cell Polarity in Type II Neuroblasts Leads to the Formation of Adult Brain Tumors
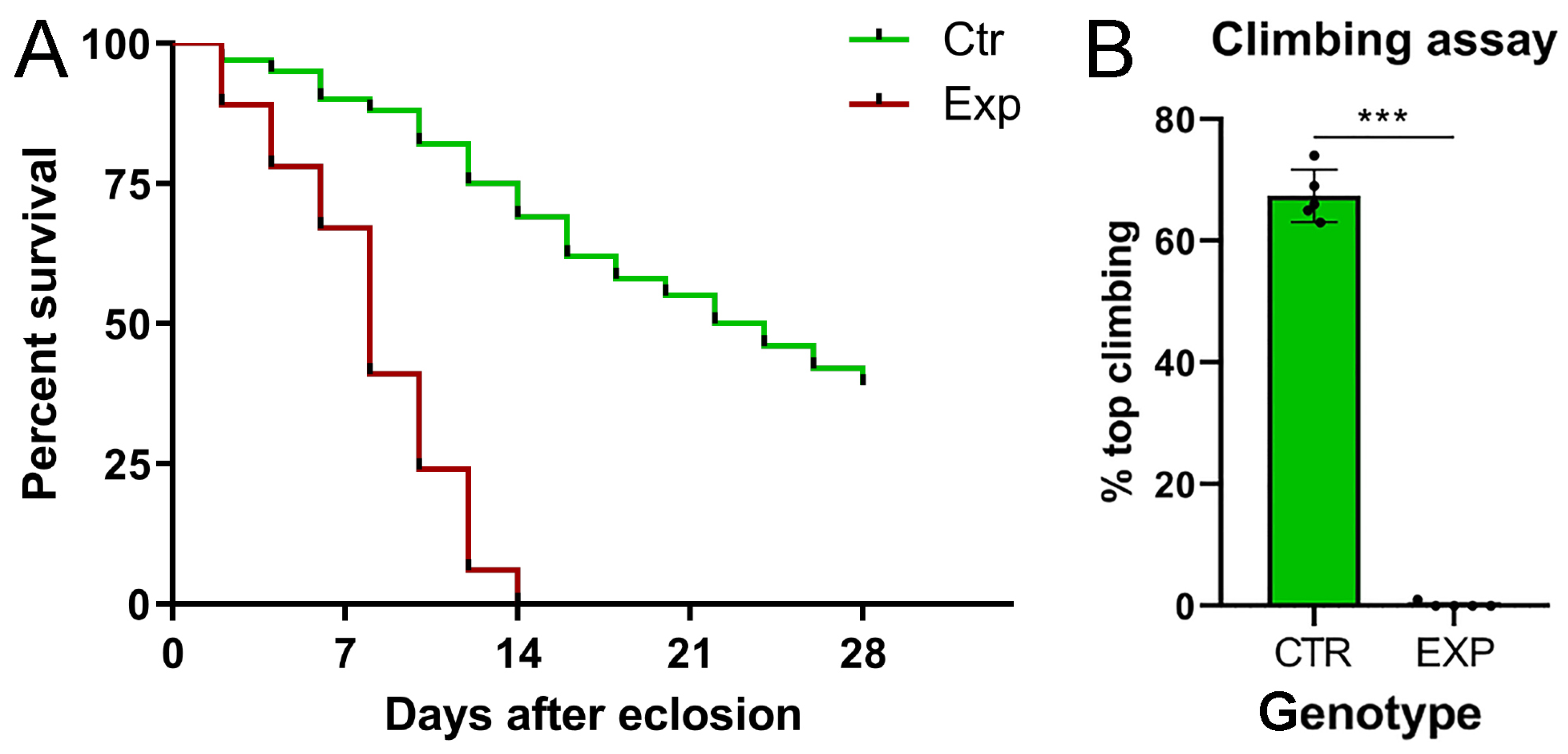
2.2. Tumor Masses Cause Neuromotor Deficits, Leading Animals to Premature Death
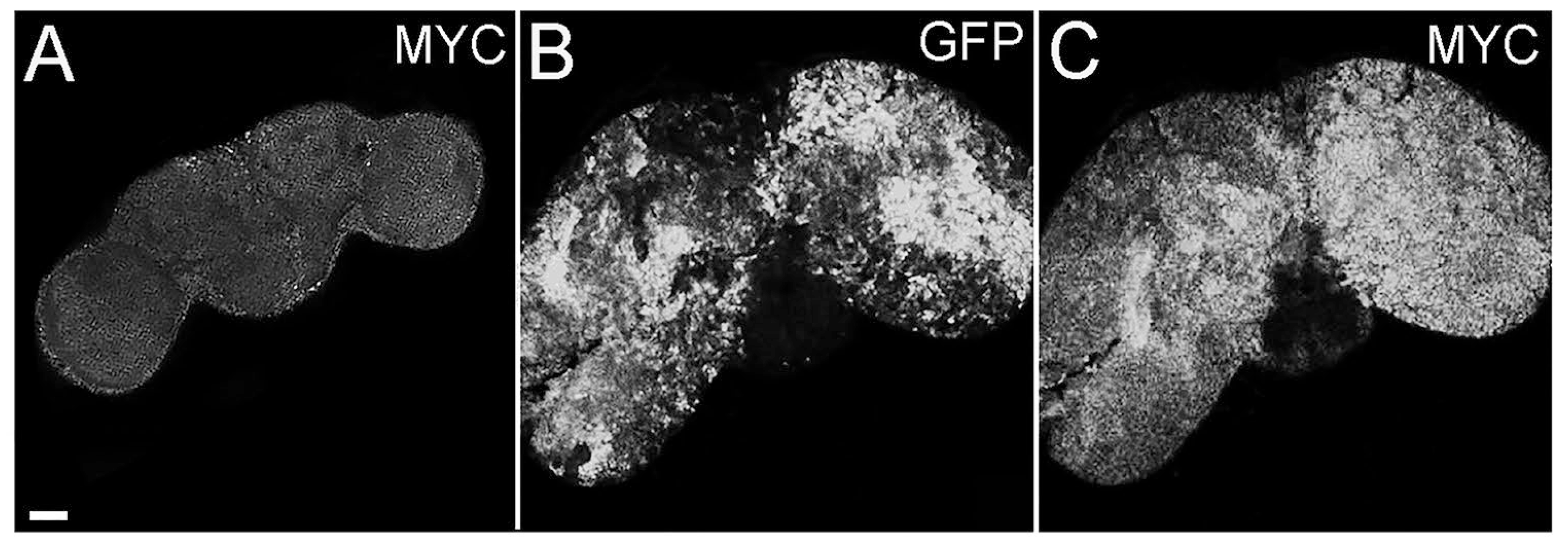
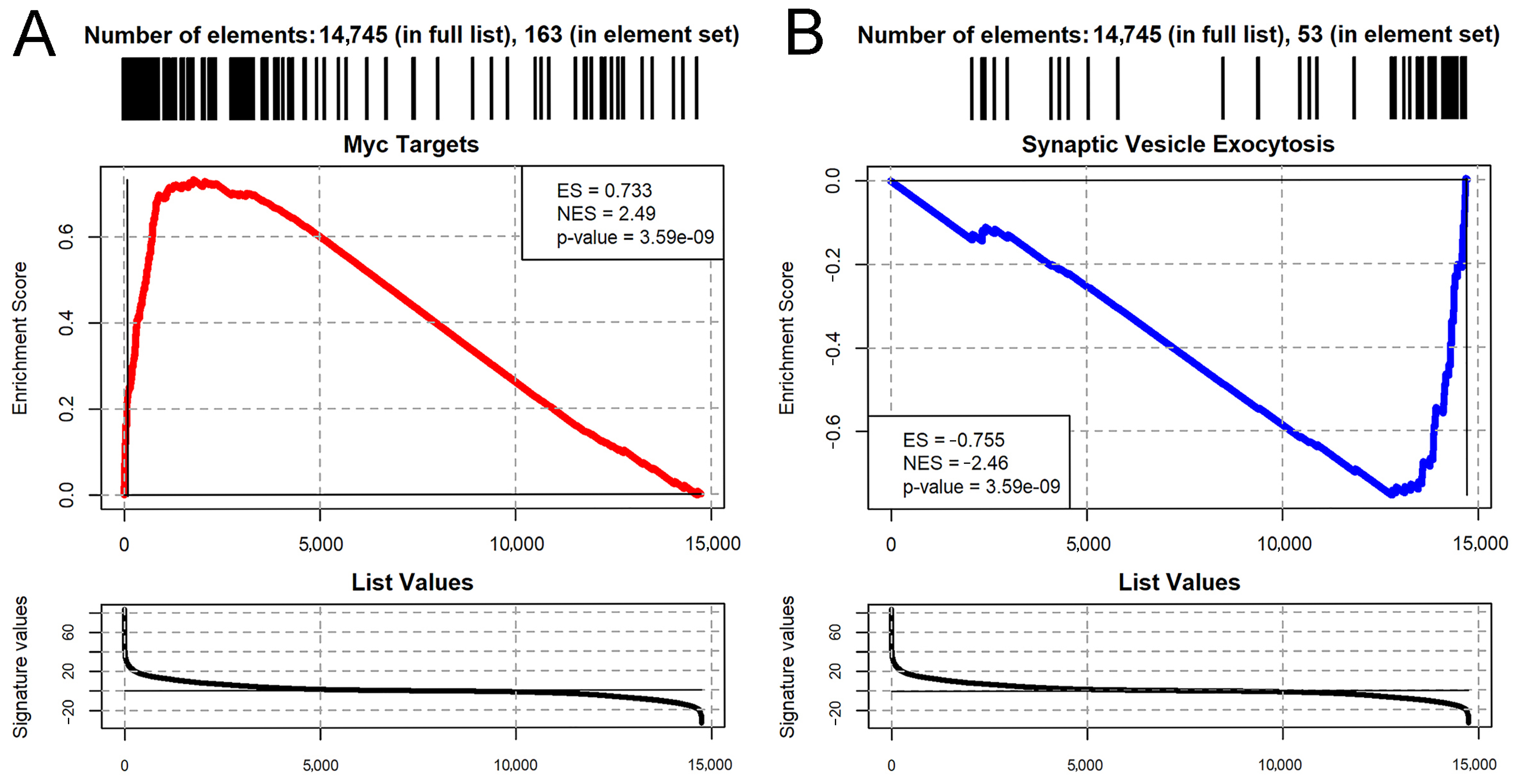
2.3. Cell Polarity Alteration in Type II NBs Causes MYC Deregulation
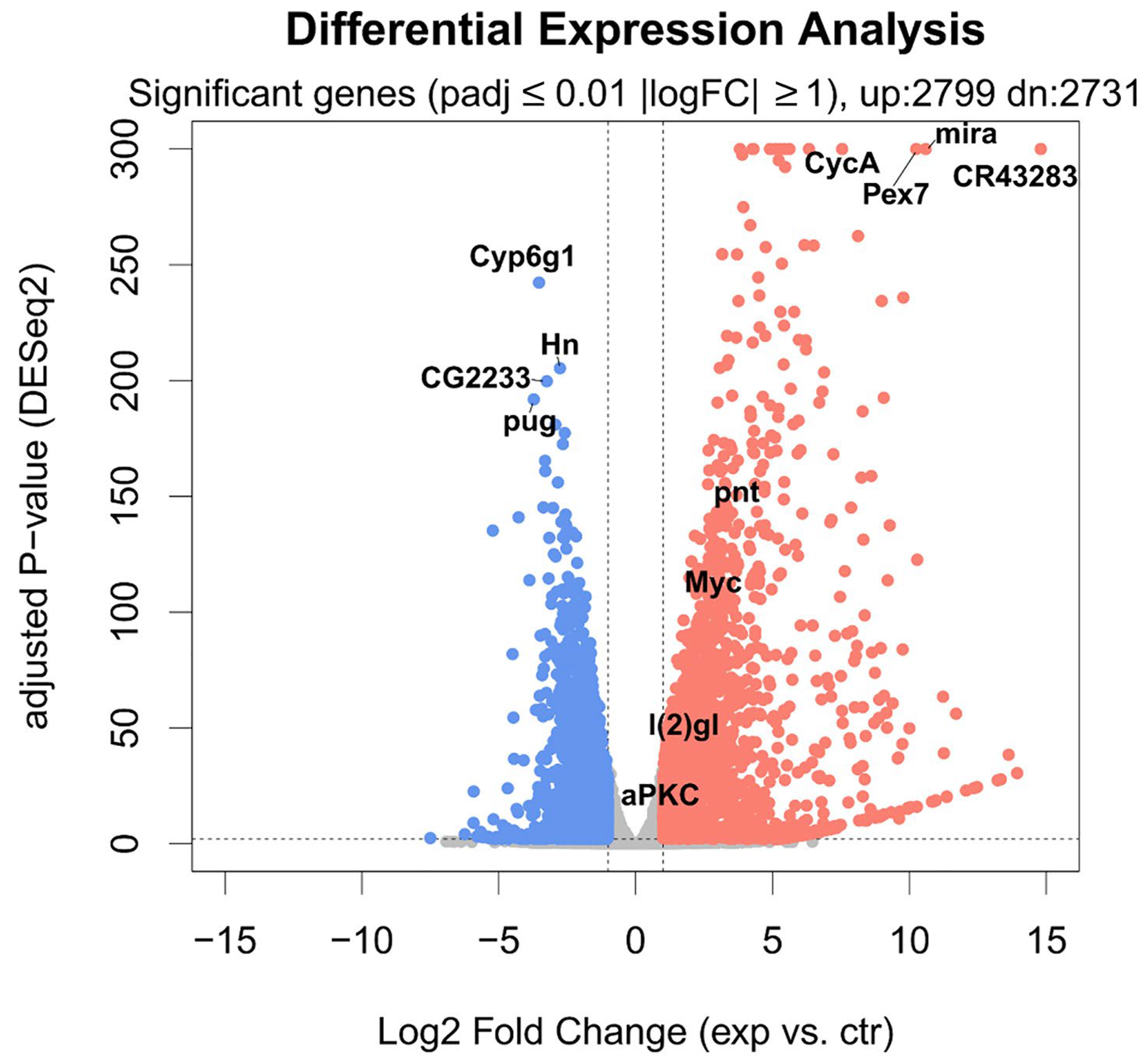
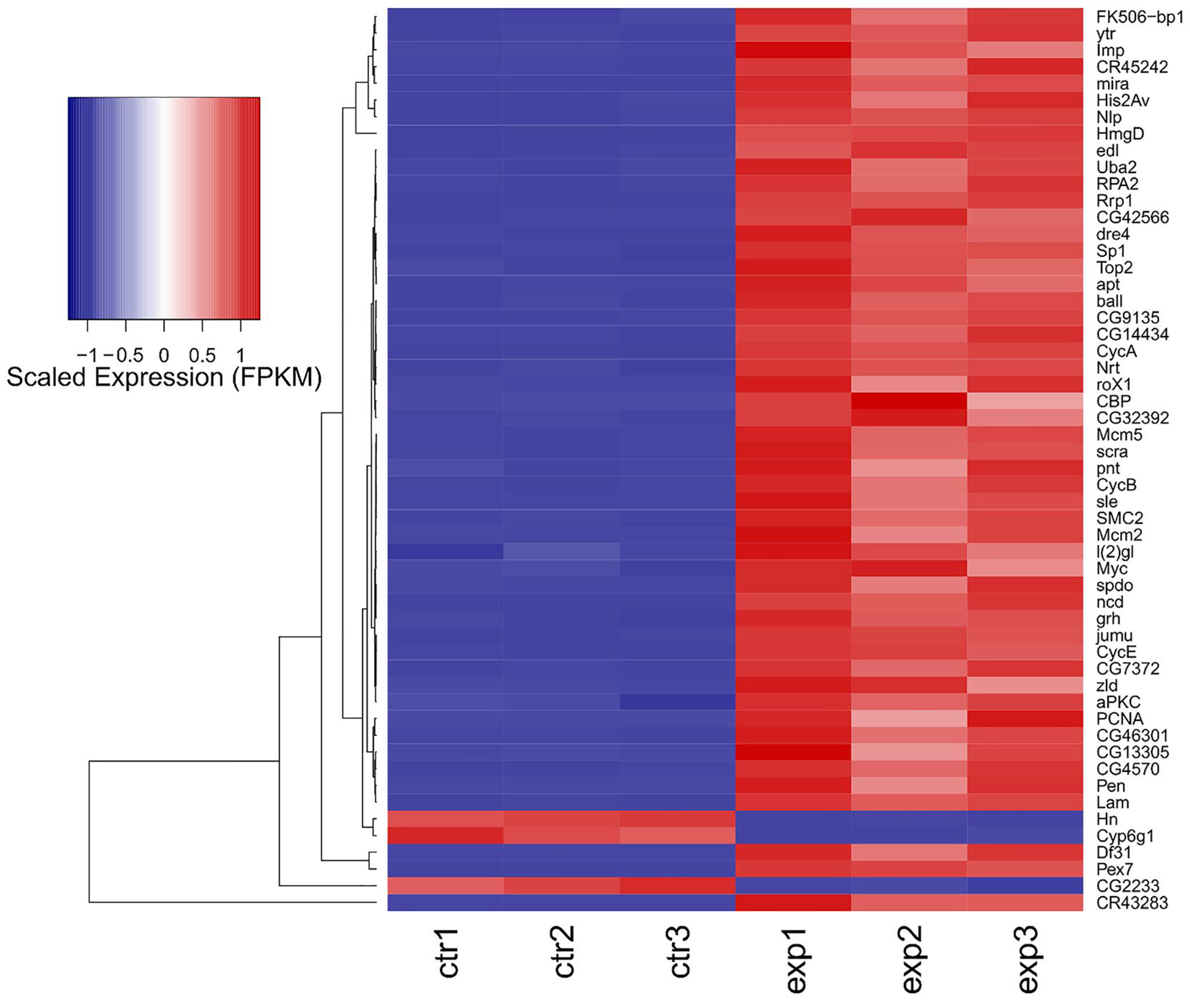
2.4. Cell Polarity Alteration in the Type II Neuroblasts Causes Significant Changes in the Adult Brain Transcriptome
2.5. About Half of the Most Deregulated Genes Are Known to Be Implicated in Human GMB

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fly Gene | Human Gene | Implication in GMB | Ref. |
|---|---|---|---|
| CycA | CCNA2 | Bad Prognosis | [51] |
| Lam | LMNB2 | [52] | |
| Pen | KPNA2 | [53] | |
| Top2 | TOP2A | [54] | |
| PCNA | PCNA | [55] | |
| ball | VRK1 | [56] | |
| His2Av | H2AFV | [57] | |
| Mcm2 | MCM2 | [58] | |
| Mcm5 | MCM5 | [58] | |
| HmgD | HMGB2 | Chemo-Resistance | [59] |
| ncd | KIFC1 | [60] | |
| CycE | CCNE1 | [61] | |
| Rrp1 | APEX1 | [62] | |
| Rrp1 | APEX1 | Radio-Resistance | [63] |
| CG9135 | RCC2 | [64] | |
| CG42566 | LITAF | [65] | |
| RPA2 | RPA2 | [66] | |
| CG46301 | NOL4 | Maintenance of GSCs | [67] |
| Imp | IGF2BP2 | [68] |
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-Cell RNA-Seq Highlights Intratumoral Heterogeneity in Primary Glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and Characterization of Tumorigenic, Stem-like Neural Precursors from Human Glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef] [PubMed]
- Demuth, T.; Berens, M.E. Molecular Mechanisms of Glioma Cell Migration and Invasion. J. Neuro-Oncol. 2004, 70, 217–228. [Google Scholar] [CrossRef]
- Friedmann-Morvinski, D.; Bushong, E.A.; Ke, E.; Soda, Y.; Marumoto, T.; Singer, O.; Ellisman, M.H.; Verma, I.M. Dedifferentiation of Neurons and Astrocytes by Oncogenes Can Induce Gliomas in Mice. Science 2012, 338, 1080–1084. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, J.E.; Kahng, J.Y.; Kim, S.H.; Park, J.S.; Yoon, S.J.; Um, J.-Y.; Kim, W.K.; Lee, J.-K.; Park, J.; et al. Human Glioblastoma Arises from Subventricular Zone Cells with Low-Level Driver Mutations. Nature 2018, 560, 243–247. [Google Scholar] [CrossRef]
- Lindberg, N.; Kastemar, M.; Olofsson, T.; Smits, A.; Uhrbom, L. Oligodendrocyte Progenitor Cells Can Act as Cell of Origin for Experimental Glioma. Oncogene 2009, 28, 2266–2275. [Google Scholar] [CrossRef]
- Tomasetti, C.; Vogelstein, B. Variation in Cancer Risk among Tissues Can Be Explained by the Number of Stem Cell Divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef]
- Llaguno, S.A.; Sun, D.; Pedraza, A.M.; Vera, E.; Wang, Z.; Burns, D.K.; Parada, L.F. Cell-of-Origin Susceptibility to Glioblastoma Formation Declines with Neural Lineage Restriction. Nat. Neurosci. 2019, 22, 545–555. [Google Scholar] [CrossRef]
