
Unearthing a Cryptic Biosynthetic Gene Cluster for the Piperazic Acid-Bearing Depsipeptide Diperamycin in the Ant-Dweller Streptomyces sp. CS113

 ,
,  ,
,  , ,
, ,  , ,
, ,  and
and
Abstract
1. Introduction
2. Results and Discussion
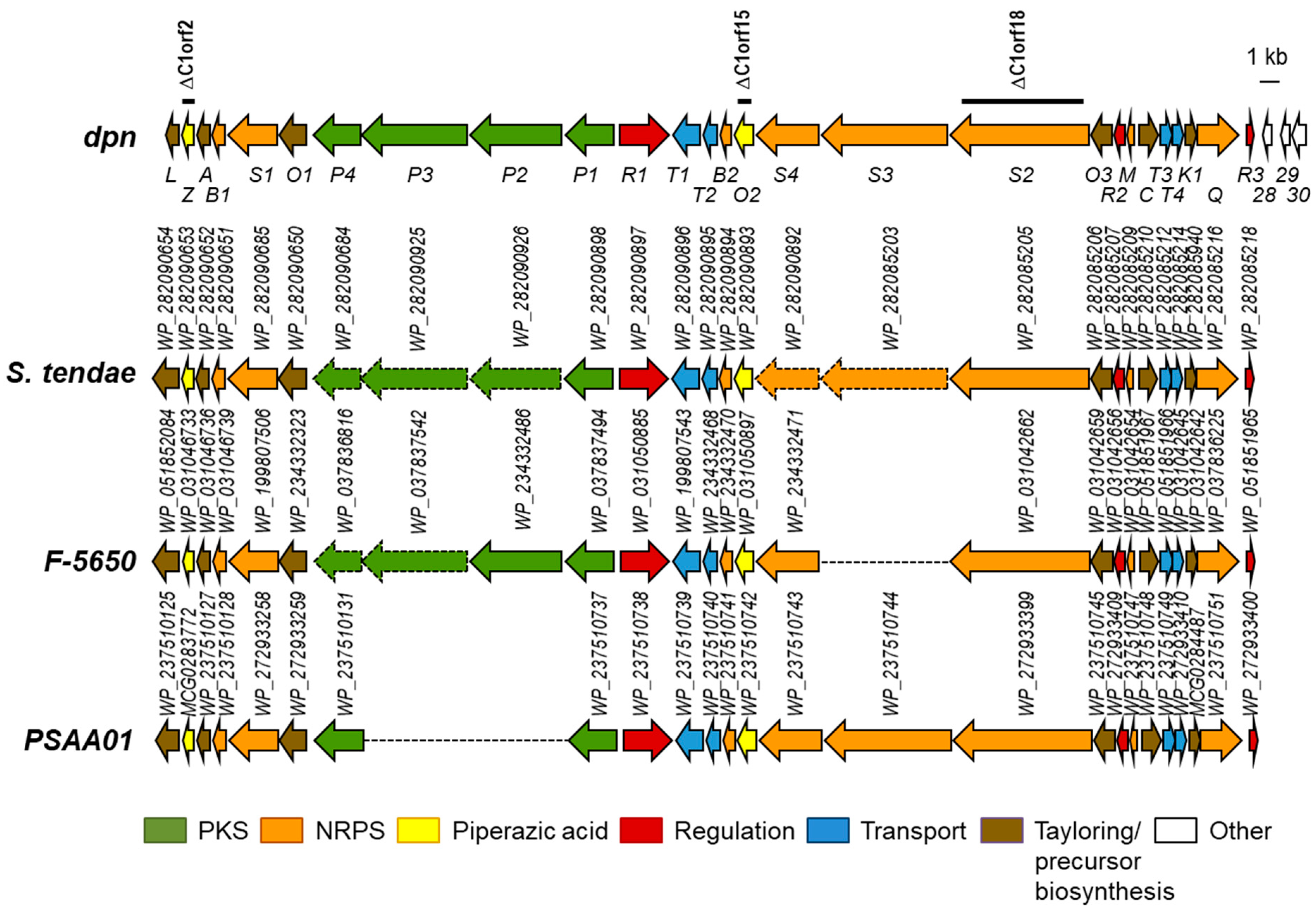
2.1. Identification of the Dpn Biosynthetic Gene Cluster
2.2. Monitoring Expression of the Dpn Biosynthetic Gene Cluster in Different Culture Media
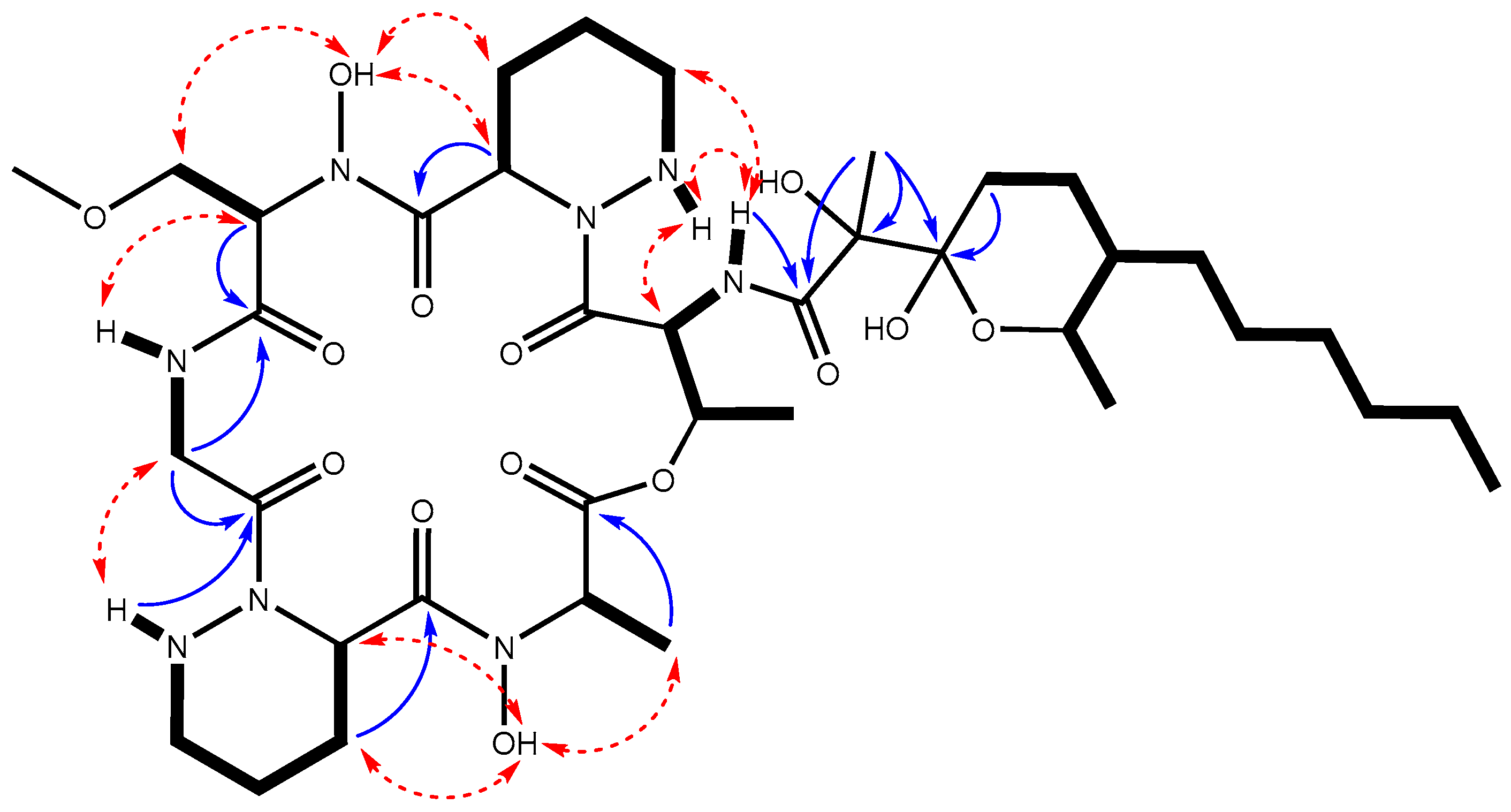
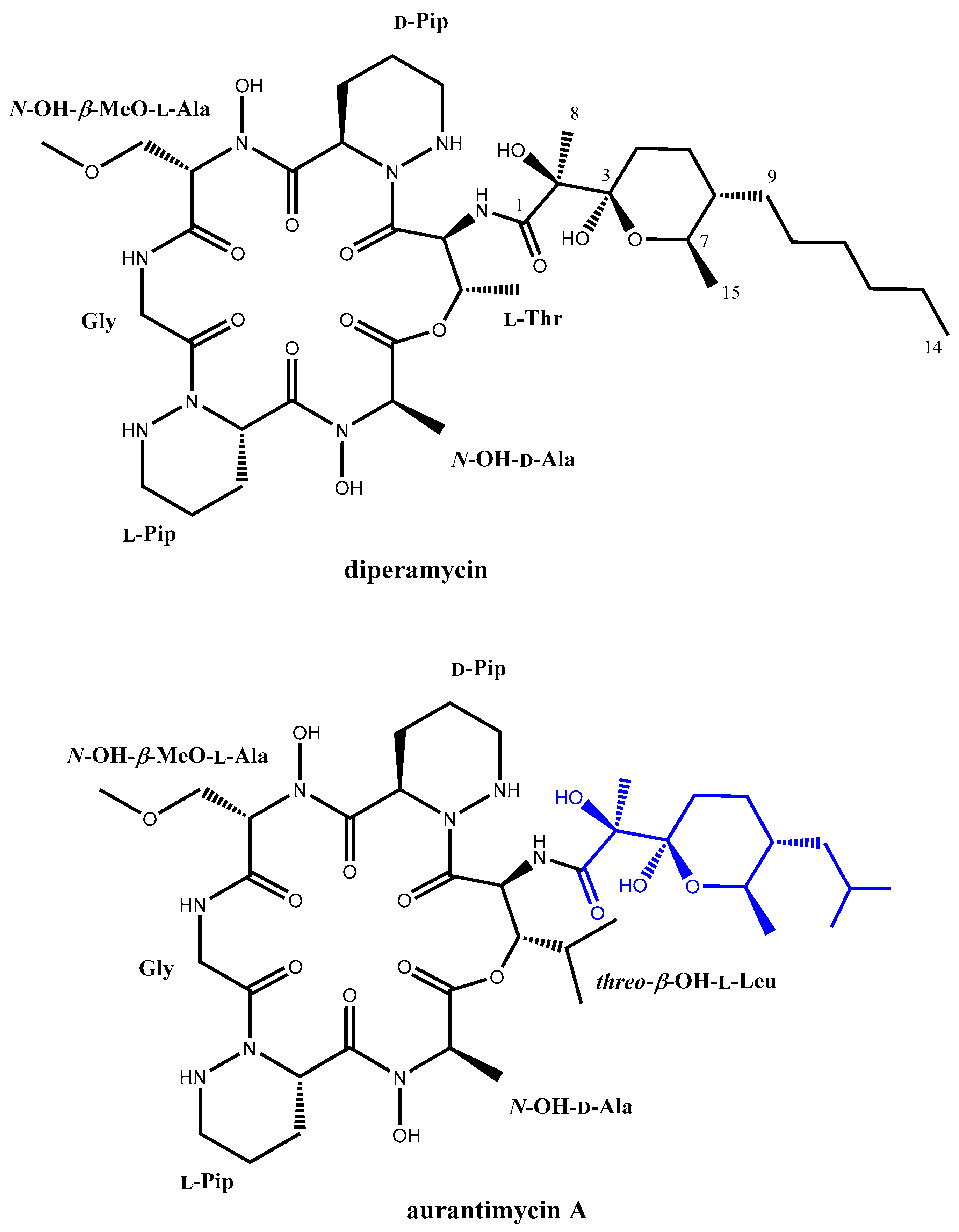
2.3. Identification and Structural Elucidation of the Dpn-Encoded Compound
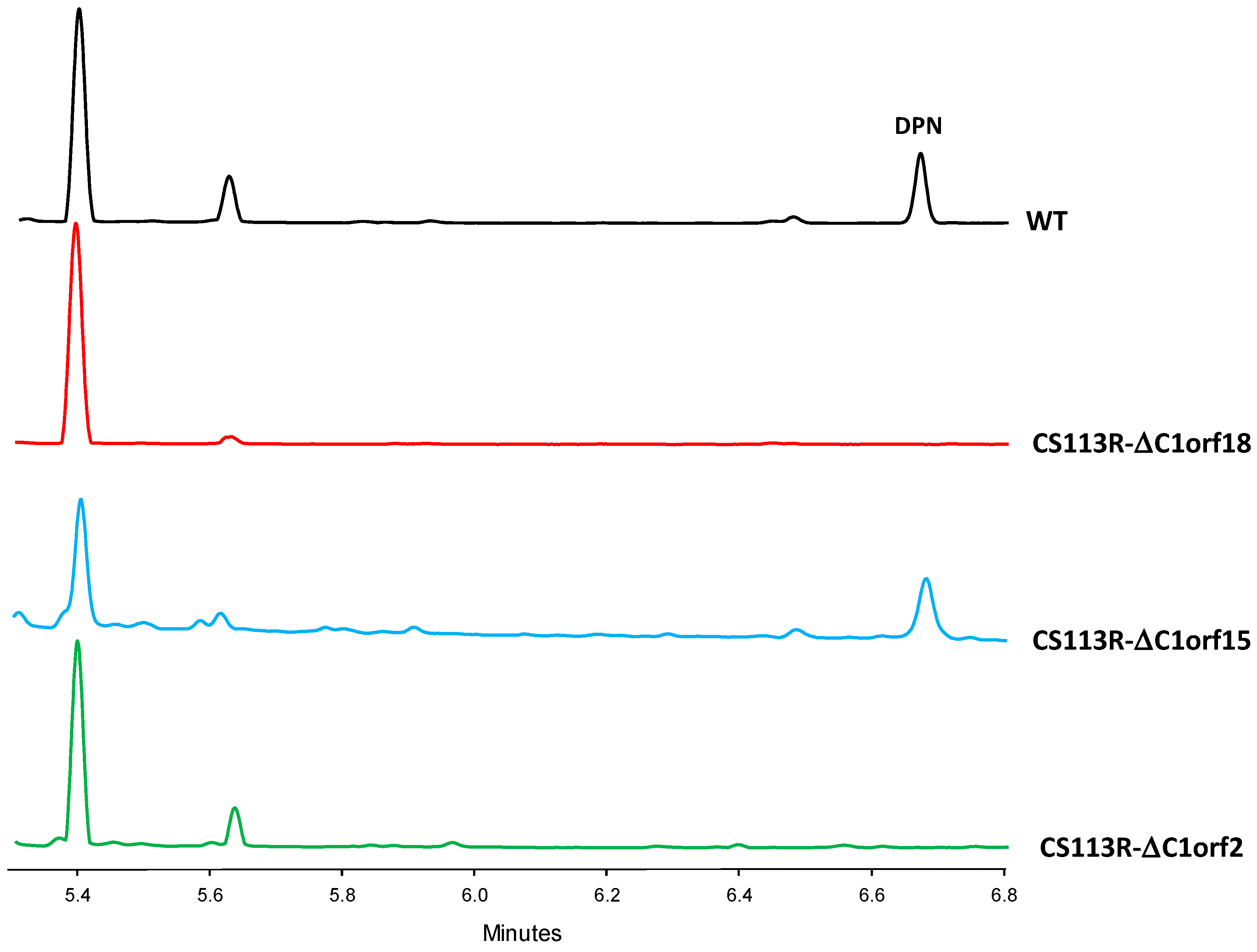
2.4. Involvement of Piperazic Acid Biosynthesis Genes in Production of Diperamycin
- Regarding the mutant in the ornithine monooxygenase-encoding gene dpnO2, the mutant CS113R-ΔC1orf15 was generated (Table 1) using pHZ-CS113-C1orf15. Its genotype was confirmed using oligonucleotides 113C1orf15-comp-F and 113C1orf15-comp-R (Table S2), which amplified a 1.5 kb DNA fragment from the mutant while also amplifying a 1.4 kb fragment from the wild type strain, confirming the deletion of dpnO2 in CS113R-ΔC1orf15 and its replacement by the apramycin resistance gene (Figure S10). The UPLC analysis of culture extracts from the resultant mutant CS113R-ΔC1orf15 (Table 1) revealed that it still produced DPN (Figure 3). This fact suggested that either the putative ornithine hydroxylase DpnO2 was not required for the production of DPN or, alternatively, that there was an additional copy of an ornithine hydroxylase-encoding gene in the CS113R genome. The existence of additional genes encoding ornithine hydroxylase has been described in other Streptomyces genomes [7]. In accordance to this, a search for additional ornithine hydroxylase-encoding genes was carried out in the CS113 genome. This resulted in the identification of an additional ornithine hydroxylase gene (WP_087804462) coding for a protein that shares 51% identical amino acids with DpnO2. The existance of this gene would explain why the CS113R-ΔC1orf15 mutant still produced DPN.
- Regarding the mutant in the piperazate synthase-encoding gene dpnZ, this gene was delated using plasmid pHZ-CS113-C1orf2 (Table 1). The resultant mutant CS113R-ΔC1orf2 was confirmed using oligonucleotides CS113C1orf2-comp-F and 113C1orf2-comp-R (Table S2), which amplified a 1.5 kb DNA fragment from the mutant strain and a 1 kb fragment from the wild type strain, confirming the replacement of dpnZ by the apramycin resistance cassette (Figure S11). The resultant mutant strain CS113R-ΔC1orf2 (Table 1) was cultivated and its metabolite profile was analyzed by UPLC. As observed in Figure 3, this mutant did not produce DPN, confirming that dpnZ was essential for DPN biosynthesis as a piperazate synthase-encoding gene, and additionally confirming that DPN is a piperazate-bearing