
Review of Recent Computational Research on the Adsorption of PFASs with a Variety of Substrates



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Adsorbents and Morphology
2.1. Activated Carbon
2.2. Graphene
2.3. Smectite Clay
2.4. Rutile TiO2
2.5. Chitosan
2.6. Micelles
2.7. Pyrophyllite Clay
2.8. Kaolinite Clay
2.9. Biochar
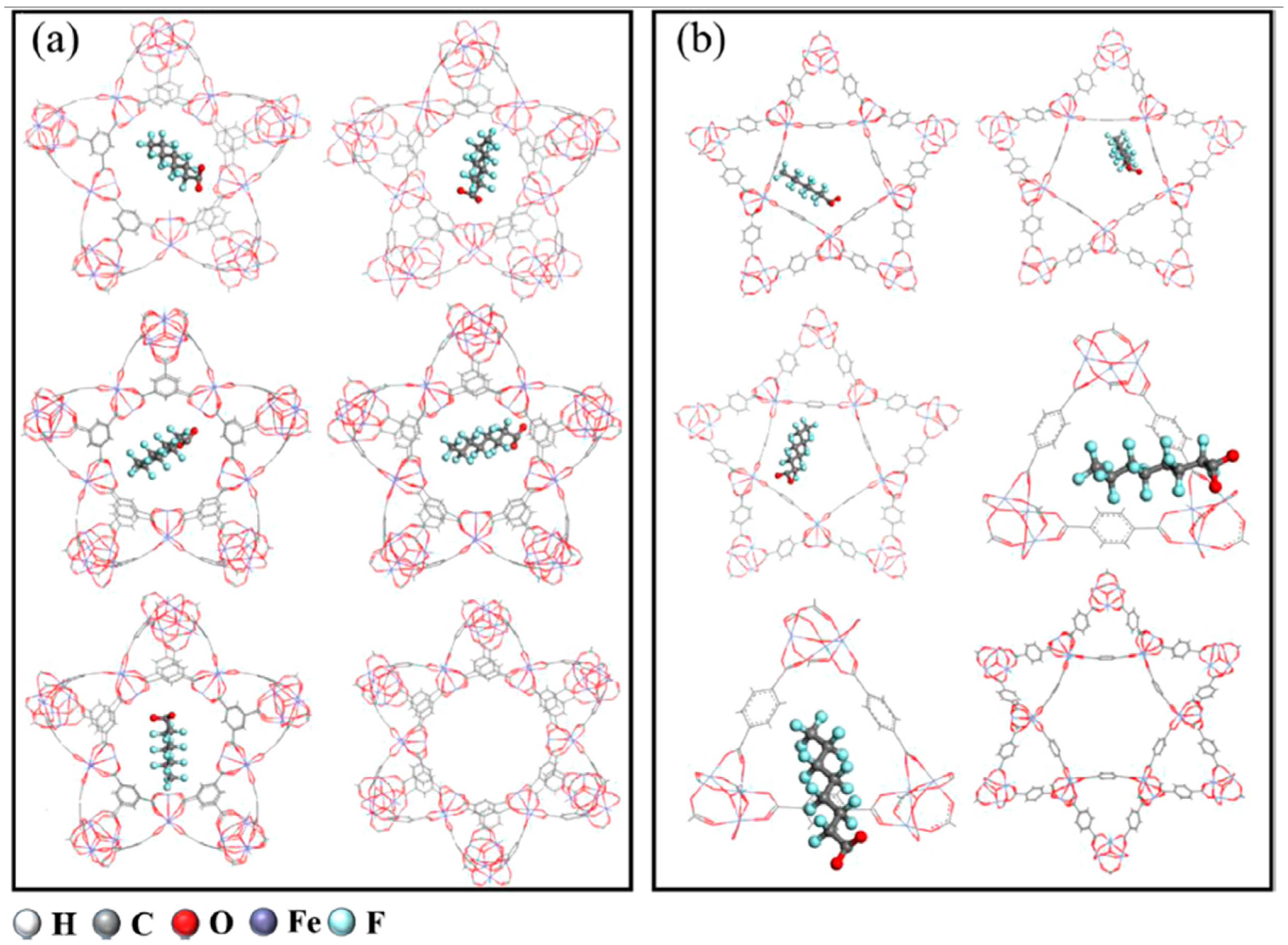
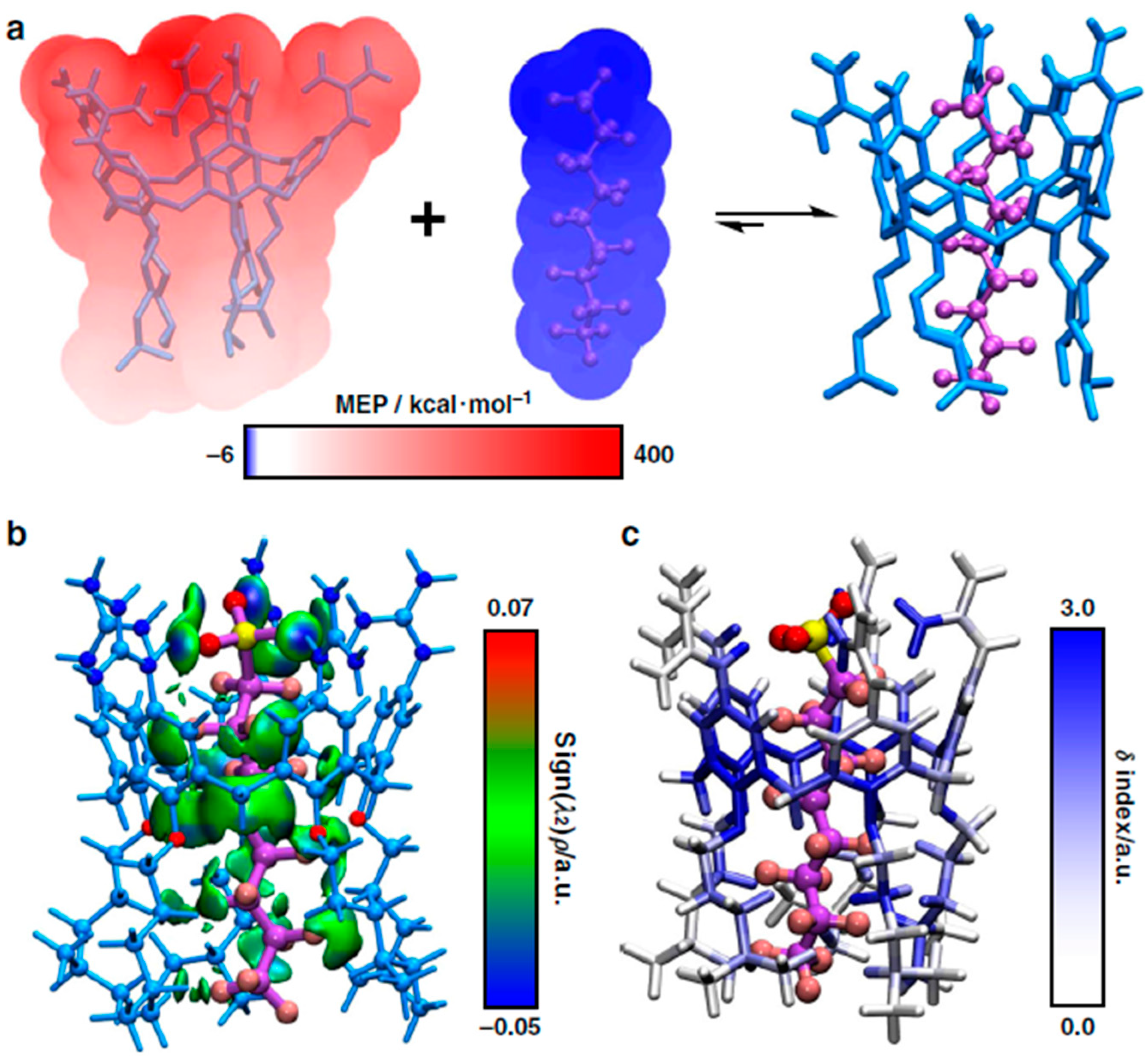
2.10. Metal–Organic Frameworks
2.11. Calixarenes
3. Conclusions
4. Future Directions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ehsan, M.N.; Riza, M.; Pervez, M.N.; Khyum, M.M.O.; Liang, Y.; Naddeo, V. Environmental and Health Impacts of PFAS: Sources, Distribution and Sustainable Management in North Carolina (USA). Sci. Total Environ. 2023, 878, 163123. [Google Scholar] [CrossRef]
- Chen, H.; He, P.; Rao, H.; Wang, F.; Liu, H.; Yao, J. Systematic Investigation of the Toxic Mechanism of PFOA and PFOS on Bovine Serum Albumin by Spectroscopic and Molecular Modeling. Chemosphere 2015, 129, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Londhe, K.; Chi, K.; Lee, C.; Venkatesan, A.K.; Hsiao, B.S. Functionalized Bio-Adsorbents for Removal of Perfluoroalkyl Substances: A Perspective. AWWA Water Sci. 2021, 3, e1258. [Google Scholar] [CrossRef]
- Gaines, L.G.T. Historical and Current Usage of Per- and Polyfluoroalkyl Substances (PFAS): A Literature Review. Am. J. Ind. Med. 2022, 66, 353–378. [Google Scholar] [CrossRef] [PubMed]
- da SFilho, A.H.; de Souza, G.L. Examining the Degradation of Environmentally-Daunting Per- and Poly-Fluoroalkyl Substances from a Fundamental Chemical Perspective. Phys. Chem. Chem. Phys. 2020, 22, 17659–17667. [Google Scholar] [CrossRef]
- Brunn, H.; Arnold, G.; Körner, W.; Rippen, G.; Steinhäuser, K.G.; Valentin, I. PFAS: Forever Chemicals—Persistent, Bioaccumulative and Mobile. Reviewing the Status and the Need for Their Phase out and Remediation of Contaminated Sites. Environ. Sci. Eur. 2023, 35, 20. [Google Scholar] [CrossRef]
- Jansen, K. “Forever Chemicals” No More? These Technologies Aim to Destroy PFAS in Water. Chemical & Engineering News. 25 March 2019. Available online: https://cen.acs.org/environment/persistent-pollutants/Forever-chemicals-technologies-aim-destroy/97/i12 (accessed on 12 October 2023).
- Domingo, J.L.; Nadal, M. Human Exposure to Per- and Polyfluoroalkyl Substances (PFAS) through Drinking Water: A Review of the Recent Scientific Literature. Environ. Res. 2019, 177, 108648. [Google Scholar] [CrossRef]
- Cerveny, D.; Grabic, R.; Fedorova, G.; Grabicova, K.; Turek, J.; Kodes, V.; Golovko, O.; Zlabek, V.; Randak, T. Perfluoroalkyl Substances in Aquatic Environment-Comparison of Fish and Passive Sampling Approaches. Environ. Res. 2016, 144, 92–98. [Google Scholar] [CrossRef]
- Long, L.; Hu, X.; Yan, J.; Zeng, Y.; Zhang, J.; Xue, Y. Novel Chitosan–Ethylene Glycol Hydrogel for the Removal of Aqueous Perfluorooctanoic Acid. J. Environ. Sci. 2019, 84, 21–28. [Google Scholar] [CrossRef]
- Krafft, M.P.; Riess, J.G. Per- and Polyfluorinated Substances (PFASs): Environmental Challenges. Curr. Opin. Colloid Interface Sci. 2015, 20, 192–212. [Google Scholar] [CrossRef]
- Lesmeister, L.; Lange, F.T.; Breuer, J.; Biegel-Engler, A.; Giese, E.; Scheurer, M. Extending the Knowledge about PFAS Bioaccumulation Factors for Agricultural Plants–A Review. Sci. Total Environ. 2021, 766, 142640. [Google Scholar] [CrossRef]
- Li, H.; Junker, A.L.; Wen, J.; Ahrens, L.; Sillanpää, M.; Tian, J.; Cui, F.; Vergeynst, L.; Wei, Z. A Recent Overview of Per- and Polyfluoroalkyl Substances (PFAS) Removal by Functional Framework Materials. Chem. Eng. J. 2023, 452, 139202. [Google Scholar] [CrossRef]
- He, C.; Yang, Y.; Hou, Y.-J.; Luan, T.; Deng, J. Chitosan-Coated Fluoro-Functionalized Covalent Organic Framework as Adsorbent for Efficient Removal of Per- and Polyfluoroalkyl Substances from Water. Sep. Purif. Technol. 2022, 294, 121195. [Google Scholar] [CrossRef]
- Lilienthal, H.; Dieter, H.H.; Hölzer, J.; Wilhelm, M. Recent Experimental Results of Effects of Perfluoroalkyl Substances in Laboratory Animals–Relation to Current Regulations and Guidance Values. Int. J. Hyg. Environ. Health 2017, 220, 766–775. [Google Scholar] [CrossRef]