- Broekman, M.L.; Maas, S.L.N.; Abels, E.R.; Mempel, T.R.; Krichevsky, A.M.; Breakefield, X.O. Multidimensional Communication in the Microenvirons of Glioblastoma. Nat. Rev. Neurol. 2018, 14, 482–495. [Google Scholar] [CrossRef]
- Read, R.D. Drosophila melanogaster as a Model System for Human Brain Cancers. Glia 2011, 59, 1364–1376. [Google Scholar] [CrossRef] [PubMed]
- Aldape, K.; Zadeh, G.; Mansouri, S.; Reifenberger, G.; von Deimling, A. Glioblastoma: Pathology, Molecular Mechanisms and Markers. Acta Neuropathol. 2015, 129, 829–848. [Google Scholar] [CrossRef] [PubMed]
- Read, R.D.; Cavenee, W.K.; Furnari, F.B.; Thomas, J.B. A Drosophila Model for EGFR-Ras and PI3K-Dependent Human Glioma. PLoS Genet. 2009, 5, e1000374. [Google Scholar] [CrossRef]
- Portela, M.; Venkataramani, V.; Fahey-Lozano, N.; Seco, E.; Losada-Perez, M.; Winkler, F.; Casas-Tintó, S. Glioblastoma Cells Vampirize WNT from Neurons and Trigger a JNK/MMP Signaling Loop That Enhances Glioblastoma Progression and Neurodegeneration. PLoS Biol. 2019, 17, e3000545. [Google Scholar] [CrossRef]
- Formica, M.; Storaci, A.M.; Bertolini, I.; Carminati, F.; Knævelsrud, H.; Vaira, V.; Vaccari, T. V-ATPase Controls Tumor Growth and Autophagy in a Drosophila Model of Gliomagenesis. Autophagy 2021, 17, 4442–4452. [Google Scholar] [CrossRef]
- Bertrand, M.; Szeremeta, F.; Hervouet-Coste, N.; Sarou-Kanian, V.; Landon, C.; Morisset-Lopez, S.; Decoville, M. An Adult Drosophila Glioma Model to Highlight Metabolic Dysfunctions and Evaluate the Role of the Serotonin 5-HT7 Receptor as a Potential Therapeutic Target. FASEB J. 2023, 37, e23230. [Google Scholar] [CrossRef]
- Gont, A.; Hanson, J.E.L.; Lavictoire, S.J.; Parolin, D.A.E.; Daneshmand, M.; Restall, I.J.; Soucie, M.; Nicholas, G.; Woulfe, J.; Kassam, A.; et al. PTEN Loss Represses Glioblastoma Tumor Initiating Cell Differentiation via Inactivation of Lgl1. Oncotarget 2013, 4, 1266–1279. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Jill, P.; Alexe, G.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Maehama, T.; Dixon, J.E. The Tumor Suppressor, PTEN/MMAC1, Dephosphorylates the Lipid Second Messenger, Phosphatidylinositol 3,4,5-Trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef]
- Le Good, J.A.; Ziegler, W.H.; Parekh, D.B.; Alessi, D.R.; Cohen, P.; Parker, P.J. Protein Kinase C Isotypes Controlled by Phosphoinositide 3-Kinase through the Protein Kinase PDK1. Science 1998, 281, 2042–2045. [Google Scholar] [CrossRef]
- Plant, P.J.; Fawcett, J.P.; Lin, D.C.C.; Holdorf, A.D.; Binns, K.; Kulkarni, S.; Pawson, T. A Polarity Complex of mPar-6 and Atypical PKC Binds, Phosphorylates and Regulates Mammalian Lgl. Nat. Cell Biol. 2003, 5, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Gateff, E. Malignant Neoplasms of Genetic Origin in Drosophila melanogaster. Science 1978, 200, 1448–1459. [Google Scholar] [CrossRef] [PubMed]