compound. The production of DPN was recovered in CS113R-ΔC1orf2 when dpnZ was expressed in trans using pSETETc-C1orf2 (Table 1; Figure S12). These and the above-mentioned results confirmed the involvement of dpn BGC in the biosynthesis of DPN.
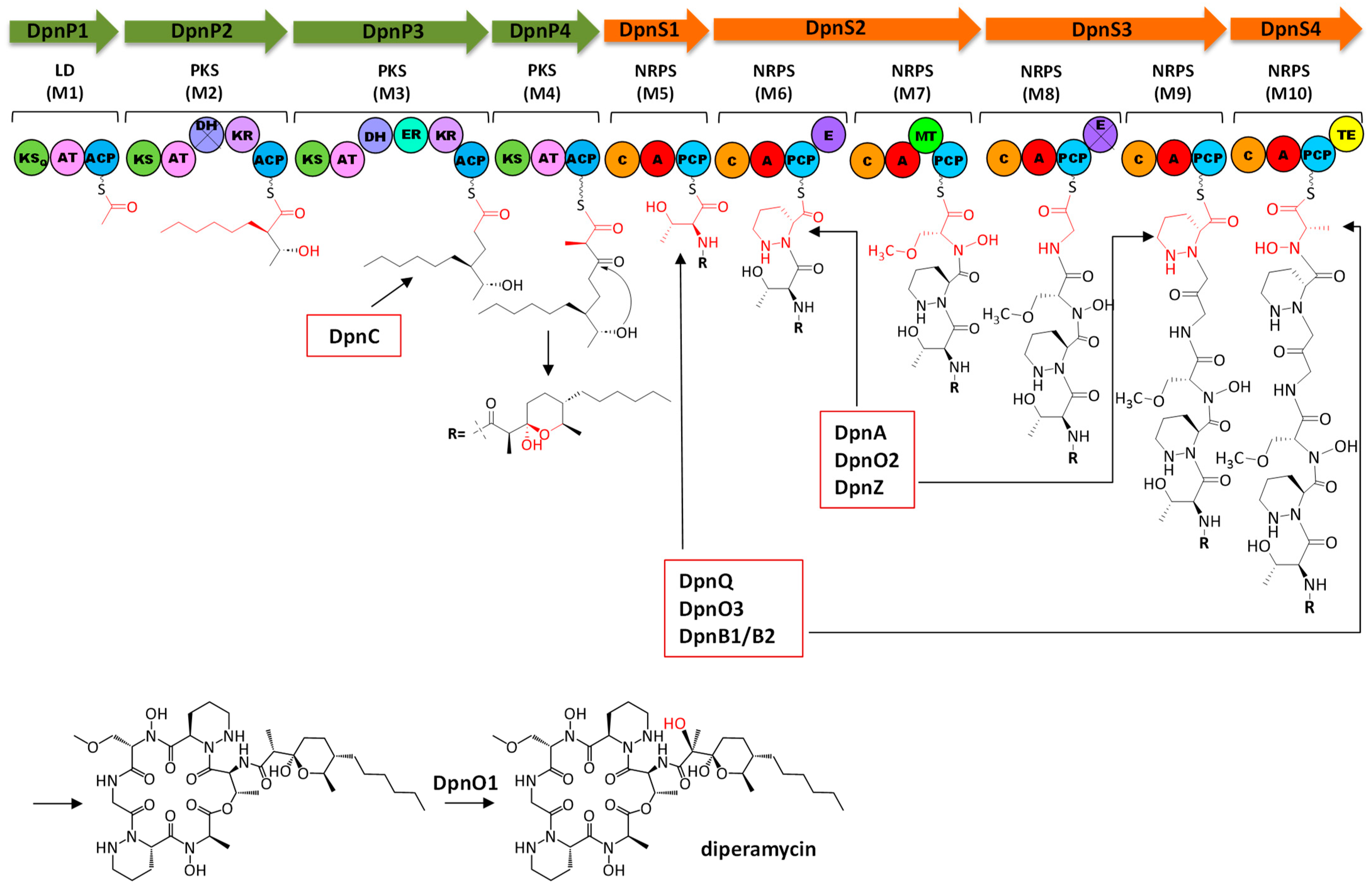
2.5. Analysis of Diperamycin Biosynthetic Gene Cluster and Proposed Biosynthesis Pathway
3. Materials and Methods
3.1. Strains and Culture Conditions, Plasmids and DNA Manipulations
3.2. DNA Manipulations and Plasmid Vectors
3.3. Construction of pOJ260ind-C1
3.4. Construction of pSETETc-C1orf2 by Gibson Assembly
3.5. Generation of Dpn Mutants
- pHZ-CS113-C1orf2. This plasmid was used to delete dpnZ, generating the mutant CS113R-ΔC1orf2. Fragment A (1.16 kb), containing dpnL and the 3′-end of dpnZ, was amplified using the oligonucleotides 113C1orf2A-F and 113C1orf2-R (Table S2). Fragment B (1.56 kb), containing the 5′-end of dpnZ, dpnA and the 3′-end of dpnB1, was amplified using the oligonucleotides 113C1orf2B-F and 113C1orf2B-R (Table S2).
- pHZ-CS113-C1orf15. This plasmid was used to delete dpnO2, generating the mutant CS113R-ΔC1orf15. Fragment A (1.84 kb), containing dpnT2, dpnB2 and the 3′-end of dpnO2, was amplified using the oligonucleotides 113C1orf15A-F and 113C1orf15A-R (Table S2). Fragment B (1.85 kb), containing the 5′-end of dpnO2 and the 3′-end of dpnS4, was amplified using the oligonucleotides 113C1orf15B-F and 113C1orf15B-R (Table S2).
- pHZ-CS113-C1orf18. This plasmid was used to delete dpnS2, generating the mutant CS113R-ΔC1orf18. Fragment A (2.01 kb), containing the 5′-end of dpnS3 and the 3′-end of dpnS2, was amplified using the oligonucleotides 113C1orf18A-F and 113C1orf18A-R (Table S2). Fragment B (2.09 kb), containing the 5′-end of dpnS2, dpnO3 and the 3′-end of dpnR2, was amplified using the oligonucleotides 113C1orf18B-F and 113C1orf18B-R (Table S2).