- Bezerra, B.; Hewage, S.A.; Kewalramani, J.A.; van Duin, A.C.; Meegoda, J.N. A ReaxFF-Based Molecular Dynamics Study of the Destruction of PFAS Due to Ultrasound. Environ. Pollut. 2023, 333, 122026. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xing, X.; Xue, M.; Bai, S.; Li, P.; Li, C.; Xia, T. Insights into Heteroaggregation of Polystyrene Nanoplastics with Hematite Nanoparticles and Configuration-Dependent Adsorption for PFOA and PFOS. Colloids Surf. A Physicochem. Eng. Asp. 2022, 649, 129467. [Google Scholar] [CrossRef]
- Lei, X.; Yao, L.; Lian, Q.; Zhang, X.; Wang, T.; Holmes, W.; Ding, G.; Gang, D.D.; Zappi, M.E. Enhanced Adsorption of Perfluorooctanoate (PFOA) onto Low Oxygen Content Ordered Mesoporous Carbon (OMC): Adsorption Behaviors and Mechanisms. J. Hazard. Mater. 2022, 421, 126810. [Google Scholar] [CrossRef]
- Van den Bergh, M.; Krajnc, A.; Voorspoels, S.; Tavares, S.F.; Mullens, S.; Beurroies, I.; Maurin, G.; Mali, G.; De Vos, D.E. Highly Selective Removal of Perfluorinated Contaminants by Adsorption on All-Silica Zeolite Beta. Angew. Chem. 2020, 59, 14086–14090. [Google Scholar] [CrossRef]
- Zenobio, J.E.; Salawu, O.A.; Han, Z.; Adeleye, A.S. Adsorption of Per- and Polyfluoroalkyl Substances (PFAS) to Containers. J. Hazard. Mater. Adv. 2022, 7, 100130. [Google Scholar] [CrossRef]
- Ezazi, M.; Shrestha, B.; Kwon, G. Lower Critical Solution Temperature-Driven Catch and Release of Perfluoroalkyl Substances from Water: Remediation and Sampling. ACS Appl. Polym. Mater. 2021, 3, 4139–4146. [Google Scholar] [CrossRef]
- Guo, H.; Liu, Y.; Ma, W.; Liu, N.; Li, K.; Lin, S. Surface Molecular Imprinting on Carbon Microspheres for Fast and Selective Adsorption of Perfluorooctane Sulfonate. J. Hazard. Mater. 2018, 348, 29–38. [Google Scholar] [CrossRef]
- Stebel, E.K.; Pike, K.A.; Nguyen, H.; Hartmann, H.A.; Klonowski, M.J.; Lawrence, M.G.; Collins, R.M.; Hefner, C.E.; Edmiston, P.L. Absorption of Short-Chain to Long-Chain Perfluoroalkyl Substances Using Swellable Organically Modified Silica. Environ. Sci. Water Res. Technol. 2019, 5, 1854–1866. [Google Scholar] [CrossRef]
- Zhang, S.; Vijayavenkataraman, S.; Lu, W.F.; Fuh, J.Y.H. A Review on the Use of Computational Methods to Characterize, Design, and Optimize Tissue Engineering Scaffolds, with a Potential in 3D Printing Fabrication. J. Biomed. Mater. Res. Part B Appl. Biomater. 2018, 107, 1329–1351. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Yu, Y.; Geng, S.; Liu, B.; Hao, Y.; Liang, G. Computational Methods for the Interaction between Cyclodextrins and Natural Compounds: Technology, Benefits, Limitations, and Trends. J. Agric. Food Chem. 2022, 70, 2466–2482. [Google Scholar] [CrossRef] [PubMed]
- Cramer, C.J. Essentials of Computational Chemistry: Theories and Models; Wiley: Chichester, UK; Hoboken, NJ, USA, 2004. [Google Scholar]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.; Liao, H.; Hu, W. Assessment of van Der Waals Inclusive Density Functional Theory Methods for Adsorption and Selective Dehydrogenation of Formic Acid on Pt(111) Surface. Phys. Chem. Chem. Phys. 2019, 21, 21049–21056. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, Z.; Zhong, W.; Liang, T. Investigating Chlorination and Migration Mechanism of NaCl on C−S−H: DFT and AIMD Study. ChemistrySelect 2023, 8, e202300911. [Google Scholar] [CrossRef]
- Grimme, S.; Hansen, A.; Brandenburg, J.G.; Bannwarth, C. Dispersion-Corrected Mean-Field Electronic Structure Methods. Chem. Rev. 2016, 116, 5105–5154. [Google Scholar] [CrossRef]
- Jurečka, P.; Černý, J.; Hobza, P.; Salahub, D.R. Density Functional Theory Augmented with an Empirical Dispersion Term. Interaction Energies and Geometries of 80 Noncovalent Complexes Compared with Ab Initio Quantum Mechanics Calculations. J. Comput. Chem. 2007, 28, 555–569. [Google Scholar] [CrossRef]
- Hermann, J.; DiStasio, R.A.; Tkatchenko, A. First-Principles Models for van Der Waals Interactions in Molecules and Materials: Concepts, Theory, and Applications. Chem. Rev. 2017, 117, 4714–4758. [Google Scholar] [CrossRef]
- van Mourik, T.; Bühl, M.; Gaigeot, M.-P. Density Functional Theory across Chemistry, Physics and Biology. Philos. Trans. Ser. A Math. Phys. Eng. Sci. 2014, 372, 20120488. [Google Scholar] [CrossRef]
- Loverde, S.M.; Klein, M.L.; Discher, D.E. Nanoparticle Shape Improves Delivery: Rational Coarse Grain Molecular Dynamics (RCG-MD) of Taxol in Worm-like PEG-PCL Micelles. Adv. Mater. 2011, 24, 3823–3830. [Google Scholar] [CrossRef]