- Wodarz, A. Tumor Suppressors: Linking Cell Polarity and Growth Control. Curr. Biol. 2000, 10, R624–R626. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.Y.; Manning, L.; Albertson, R.; Doe, C.Q. The Tumour-Suppresor Genes Lgl and Dlg Regulate Basal Protein Targeting in Drosophila Neuroblasts. Nature 2000, 408, 596–600. [Google Scholar] [CrossRef]
- Grifoni, D.; Garoia, F.; Schimanski, C.C.; Schmitz, G.; Laurenti, E.; Galle, P.R.; Pession, A.; Cavicchi, S.; Strand, D. The Human Protein Hugl-1 Substitutes for Drosophila Lethal Giant Larvae Tumour Suppressor Function in Vivo. Oncogene 2004, 23, 8688–8694. [Google Scholar] [CrossRef]
- Kuphal, S.; Wallner, S.; Schimanski, C.C.; Bataille, F.; Hofer, P.; Strand, S.; Strand, D.; Bosserhoff, A.K. Expression of Hugl-1 Is Strongly Reduced in Malignant Melanoma. Oncogene 2006, 25, 103–110. [Google Scholar] [CrossRef]
- Grifoni, D.; Garoia, F.; Bellosta, P.; Parisi, F.; De Biase, D.; Collina, G.; Strand, D.; Cavicchi, S.; Pession, A. aPKCζ Cortical Loading Is Associated with Lgl Cytoplasmic Release and Tumor Growth in Drosophila and Human Epithelia. Oncogene 2007, 26, 5960–5965. [Google Scholar] [CrossRef]
- Lu, X.; Feng, X.; Man, X.; Yang, G.; Tang, L.; Du, D.; Zhang, F.; Yuan, H.; Huang, Q.; Zhang, Z.; et al. Aberrant Splicing of Hugl-1 Is Associated with Hepatocellular Carcinoma Progression. Clin. Cancer Res. 2009, 15, 3287–3296. [Google Scholar] [CrossRef]
- Schimanski, C.C.; Schmitz, G.; Kashyap, A.; Bosserhoff, A.K.; Bataille, F.; Schäfer, S.C.; A Lehr, H.; Berger, M.R.; Galle, P.R.; Strand, S.; et al. Reduced Expression of Hugl-1, the Human Homologue of Drosophila Tumour Suppressor Gene Lgl, Contributes to Progression of Colorectal Cancer. Oncogene 2005, 24, 3100–3109. [Google Scholar] [CrossRef]
- Bilder, D. Epithelial Polarity and Proliferation Control: Links from the Drosophila Neoplastictumor Suppressors. Genes Dev. 2004, 18, 1909–1925. [Google Scholar] [CrossRef]
- Gont, A.; Hanson, J.E.L.; Lavictoire, S.J.; Daneshmand, M.; Nicholas, G.; Woulfe, J.; Kassam, A.; Da Silva, V.F.; Lorimer, I.A.J. Inhibition of Glioblastoma Malignancy by Lgl1. Oncotarget 2014, 5, 11541–11551. [Google Scholar] [CrossRef] [PubMed]
- Paglia, S.; Sollazzo, M.; Di Giacomo, S.; De Biase, D.; Pession, A.; Grifoni, D. Failure of the PTEN/aPKC/Lgl Axis Primes Formation of Adult Brain Tumours in Drosophila. BioMed Res. Int. 2017, 2017, 2690187. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Barshow, S.; Wildonger, J.; Jan, L.Y.; Jan, Y.N. Ets Transcription Factor Pointed Promotes the Generation of Intermediate Neural Progenitors in Drosophila Larval Brains. Proc. Natl. Acad. Sci. USA 2011, 108, 20615–20620. [Google Scholar] [CrossRef]