3.6. Extraction, Ultra-Performance Liquid Chromatography Analysis and Purification of Diperamycin
3.7. Structural Elucidation of Diperamycin
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Blair, L.M.; Sperry, J. Natural products containing a nitrogen-nitrogen bond. J. Nat. Prod. 2013, 76, 794–812. [Google Scholar] [CrossRef]
- Chen, L.; Deng, Z.; Zhao, C. Nitrogen-Nitrogen Bond Formation Reactions Involved in Natural Product Biosynthesis. ACS Chem. Biol. 2021, 16, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Le Goff, G.; Ouazzani, J. Natural hydrazine-containing compounds: Biosynthesis, isolation, biological activities and synthesis. Bioorg. Med. Chem. 2014, 22, 6529–6544. [Google Scholar] [CrossRef] [PubMed]
- Morgan, K.D.; Andersen, R.J.; Ryan, K.S. Piperazic acid-containing natural products: Structures and biosynthesis. Nat. Prod. Rep. 2019, 36, 1628–1653. [Google Scholar] [CrossRef] [PubMed]
- Neumann, C.S.; Jiang, W.; Heemstra, J.R., Jr.; Gontang, E.A.; Kolter, R.; Walsh, C.T. Biosynthesis of piperazic acid via N5-hydroxy-ornithine in Kutzneria spp. 744. ChemBioChem 2012, 13, 972–976. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.L.; He, H.Y.; Higgins, M.A.; Ryan, K.S. A heme-dependent enzyme forms the nitrogen-nitrogen bond in piperazate. Nat. Chem. Biol. 2017, 13, 836–838. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Qi, Y.; Stumpf, S.D.; D’Alessandro, J.M.; Blodgett, J.A.V. Bioinformatic and Functional Evaluation of Actinobacterial Piperazate Metabolism. ACS Chem. Biol. 2019, 14, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Byun, W.S.; Kang, S.; Kang, I.; Bae, E.S.; An, J.S.; Im, J.H.; Park, J.; Kim, E.; Ko, K.; et al. Targeted and Logical Discovery of Piperazic Acid-Bearing Natural Products Based on Genomic and Spectroscopic Signatures. J. Am. Chem. Soc. 2023, 145, 19676–19690. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Augustijn, H.E.; Reitz, Z.L.; Biermann, F.; Alanjary, M.; Fetter, A.; Terlouw, B.R.; Metcalf, W.W.; Helfrich, E.J.N.; et al. antiSMASH 7.0: New and improved predictions for detection, regulation, chemical structures and visualisation. Nucleic Acids Res. 2023, 51, W46–W50. [Google Scholar] [CrossRef]
- Batey, S.F.D.; Greco, C.; Hutchings, M.I.; Wilkinson, B. Chemical warfare between fungus-growing ants and their pathogens. Curr. Opin. Chem. Biol. 2020, 59, 172–181. [Google Scholar] [CrossRef]
- Ossai, J.; Khatabi, B.; Nybo, S.E.; Kharel, M.K. Renewed interests in the discovery of bioactive actinomycete metabolites driven by emerging technologies. J. Appl. Microbiol. 2022, 132, 59–77. [Google Scholar] [CrossRef] [PubMed]
- Van Moll, L.; De Smet, J.; Cos, P.; Van Campenhout, L. Microbial symbionts of insects as a source of new antimicrobials: A review. Crit. Rev. Microbiol. 2021, 47, 562–579. [Google Scholar] [CrossRef] [PubMed]
- Malmierca, M.G.; Gonzalez-Montes, L.; Perez-Victoria, I.; Sialer, C.; Brana, A.F.; Garcia Salcedo, R.; Martin, J.; Reyes, F.; Mendez, C.; Olano, C.; et al. Searching for glycosylated natural products in Actinomycetes and identification of novel macrolactams and angucyclines. Front. Microbiol. 2018, 9, 39. [Google Scholar] [CrossRef] [PubMed]
- Prado-Alonso, L.; Pérez-Victoria, I.; Malmierca, M.G.; Montero, I.; Rioja-Blanco, E.; Martín, J.; Reyes, F.; Méndez, C.; Salas, J.A.; Olano, C. Colibrimycins, Novel Halogenated Hybrid Polyketide Synthase-Nonribosomal Peptide Synthetase (PKS-NRPS) Compounds Produced by Streptomyces sp. Strain CS147. Appl. Environ. Microbiol. 2022, 88, e0183921. [Google Scholar] [CrossRef] [PubMed]
- Morgan, K.D.; Williams, D.E.; Patrick, B.O.; Remigy, M.; Banuelos, C.A.; Sadar, M.D.; Ryan, K.S.; Andersen, R.J. Incarnatapeptins A and B, Nonribosomal Peptides Discovered Using Genome Mining and 1H/15N HSQC-TOCSY. Org. Lett. 2020, 22, 4053–4057. [Google Scholar] [CrossRef]
- Bode, H.B.; Bethe, B.; Höfs, R.; Zeeck, A. Big effects from small changes: Possible ways to explore nature’s chemical diversity. ChemBioChem 2002, 3, 619–627. [Google Scholar] [CrossRef]
- Olano, C.; García, I.; González, A.; Rodriguez, M.; Rozas, D.; Rubio, J.; Sánchez-Hidalgo, M.; Braña, A.F.; Méndez, C.; Salas, J.A. Activation and identification of five clusters for secondary metabolites in Streptomyces albus J1074. Microb. Biotechnol. 2014, 7, 242–256. [Google Scholar] [CrossRef]