- Sadeghi, M.S.; Moghbeli, M.R.; Goddard, W.A. A Coarse-Grain Force Field Based on Quantum Mechanics (CGq FF) for Molecular Dynamics Simulation of Poly(Ethylene Glycol)-Block-Poly(ε-Caprolactone) (PEG-b-PCL) Micelles. Phys. Chem. Chem. Phys. 2020, 22, 24028–24040. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, K.R.; Koo, B.; Khafagy, K.H.; Chattopadhyay, A. Multiscale Modeling of Carbon Nanotube-Reinforced Polymer with Coarse-Grain Molecular Dynamics Informed Morphology. Compos. Sci. Technol. 2022, 223, 109412. [Google Scholar] [CrossRef]
- Kuo, A.-T.; Okazaki, S.; Shinoda, W. Transferable Coarse-Grained Model for Perfluorosulfonic Acid Polymer Membranes. J. Chem. Phys. 2017, 147, 094904. [Google Scholar] [CrossRef]
- Kuo, A.-T.; Urata, S.; Nakabayashi, K.; Watabe, H.; Honmura, S. Coarse-Grained Molecular Dynamics Simulation of Perfluorosulfonic Acid Polymer in Water–Ethanol Mixtures. Macromolecules 2021, 54, 609–620. [Google Scholar] [CrossRef]
- Tarokh, A.; Karan, K.; Ponnurangam, S. Atomistic MD Study of Nafion Dispersions: Role of Solvent and Counterion in the Aggregate Structure, Ionic Clustering, and Acid Dissociation. Macromolecules 2019, 53, 288–301. [Google Scholar] [CrossRef]
- Feng, H.; Zhang, H.; Cao, H.; Sun, Y.; Zhang, A.; Fu, J. Application of a Novel Coarse-Grained Soil Organic Matter Model in the Environment. Environ. Sci. Technol. 2018, 52, 14228–14234. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, L.; Moldabekov, Z.A.; Shao, X.; Jiang, K.; Dornheim, T.; Pavanello, M.; Cangi, A. Accelerating Equilibration in First-Principles Molecular Dynamics with Orbital-Free Density Functional Theory. Phys. Rev. Res. 2022, 4, 043033. [Google Scholar] [CrossRef]
- da Silva, A.J.R.; Cheng, H.-Y.; Gibson, D.A.; Sorge, K.L.; Liu, Z.; Carter, E.A. Limitations of Ab Initio Molecular Dynamics Simulations of Simple Reactions: F + H2 as a Prototype. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 1997, 53, 1285–1299. [Google Scholar] [CrossRef]
- Kongsted, J.; Osted, A.; Mikkelsen, K.V.; Åstrand, P.-O.; Christiansen, O. Solvent Effects on the N→π* Electronic Transition in Formaldehyde: A Combined Coupled Cluster/Molecular Dynamics Study. J. Chem. Phys. 2004, 121, 8435. [Google Scholar] [CrossRef]
- Steele, R.P. Communication: Multiple-Timestep Ab Initio Molecular Dynamics with Electron Correlation. J. Chem. Phys. 2013, 139, 011102. [Google Scholar] [CrossRef] [PubMed]
- Bussai, C.; Fritzsche, S.; Haberlandt, R.; Hannongbua, S. Concentration Dependence of the Methane Structure in Silicalite-1: A Molecular Dynamics Study Using the Møller−Plesset-Based Potential. Langmuir 2005, 21, 5847–5851. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Fan, C.; Cheng, L.; Shan, Y. Adsorption of Perfluoroalkyl Substances on Deep Eutectic Solvent-Based Amorphous Metal-Organic Framework: Structure and Mechanism. Environ. Res. 2024, 248, 118261. [Google Scholar] [CrossRef] [PubMed]
- Ganji, M.D.; Mirzaei, S.; Dalirandeh, Z. Molecular Origin of Drug Release by Water Boiling inside Carbon Nanotubes from Reactive Molecular Dynamics Simulation and DFT Perspectives. Sci. Rep. 2017, 7, 4669. [Google Scholar] [CrossRef]
- Yan, B.; Wang, J.; Liu, J. STXM-XANES and Computational Investigations of Adsorption of Per- and Polyfluoroalkyl Substances on Modified Clay. Water Res. 2021, 201, 117371. [Google Scholar] [CrossRef]
- Saha, S.K.; Hens, A.; Murmu, N.C.; Banerjee, P. A Comparative Density Functional Theory and Molecular Dynamics Simulation Studies of the Corrosion Inhibitory Action of Two Novel N-Heterocyclic Organic Compounds along with a Few Others over Steel Surface. J. Mol. Liq. 2016, 215, 486–495. [Google Scholar] [CrossRef]
- Gar Alalm, M.; Boffito, D.C. Mechanisms and Pathways of PFAS Degradation by Advanced Oxidation and Reduction Processes: A Critical Review. Chem. Eng. J. 2022, 450, 138352. [Google Scholar] [CrossRef]
- Peritore, A.F.; Gugliandolo, E.; Cuzzocrea, S.; Crupi, R.; Britti, D. Current Review of Increasing Animal Health Threat of Per- and Polyfluoroalkyl Substances (PFAS): Harms, Limitations, and Alternatives to Manage Their Toxicity. Int. J. Mol. Sci. 2023, 24, 11707. [Google Scholar] [CrossRef] [PubMed]
- EPA Regulation Targets Phased-Out PFAS. Chemical & Engineering News. Available online: https://cen.acs.org/environment/persistent-pollutants/EPA-regulation-targets-phasedPFAS/101/i5?utm_source=LatestNews&utm_medium=LatestNews&utm_campaign=CENRSS (accessed on 12 October 2023).