- Bello, B.C.; Izergina, N.; Caussinus, E.; Reichert, H. Amplification of Neural Stem Cell Proliferation by Intermediate Progenitor Cells in Drosophila Brain Development. Neural Dev. 2008, 3, 5. [Google Scholar] [CrossRef]
- Weng, R.; Cohen, S.M. Control of Drosophila Type I and Type II Central Brain Neuroblast Proliferation by Bantam microRNA. Development 2015, 142, 3713–3720. [Google Scholar] [CrossRef]
- Boone, J.Q.; Doe, C.Q. Identification of Drosophila Type II Neuroblast Lineages Containing Transit Amplifying Ganglion Mother Cells. Dev. Neurobiol. 2008, 68, 1185–1195. [Google Scholar] [CrossRef]
- Zhu, S.; Wildonger, J.; Barshow, S.; Younger, S.; Huang, Y.; Lee, T. The bHLH Repressor Deadpan Regulates the Self-Renewal and Specification of Drosophila Larval Neural Stem Cells Independently of Notch. PLoS ONE 2012, 7, e46724. [Google Scholar] [CrossRef]
- Wang, H.; Chia, W. Drosophila Neural Progenitor Polarity and Asymmetric Division. Biol. Cell 2005, 97, 63–74. [Google Scholar] [CrossRef]
- Hans, F.; Dimitrov, S. Histone H3 Phosphorylation and Cell Division. Oncogene 2001, 20, 3021–3027. [Google Scholar] [CrossRef]
- Li, G.; Hidalgo, A. Adult Neurogenesis in the Drosophila Brain: The Evidence and the Void. Int. J. Mol. Sci. 2020, 21, 6653. [Google Scholar] [CrossRef]
- Dhar, G.; Mukherjee, S.; Nayak, N.; Sahu, S.; Bag, J.; Rout, R.; Mishra, M. Various Behavioural Assays to Detect the Neuronal Abnormality in Flies. In Fundamental Approaches to Screen Abnormalities in Drosophila; Mishra, M., Ed.; Springer: New York, NY, USA, 2020; pp. 223–251. [Google Scholar] [CrossRef]
- Dang, C.V. C-Myc Target Genes Involved in Cell Growth, Apoptosis, and Metabolism. Mol. Cell. Biol. 1999, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kalkat, M.; De Melo, J.; Hickman, K.A.; Lourenco, C.; Redel, C.; Resetca, D.; Tamachi, A.; Tu, W.B.; Penn, L.Z. MYC Deregulation in Primary Human Cancers. Genes 2017, 8, 151. [Google Scholar] [CrossRef] [PubMed]
- Borgenvik, A.; Čančer, M.; Hutter, S.; Swartling, F.J. Targeting MYCN in Molecularly Defined Malignant Brain Tumors. Front. Oncol. 2021, 10, 626751. [Google Scholar] [CrossRef]
- Herms, J.W.; Von Loewenich, F.D.; Behnke, J.; Markakis, E.; Kretzschmar, H.A. C-MYC Oncogene Family Expression in Glioblastoma and Survival. Surg. Neurol. 1999, 51, 536–542. [Google Scholar] [CrossRef]
- Faria, M.H.; Khayat, A.S.; Burbano, R.R.; Rabenhorst, S.H. C-MYC Amplification and Expression in Astrocytic Tumors. Acta Neuropathol. 2008, 116, 87–95. [Google Scholar] [CrossRef]
- Hofstetter, J.; Ogunleye, A.; Kutschke, A.; Buchholz, L.M.; Wolf, E.; Raabe, T.; Gallant, P. Spt5 Interacts Genetically with Myc and Is Limiting for Brain Tumor Growth in Drosophila. Life Sci. Alliance 2024, 7, e202302130. [Google Scholar] [CrossRef]