- Gräfe, U.; Schlegel, R.; Ritzau, M.; Ihn, W.; Dornberger, K.; Stengel, C.; Fleck, W.F.; Gutsche, W.; Härtl, A.; Paulus, E.F. Aurantimycins, new depsipeptide antibiotics from Streptomyces aurantiacus IMET 43917. Production, isolation, structure elucidation, and biological activity. J. Antibiot. 1995, 48, 119–125. [Google Scholar] [CrossRef]
- Matsumoto, N.; Momose, I.; Umekita, M.; Kinoshita, N.; Chino, M.; Iinuma, H.; Sawa, T.; Hamada, M.; Takeuchi, T. Diperamycin, a new antimicrobial antibiotic produced by Streptomyces griseoaurantiacus MK393-AF2. I. Taxonomy, fermentation, isolation, physico-chemical properties and biological activities. J. Antibiot. 1998, 51, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wang, L.; Wan, D.; Qi, J.; Gong, R.; Deng, Z.; Chen, W. Characterization of the aurantimycin biosynthetic gene cluster and enhancing its production by manipulating two pathway-specific activators in Streptomyces aurantiacus JA 4570. Microb. Cell Fact. 2016, 15, 160. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Donadio, S.; Katz, L. Organization of the enzymatic domains in the multifunctional polyketide synthase involved in erythromycin formation in Saccharopolyspora erythraea. Gene 1992, 111, 51–60. [Google Scholar] [CrossRef]
- Xu, W.; Qiao, K.; Tang, Y. Structural analysis of protein-protein interactions in type I polyketide synthases. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 98–122. [Google Scholar] [CrossRef]
- Bisang, C.; Long, P.F.; Cortés, J.; Westcott, J.; Crosby, J.; Matharu, A.L.; Cox, R.J.; Simpson, T.J.; Staunton, J.; Leadlay, P.F. A chain initiation factor common to both modular and aromatic polyketide synthases. Nature 1999, 401, 502–505. [Google Scholar] [CrossRef] [PubMed]
- Keatinge-Clay, A.T. The structures of type I polyketide synthases. Nat. Prod. Rep. 2012, 29, 1050–1073. [Google Scholar] [CrossRef]
- Wilson, M.C.; Moore, B.S. Beyond ethylmalonyl-CoA: The functional role of crotonyl-CoA carboxylase/reductase homologs in expanding polyketide diversity. Nat. Prod. Rep. 2012, 29, 72–86. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, H.; Kazuo, S.Y.; Omura, S. Genome mining of the Streptomyces avermitilis genome and development of genome-minimized hosts for heterologous expression of biosynthetic gene clusters. J. Ind. Microbiol. Biotechnol. 2014, 41, 233–250. [Google Scholar] [CrossRef] [PubMed]
- Rachid, S.; Huo, L.; Herrmann, J.; Stadler, M.; Köpcke, B.; Bitzer, J.; Müller, R. Mining the cinnabaramide biosynthetic pathway to generate novel proteasome inhibitors. ChemBioChem 2011, 12, 922–931. [Google Scholar] [CrossRef] [PubMed]
- Keatinge-Clay, A.T. A tylosin ketoreductase reveals how chirality is determined in polyketides. Chem. Biol. 2007, 14, 898–908. [Google Scholar] [CrossRef] [PubMed]
- Kwan, D.H.; Schulz, F. The stereochemistry of complex polyketide biosynthesis by modular polyketide synthases. Molecules 2011, 16, 6092–6115. [Google Scholar] [CrossRef]
- Kwan, D.H.; Sun, Y.; Schulz, F.; Hong, H.; Popovic, B.; Sim-Stark, J.C.; Haydock, S.F.; Leadlay, P.F. Prediction and manipulation of the stereochemistry of enoylreduction in modular polyketide synthases. Chem. Biol. 2008, 15, 1231–1240. [Google Scholar] [CrossRef]
- De Crécy-Lagard, V.; Marlière, P.; Saurin, W. Multienzymatic non ribosomal peptide biosynthesis: Identification of the functional domains catalysing peptide elongation and epimerisation. C. R. Acad. Sci. III 1995, 318, 927–936. [Google Scholar] [PubMed]
- Bloudoff, K.; Schmeing, T.M. Structural and functional aspects of the nonribosomal peptide synthetase condensation domain superfamily: Discovery, dissection and diversity. Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 1587–1604. [Google Scholar] [CrossRef] [PubMed]
- Lambalot, R.H.; Gehring, A.M.; Flugel, R.S.; Zuber, P.; LaCelle, M.; Marahiel, M.A.; Reid, R.; Khosla, C.; Walsh, C.T. A new enzyme superfamily—the phosphopantetheinyl transferases. Chem. Biol. 1996, 3, 923–936. [Google Scholar] [CrossRef] [PubMed]