- Buck, R.C.; Korzeniowski, S.H.; Laganis, E.; Adamsky, F. Identification and Classification of Commercially Relevant Per- and Poly-Fluoroalkyl Substances (PFAS). Integr. Environ. Assess. Manag. 2021, 17, 1045–1055. [Google Scholar] [CrossRef] [PubMed]
- Saha, D.; Khan, S.; Van Bramer, S.E. Can Porous Carbons Be a Remedy for PFAS Pollution in Water? A Perspective. J. Environ. Chem. Eng. 2021, 9, 106665. [Google Scholar] [CrossRef]
- Park, M.; Daniels, K.D.; Wu, S.; Ziska, A.D.; Snyder, S.A. Magnetic Ion-Exchange (MIEX) Resin for Perfluorinated Alkylsubstance (PFAS) Removal in Groundwater: Roles of Atomic Charges for Adsorption. Water Res. 2020, 181, 115897. [Google Scholar] [CrossRef] [PubMed]
- He, A.; Lu, Y.; Chen, F.; Li, F.; Lv, K.; Cao, H.; Sun, Y.; Liang, Y.; Li, J.; Zhao, L.; et al. Exploring the Origin of Efficient Adsorption of Poly- and Perfluoroalkyl Substances in Household Point-of-Use Water Purifiers: Deep Insights from a Joint Experimental and Computational Study. Sci. Total Environ. 2022, 831, 154988. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Wang, X.; Jiang, Z.; Zhang, H.; Yuan, S. Contribution of Air-Water Interface in Removing PFAS from Drinking Water: Adsorption, Stability, Interaction and Machine Learning Studies. Water Res. 2023, 236, 119947. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Qiao, M.; Zhao, H.; Ran, Q.; Yuan, S. Effect of Sodium Alkyl Sulfate Chain Length on Foam Stability: A Molecular Dynamics Study. Colloids Surf. A Physicochem. Eng. Asp. 2023, 656, 130394. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, W.; Yu, G.; Deng, S. Contribution of Nanobubbles for PFAS Adsorption on Graphene and OH- and NH2-Functionalized Graphene: Comparing Simulations with Experimental Results. Environ. Sci. Technol. 2021, 55, 13254–13263. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, J.A.R.; Bourg, I.C. Molecular Dynamics Simulation of the Adsorption of Per- and Polyfluoroalkyl Substances (PFASs) on Smectite Clay. J. Colloid Interface Sci. 2021, 585, 337–346. [Google Scholar] [CrossRef]
- He, G.; Zhang, M.; Zhou, Q.; Pan, G. Molecular Dynamics Simulations of Structural Transformation of Perfluorooctane Sulfonate (PFOS) at Water/Rutile Interfaces. Chemosphere 2015, 134, 272–278. [Google Scholar] [CrossRef]
- He, G.; Pan, G.; Zhang, M. Assembling Structures and Dynamics Properties of Perfluorooctane Sulfonate (PFOS) at Water–Titanium Oxide Interfaces. J. Colloid Interface Sci. 2013, 405, 189–194. [Google Scholar] [CrossRef]