- Jarabo, P.; de Pablo, C.; González-Blanco, A.; Casas-Tintó, S. Circadian Gene Cry Controls Tumorigenesis through Modulation of Myc Accumulation in Glioblastoma Cells. Int. J. Mol. Sci. 2022, 23, 2043. [Google Scholar] [CrossRef]
- Morciano, P.; Grifoni, D. Breaking the Brain Barrier: Cell Competition in Neural Development and Disease. Neural Regen. Res. 2023, 19, 1863–1864. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Chen, C.; Sun, C.; Tang, D.; Yang, G.; Zhou, X.; Wang, D. Identification of Key Genes in Glioblastoma-Associated Stromal Cells Using Bioinformatics Analysis. Oncol. Lett. 2016, 11, 3999–4007. [Google Scholar] [CrossRef]
- Pei, S.; Wang, X.; Wang, X.; Huang, H.; Tao, H.; Xie, B.; Yang, A.; Qiu, M.; Tan, Z. Aberrant Nuclear Lamina Contributes to the Malignancy of Human Gliomas. J. Genet. Genom. 2022, 49, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Gousias, K.; Becker, A.J.; Simon, M.; Niehusmann, P. Nuclear Karyopherin A2: A Novel Biomarker for Infiltrative Astrocytomas. J. Neuro-Oncol. 2012, 109, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Wang, Y.; Qian, D.; Liang, Q.; Wang, B. Over-Expression of TOP2A as a Prognostic Biomarker in Patients with Glioma. Int. J. Clin. Exp. Pathol. 2018, 11, 1228. [Google Scholar]
- Kayaselçuk, F.; Zorludemir, S.; Gümürdülü, D.; Zeren, H.; Erman, T. PCNA and Ki-67 in Central Nervous System Tumors: Correlation with the Histological Type and Grade. J. Neuro-Oncol. 2002, 57, 115–121. [Google Scholar] [CrossRef]
- Ben, Z.; Gong, L.; Qiu, Y. High Expression of VRK1 Is Related to Poor Prognosis in Glioma. Pathol. Res. Pract. 2018, 214, 112–118. [Google Scholar] [CrossRef]
- Lee, H.-H.; Lin, C.-H.; Lin, H.-Y.; Kuei, C.-H.; Zheng, J.-Q.; Wang, Y.-H.; Lu, L.-S.; Lee, F.-P.; Hu, C.-J.; Wu, D.; et al. Histone 2A Family Member j Drives Mesenchymal Transition and Temozolomide Resistance in Glioblastoma Multiforme. Cancers 2020, 12, 98. [Google Scholar] [CrossRef]
- Hua, C.; Zhao, G.; Li, Y.; Bie, L. Minichromosome Maintenance (MCM) Family as Potential Diagnostic and Prognostic Tumor Markers for Human Gliomas. BMC Cancer 2014, 14, 526. [Google Scholar] [CrossRef]
- Wu, Z.B.; Cai, L.; Lin, S.J.; Xiong, Z.K.; Lu, J.L.; Mao, Y.; Yao, Y.; Zhou, L.F. High-Mobility Group Box 2 Is Associated with Prognosis of Glioblastoma by Promoting Cell Viability, Invasion, and Chemotherapeutic Resistance. Neuro-Oncology 2013, 15, 1264–1275. [Google Scholar] [CrossRef]
- Xue, P.; Zheng, J.; Li, R.; Yan, L.; Wang, Z.; Jia, Q.; Zhang, L.; Li, X. High Expression of KIFC1 in Glioma Correlates with Poor Prognosis. J. Korean Neurosurg. Soc. 2024, 67, 364–375. [Google Scholar] [CrossRef]
- Liang, H.; Chen, Z.; Sun, L. Inhibition of Cyclin E1 Overcomes Temozolomide Resistance in Glioblastoma by Mcl-1 Degradation. Mol. Carcinog. 2019, 58, 1502–1511. [Google Scholar] [CrossRef]