- Liu, W.; Lin, T.; Zhang, X.; Jiang, F.; Yan, X.; Chen, H. Adsorption of Perfluoroalkyl Acids on Granular Activated Carbon Supported Chitosan: Role of Nanobubbles. Chemosphere 2022, 309, 136733. [Google Scholar] [CrossRef]
- Zhang, Q.; Deng, S.; Yu, G.; Huang, J. Removal of Perfluorooctane Sulfonate from Aqueous Solution by Crosslinked Chitosan Beads: Sorption Kinetics and Uptake Mechanism. Bioresour. Technol. 2011, 102, 2265–2271. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Kancharla, S.; Hooper, J.B.; Tsianou, M.; Bedrov, D.; Alexandridis, P. Controlling the Self-Assembly of Perfluorinated Surfactants in Aqueous Environments. Phys. Chem. Chem. Phys. 2021, 23, 10029–10039. [Google Scholar] [CrossRef] [PubMed]
- Kancharla, S.; Dong, D.; Bedrov, D.; Tsianou, M.; Alexandridis, P. Structure and Interactions in Perfluorooctanoate Micellar Solutions Revealed by Small-Angle Neutron Scattering and Molecular Dynamics Simulations Studies: Effect of Urea. Langmuir 2021, 37, 5339–5347. [Google Scholar] [CrossRef] [PubMed]
- Luft, C.M.; Schutt, T.C.; Shukla, M.K. Properties and Mechanisms for PFAS Adsorption to Aqueous Clay and Humic Soil Components. Environ. Sci. Technol. 2022, 56, 10053–10061. [Google Scholar] [CrossRef] [PubMed]
- Loganathan, N.; Wilson, A.K. Adsorption, Structure, and Dynamics of Short- and Long-Chain PFAS Molecules in Kaolinite: Molecular-Level Insights. Environ. Sci. Technol. 2022, 56, 8043–8052. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tan, X.; Lü, R.; Tang, Y.; Qie, H.; Huang, Z.; Zhao, J.; Cui, J.; Yang, W.; Lin, A. Enhanced Removal of Polyfluoroalkyl Substances by Simple Modified Biochar: Adsorption Performance and Theoretical Calculation. ACS ES&T Water 2023, 3, 817–826. [Google Scholar] [CrossRef]
- Yang, Y.; Zheng, Z.; Ji, W.; Xu, J.; Zhang, X. Insights to Perfluorooctanoic Acid Adsorption Micro-Mechanism over Fe-Based Metal Organic Frameworks: Combining Computational Calculation with Response Surface Methodology. J. Hazard. Mater. 2020, 395, 122686. [Google Scholar] [CrossRef]
- Erkal, T.S.; Shamsuddin, N.; Kırmızıaltın, S.; Yazaydın, A.Ö. Computational Investigation of Structure–Function Relationship in Fluorine-Functionalized MOFs for PFOA Capture from Water. J. Phys. Chem. C 2023, 127, 3204–3216. [Google Scholar] [CrossRef]
- Zheng, Z.; Yu, H.; Geng, W.-C.; Hu, X.-Y.; Wang, Y.-Y.; Li, Z.; Wang, Y.; Guo, D.-S. Guanidinocalix[5]Arene for Sensitive Fluorescence Detection and Magnetic Removal of Perfluorinated Pollutants. Nat. Commun. 2019, 10, 5762. [Google Scholar] [CrossRef]


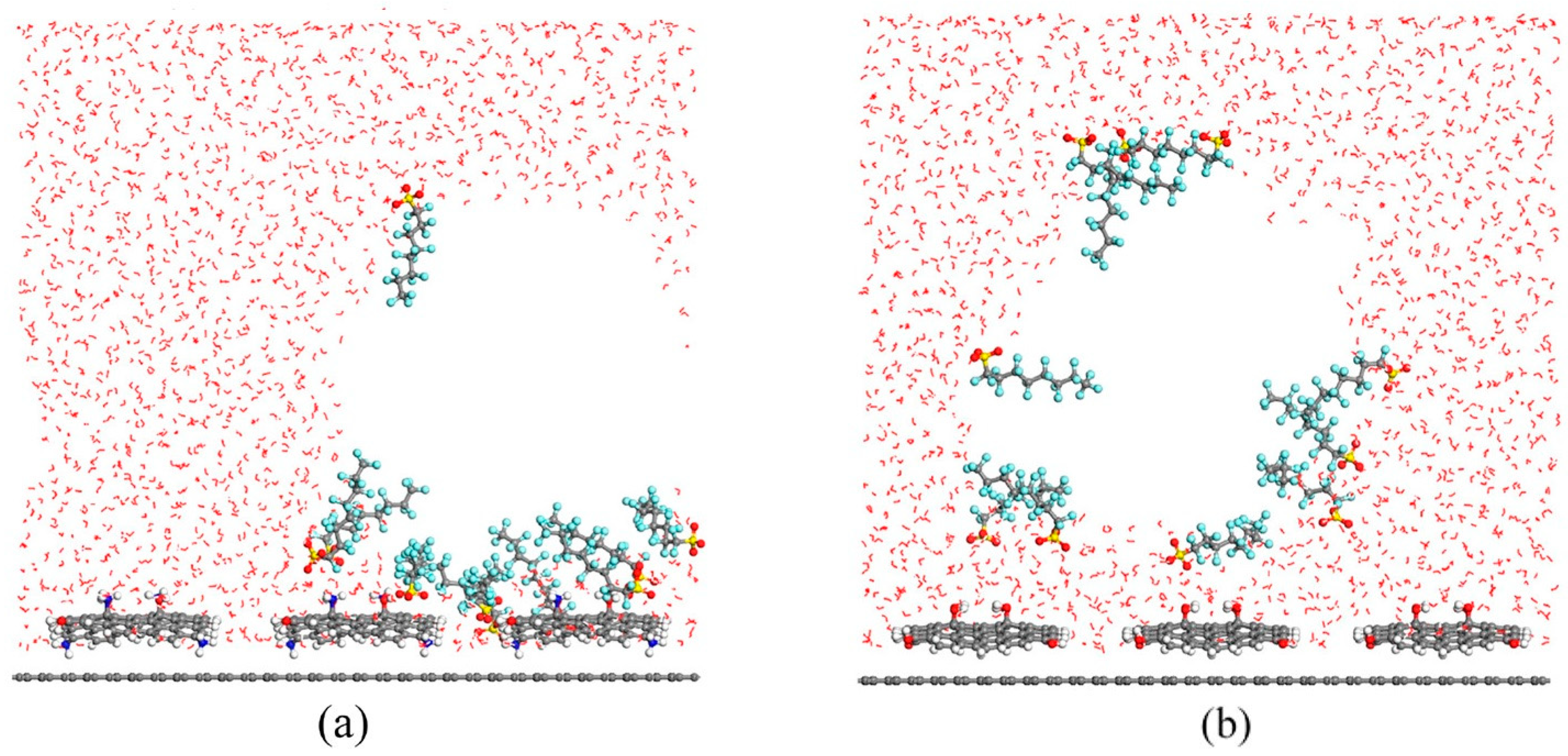