- Montaldi, A.P.; Godoy, P.R.D.V.; Sakamoto-Hojo, E.T. APE1/REF-1 down-Regulation Enhances the Cytotoxic Effects of Temozolomide in a Resistant Glioblastoma Cell Line. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2015, 793, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Naidu, M.D.; Mason, J.M.; Pica, R.V.; Hua, F.; Peña, L.A. Radiation Resistance in Glioma Cells Determined by DNA Damage Repair Activity of Ape1/Ref-1. J. Radiat. Res. 2010, 51, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Zhang, S.; Ibrahim, A.N.; Wang, J.; Deng, Z.; Wang, M. RCC2 Promotes Proliferation and Radio-Resistance in Glioblastoma via Activating Transcription of DNMT1. Biochem. Biophys. Res. Commun. 2019, 516, 999–1006. [Google Scholar] [CrossRef]
- Huang, C.; Chen, D.; Zhu, H.; Lv, S.; Li, Q.; Li, G. LITAF Enhances Radiosensitivity of Human Glioma Cells via the FoxO1 Pathway. Cell. Mol. Neurobiol. 2019, 39, 871–882. [Google Scholar] [CrossRef]
- Pedersen, H.; Obara, E.A.A.; Elbæk, K.J.; Vitting-Serup, K.; Hamerlik, P. Replication Protein a (RPA) Mediates Radio-Resistance of Glioblastoma Cancer Stem-like Cells. Int. J. Mol. Sci. 2020, 21, 1588. [Google Scholar] [CrossRef]
- Stangeland, B.; Mughal, A.A.; Grieg, Z.; Sandberg, C.J.; Joel, M.; Nygård, S.; Meling, T.; Murrell, W.; Vik Mo, E.O.; Langmoen, I.A. Combined Expressional Analysis, Bioinformatics and Targeted Proteomics Identify New Potential Therapeutic Targets in Glioblastoma Stem Cells. Oncotarget 2015, 6, 26192–26215. [Google Scholar] [CrossRef]
- Janiszewska, M.; Suvà, M.L.; Riggi, N.; Houtkooper, R.H.; Auwerx, J.; Clément-Schatlo, V.; Radovanovic, I.; Rheinbay, E.; Provero, P.; Stamenkovic, I. Imp2 Controls Oxidative Phosphorylation and Is Crucial for Preservin Glioblastoma Cancer Stem Cells. Genes Dev. 2012, 26, 1926–1944. [Google Scholar] [CrossRef]
- Tang, X.; Zuo, C.; Fang, P.; Liu, G.; Qiu, Y.; Huang, Y.; Tang, R. Targeting Glioblastoma Stem Cells: A Review on Biomarkers, Signal Pathways and Targeted Therapy. Front. Oncol. 2021, 11, 701291. [Google Scholar] [CrossRef]
- Carney, T.D.; Miller, M.R.; Robinson, K.J.; Bayraktar, O.A.; Osterhout, J.A.; Doe, C.Q. Functional Genomics Identifies Neural Stem Cell Sub-Type Expression Profiles and Genes Regulating Neuroblast Homeostasis. Dev. Biol. 2012, 361, 137–146. [Google Scholar] [CrossRef]
- Walsh, K.T.; Doe, C.Q. Drosophila Embryonic Type II Neuroblasts: Origin, Temporal Patterning, and Contribution to the Adult Central Complex. Development 2017, 144, 4552–4562. [Google Scholar] [CrossRef]
- Brennan, C.; Momota, H.; Hambardzumyan, D.; Ozawa, T.; Tandon, A.; Pedraza, A.; Holland, E. Glioblastoma Subclasses Can Be Defined by Activity among Signal Transduction Pathways and Associated Genomic Alterations. PLoS ONE 2009, 4, e7752. [Google Scholar] [CrossRef] [PubMed]