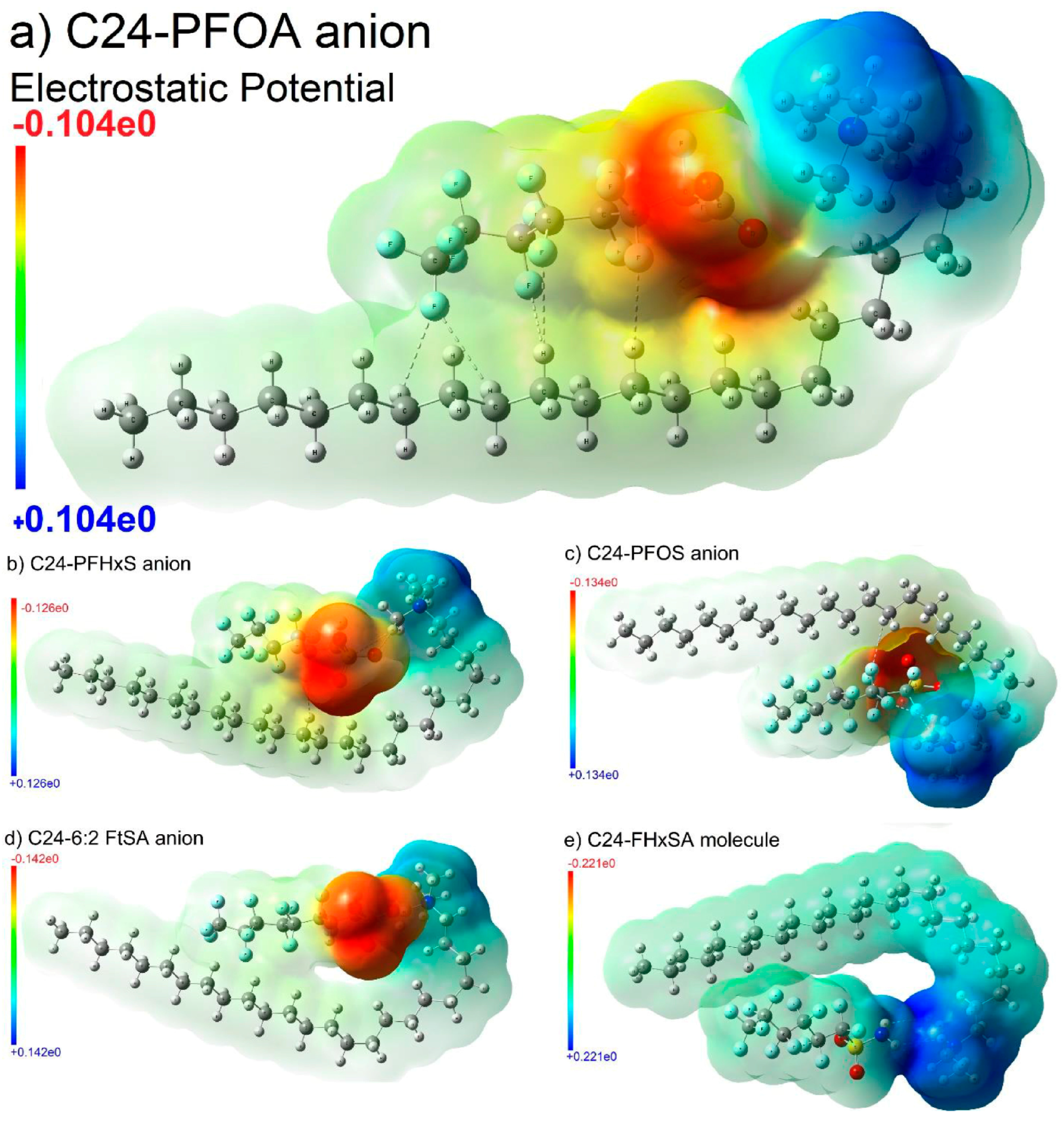
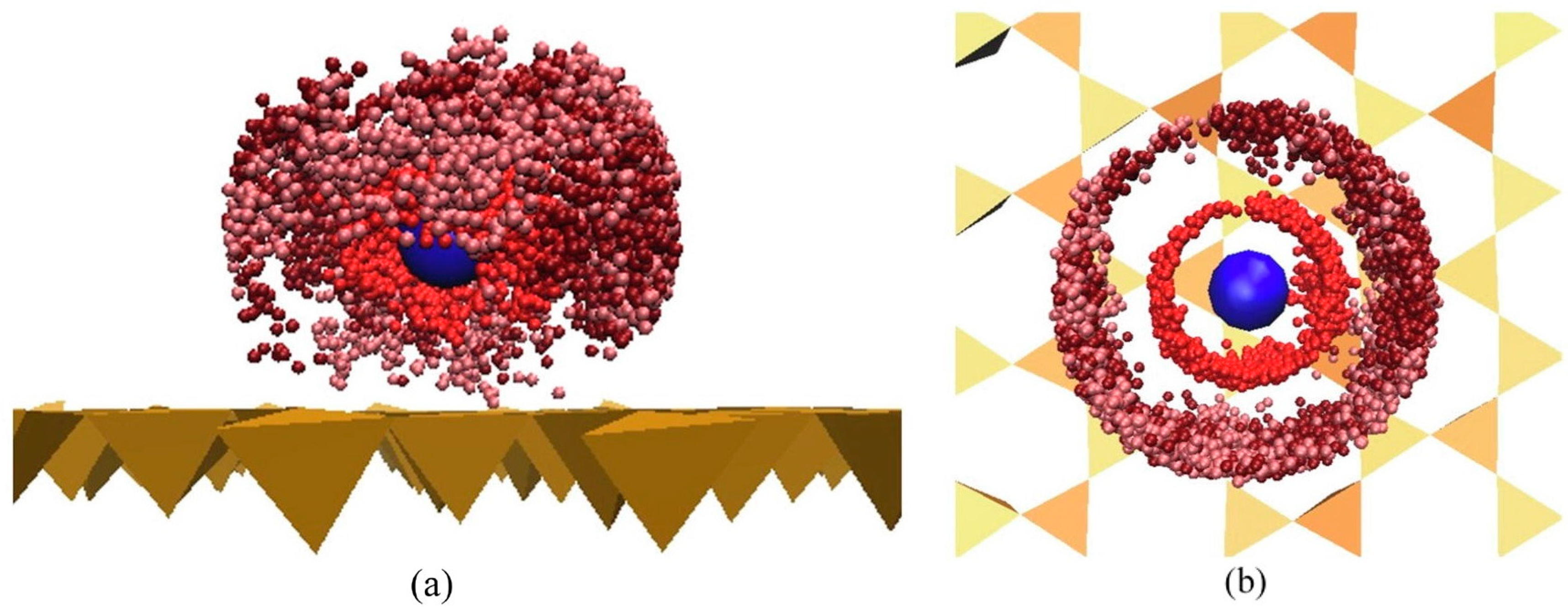
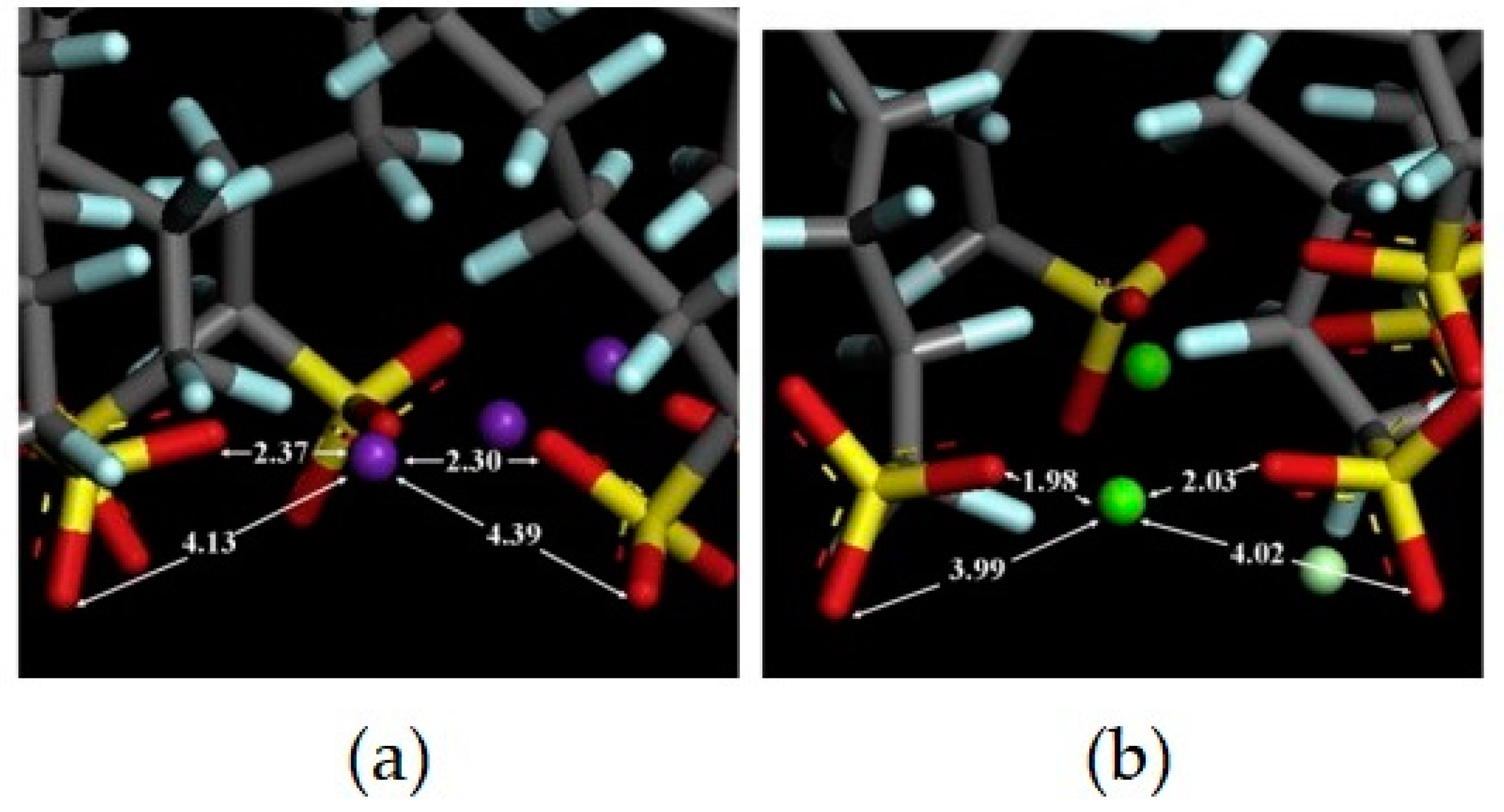

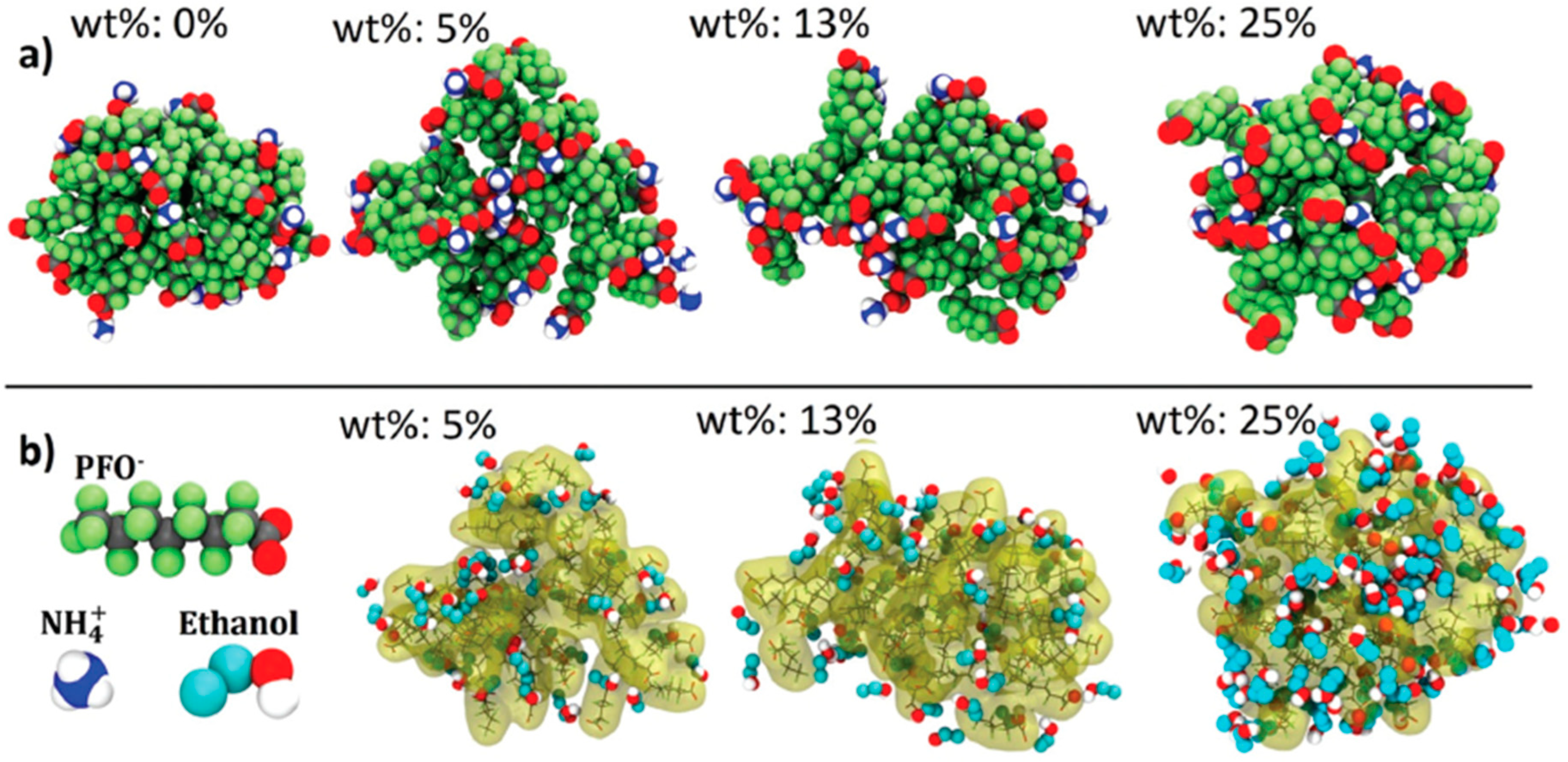
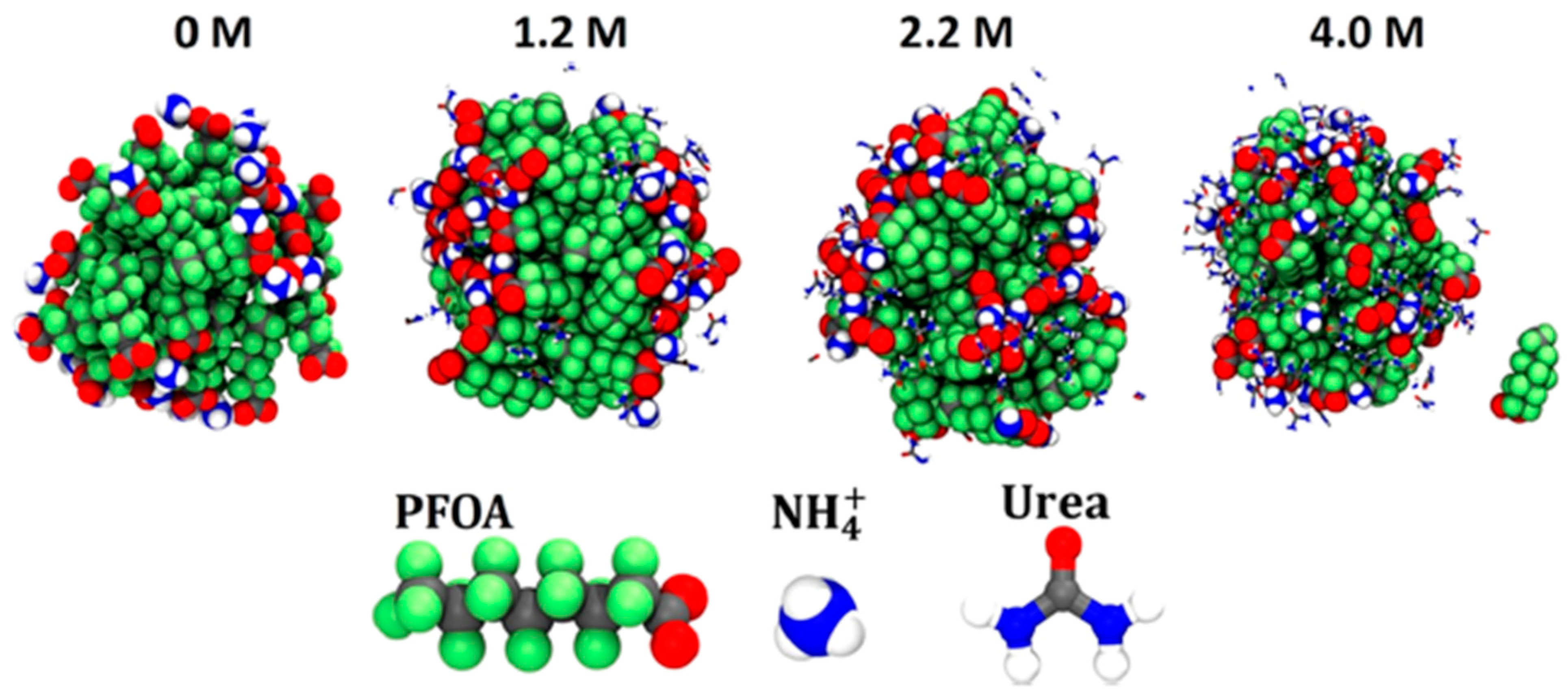
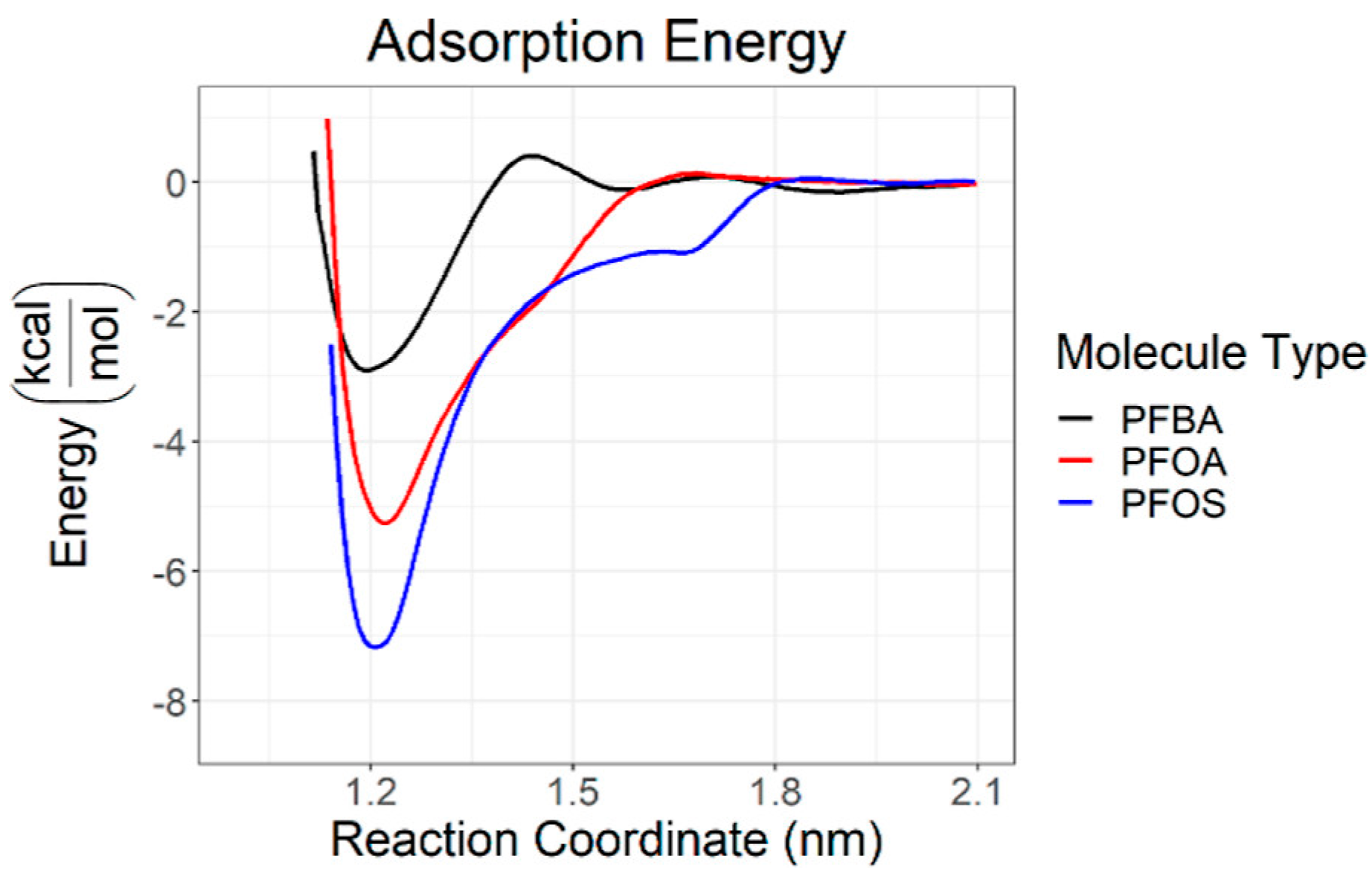

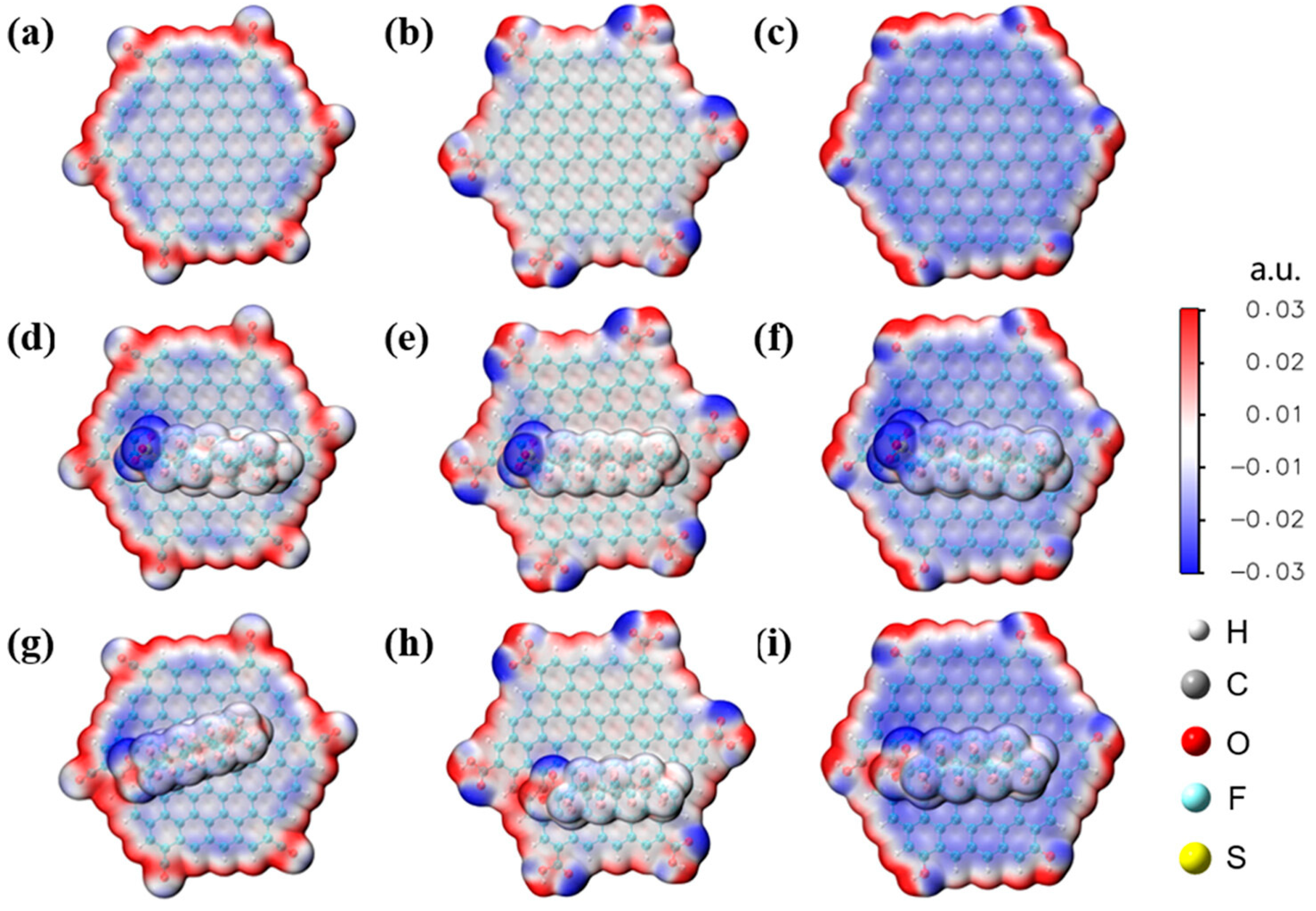
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minervino, A.; Belfield, K.D. Review of Recent Computational Research on the Adsorption of PFASs with a Variety of Substrates. Int. J. Mol. Sci. 2024, 25, 3445. https://doi.org/10.3390/ijms25063445
Minervino A, Belfield KD. Review of Recent Computational Research on the Adsorption of PFASs with a Variety of Substrates. International Journal of Molecular Sciences. 2024; 25(6):3445. https://doi.org/10.3390/ijms25063445
Chicago/Turabian StyleMinervino, Alfonso, and Kevin D. Belfield. 2024. "Review of Recent Computational Research on the Adsorption of PFASs with a Variety of Substrates" International Journal of Molecular Sciences 25, no. 6: 3445. https://doi.org/10.3390/ijms25063445
APA StyleMinervino, A., & Belfield, K. D. (2024). Review of Recent Computational Research on the Adsorption of PFASs with a Variety of Substrates. International Journal of Molecular Sciences, 25(6), 3445. https://doi.org/10.3390/ijms25063445