- Zaytseva, O.; Kim, N.H.; Quinn, L.M. Myc in Brain Development and Cancer. Int. J. Mol. Sci. 2020, 21, 7742. [Google Scholar] [CrossRef] [PubMed]
- Betschinger, J.; Mechtler, K.; Knoblich, J.A. Asymmetric Segregation of the Tumor Suppressor Brat Regulates Self-Renewal in Drosophila Neural Stem Cells. Cell 2006, 124, 1241–1253. [Google Scholar] [CrossRef]
- Wang, J.; Wang, H.; Li, Z.; Wu, Q.; Lathia, J.D.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. C-Myc Is Required for Maintenance of Glioma Cancer Stem Cells. PLoS ONE 2008, 3, e3769. [Google Scholar] [CrossRef]
- Junttila, M.R.; Westermarck, J. Mechanisms of MYC Stabilization in Human Malignancies. Cell Cycle 2008, 7, 592–596. [Google Scholar] [CrossRef]
- Pulverer, B.J.; Fisher, C.; Vousden, K.; Littlewood, T.; Evan, G.; Woodgett, J.R. Site-Specific Modulation of c-Myc Cotransformation by Residues Phosphorylated in Vivo. Oncogene 1994, 9, 59–70. [Google Scholar]
- Gregory, M.A.; Qi, Y.; Hann, S.R. Phosphorylation by Glycogen Synthase Kinase-3 Controls c-Myc Proteolysis and Subnuclear Localization. J. Biol. Chem. 2003, 278, 51606–51612. [Google Scholar] [CrossRef]
- Goode, N.; Hughes, K.; Woodgettq, J.R.; Parkersli, P.J. Differential Regulation of Glycogen Synthase Kinase-3P by Protein Kinase C Isotypes*. J. Biol. Chem. 1992, 267, 16878–16882. [Google Scholar] [CrossRef]
- Mu, Q.; Wang, L.; Yu, F.; Gao, H.; Lei, T.; Li, P.; Liu, P.; Zheng, X.; Hu, X.; Chen, Y.; et al. Imp2 Regulates GBM Progression by Activating IGF2/PI3K/Akt Pathway. Cancer Biol. Ther. 2015, 16, 623–633. [Google Scholar] [CrossRef]
- Degrauwe, N.; Schlumpf, T.B.; Janiszewska, M.; Martin, P.; Cauderay, A.; Provero, P.; Riggi, N.; Suvà, M.L.; Paro, R.; Stamenkovic, I. The RNA Binding Protein IMP2 Preserves Glioblastoma Stem Cells by Preventing Let-7 Target Gene Silencing. Cell Rep. 2016, 15, 1634–1647. [Google Scholar] [CrossRef]
- Li, J.; Liu, Q.; Liu, Z.; Xia, Q.; Zhang, Z.; Zhang, R.; Gao, T.; Gu, G.; Wang, Y.; Wang, D.; et al. KPNA2 Promotes Metabolic Reprogramming in Glioblastomas by Regulation of C-Myc. J. Exp. Clin. Cancer Res. 2018, 37, 194. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, A.L.; Walton, Z.E.; Altman, B.J.; Stine, Z.E.; Dang, C.V. MYC and Metabolism on the Path to Cancer. Semin. Cell Dev. Biol. 2015, 43, 11–21. [Google Scholar] [CrossRef]
- Samuels, T.J.; Järvelin, A.I.; Ish-Horowicz, D.; Davis, I. Imp/IGF2BP Levels Modulate Individual Neural Stem Cell Growth and Division through Myc mRNA Stability. eLife 2020, 9, e51529. [Google Scholar] [CrossRef]
- Bell, J.L.; Wächter, K.; Mühleck, B.; Pazaitis, N.; Köhn, M.; Lederer, M.; Hüttelmaier, S. Insulin-like Growth Factor 2 mRNA-Binding Proteins (IGF2BPs): Post-Transcriptional Drivers of Cancer Progression? Cell. Mol. Life Sci. 2013, 70, 2657–2675. [Google Scholar] [CrossRef]
- Quinones, A.; Le, A. The Multifaceted Glioblastoma: From Genomic Alterations to Metabolic Adaptations. Adv. Exp. Med. Biol. 2021, 1311, 59–76. [Google Scholar] [CrossRef]
- Del Fabbro, C.; Scalabrin, S.; Morgante, M.; Giorgi, F.M. An Extensive Evaluation of Read Trimming Effects on Illumina NGS Data Analysis. PLoS ONE 2013, 8, e85024. [Google Scholar] [CrossRef]
- Thurmond, J.; Goodman, J.L.; Strelets, V.B.; Attrill, H.; Gramates, L.S.; Marygold, S.J.; Matthews, B.B.; Millburn, G.; Antonazzo, G.; Trovisco, V.; et al. FlyBase 2.0: The next Generation. Nucleic Acids Res. 2019, 47, D759–D765. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A Fast Spliced Aligner with Low Memory Requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Yates, A.D.; Achuthan, P.; Akanni, W.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2020. Nucleic Acids Res. 2020, 48, D682–D688. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Drai, D.; Elmer, G.; Kafkafi, N.; Golani, I. Controlling the False Discovery Rate in Behavior Genetics Research. Behav. Brain Res. 2001, 125, 279–284. [Google Scholar] [CrossRef]
- Mercatelli, D.; Lopez-Garcia, G.; Giorgi, F.M. Corto: A Lightweight R Package for Gene Network Inference and Master Regulator Analysis. Bioinformatics 2020, 36, 3916–3917. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Hu, Y.; Flockhart, I.; Vinayagam, A.; Bergwitz, C.; Berger, B.; Perrimon, N.; Mohr, S.E. An Integrative Approach to Ortholog Prediction for Disease-Focused and Other Functional Studies. BMC Bioinform. 2011, 12, 357. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paglia, S.; Morciano, P.; de Biase, D.; Giorgi, F.M.; Pession, A.; Grifoni, D. Transcriptome-Wide Analysis of Brain Cancer Initiated by Polarity Disruption in Drosophila Type II Neuroblasts. Int. J. Mol. Sci. 2025, 26, 5115. https://doi.org/10.3390/ijms26115115
Paglia S, Morciano P, de Biase D, Giorgi FM, Pession A, Grifoni D. Transcriptome-Wide Analysis of Brain Cancer Initiated by Polarity Disruption in Drosophila Type II Neuroblasts. International Journal of Molecular Sciences. 2025; 26(11):5115. https://doi.org/10.3390/ijms26115115
Chicago/Turabian StylePaglia, Simona, Patrizia Morciano, Dario de Biase, Federico Manuel Giorgi, Annalisa Pession, and Daniela Grifoni. 2025. "Transcriptome-Wide Analysis of Brain Cancer Initiated by Polarity Disruption in Drosophila Type II Neuroblasts" International Journal of Molecular Sciences 26, no. 11: 5115. https://doi.org/10.3390/ijms26115115
APA StylePaglia, S., Morciano, P., de Biase, D., Giorgi, F. M., Pession, A., & Grifoni, D. (2025). Transcriptome-Wide Analysis of Brain Cancer Initiated by Polarity Disruption in Drosophila Type II Neuroblasts. International Journal of Molecular Sciences, 26(11), 5115. https://doi.org/10.3390/ijms26115115