- Challis, G.L.; Ravel, J.; Townsend, C.A. Predictive, structure-based model of amino acid recognition by nonribosomal peptide synthetase adenylation domains. Chem. Biol. 2000, 7, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Stachelhaus, T.; Mootz, H.D.; Marahiel, M.A. The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. Chem. Biol. 1999, 6, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.W.; Niikura, H.; Morgan, K.D.; Vacariu, C.M.; Andersen, R.J.; Ryan, K.S. Free Piperazic Acid as a Precursor to Nonribosomal Peptides. J. Am. Chem. Soc. 2022, 144, 13556–13564. [Google Scholar] [CrossRef]
- Süssmuth, R.D.; Mainz, A. Nonribosomal Peptide Synthesis-Principles and Prospects. Angew. Chem. Int. Ed. Engl. 2017, 56, 3770–3821. [Google Scholar] [CrossRef]
- Du, Y.; Wang, Y.; Huang, T.; Tao, M.; Deng, Z.; Lin, S. Identification and characterization of the biosynthetic gene cluster of polyoxypeptin A, a potent apoptosis inducer. BMC Microbiol. 2014, 14, 30. [Google Scholar] [CrossRef]
- Patel, K.D.; d’Andrea, F.B.; Gaudelli, N.M.; Buller, A.R.; Townsend, C.A.; Gulick, A.M. Structure of a bound peptide phosphonate reveals the mechanism of nocardicin bifunctional thioesterase epimerase-hydrolase half-reactions. Nat. Commun. 2019, 10, 3868. [Google Scholar] [CrossRef]
- Yu, J.; Song, J.; Chi, C.; Liu, T.; Geng, T.; Cai, Z.; Dong, W.; Shi, C.; Ma, X.; Zhang, Z.; et al. Functional Characterization and Crystal Structure of the Bifunctional Thioesterase Catalyzing Epimerization and Cyclization in Skyllamycin Biosynthesis. ACS Catal. 2021, 11, 11733–11741. [Google Scholar] [CrossRef]
- Pohle, S.; Appelt, C.; Roux, M.; Fiedler, H.P.; Süssmuth, R.D. Biosynthetic gene cluster of the non-ribosomally synthesized cyclodepsipeptide skyllamycin: Deciphering unprecedented ways of unusual hydroxylation reactions. J. Am. Chem. Soc. 2011, 133, 6194–6205. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.T.; O’Brien, R.V.; Khosla, C. Nonproteinogenic amino acid building blocks for nonribosomal peptide and hybrid polyketide scaffolds. Angew. Chem. Int. Ed. Engl. 2013, 52, 7098–7124. [Google Scholar] [CrossRef] [PubMed]
- Kresna, I.D.M.; Wuisan, Z.G.; Pohl, J.M.; Mettal, U.; Otoya, V.L.; Gand, M.; Marner, M.; Otoya, L.L.; Böhringer, N.; Vilcinskas, A.; et al. Genome-Mining-Guided Discovery and Characterization of the PKS-NRPS-Hybrid Polyoxyperuin Produced by a Marine-Derived Streptomycete. J. Nat. Prod. 2022, 85, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Becerril, A.; Pérez-Victoria, I.; Ye, S.; Braña, A.F.; Martín, J.; Reyes, F.; Salas, J.A.; Méndez, C. Discovery of Cryptic Largimycins in Streptomyces Reveals Novel Biosynthetic Avenues Enriching the Structural Diversity of the Leinamycin Family. ACS Chem. Biol. 2020, 15, 1541–1553. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, L.; Malmierca, M.G.; García-Salcedo, R.; Méndez, C.; Salas, J.A.; Olano, C.; Ceniceros, A. Co-Expression of Transcriptional Regulators and Housekeeping Genes in Streptomyces spp.: A Strategy to Optimize Metabolite Production. Microorganisms 2023, 11, 1585. [Google Scholar] [CrossRef] [PubMed]
- Kieser, T.; Bibb, M.J.; Buttner, M.J.; Chater, K.F.; Hopwood, D.A. Practical Streptomyces Genetics; The John Innes Foundation: Norwich, UK, 2000. [Google Scholar]
- Fernández, E.; Weissbach, U.; Sánchez Reillo, C.; Braña, A.F.; Méndez, C.; Rohr, J.; Salas, J.A. Identification of two genes from Streptomyces argillaceus encoding glycosyltransferases involved in transfer of a disaccharide during biosynthesis of the antitumor drug mithramycin. J. Bacteriol. 1998, 180, 4929–4937. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, G.; Frazer, C.M.; Gardner, D.C.J.; Cullum, J.A.; Oliver, S.G. Dispersed growth of Streptomyces in liquid culture. Appl. Microbiol. Biotechnol. 1989, 31, 272–277. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- Altschul, S.F.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Bachmann, B.O.; Ravel, J. Methods for in silico prediction of microbial polyketide and nonribosomal peptide biosynthetic pathways from DNA sequence data. Methods Enzymol. 2009, 458, 181–217. [Google Scholar] [CrossRef]
- Khater, S.; Gupta, M.; Agrawal, P.; Sain, N.; Prava, J.; Gupta, P.; Grover, M.; Kumar, N.; Mohanty, D. SBSPKSv2: Structure-based sequence analysis of polyketide synthases and non-ribosomal peptide synthetases. Nucleic Acids Res. 2017, 45, W72–W79. [Google Scholar] [CrossRef]
- Sun, Y.; Zhou, X.; Liu, J.; Bao, K.; Zhang, G.; Tu, G.; Kieser, T.; Deng, Z. ‘Streptomyces nanchangensis’, a producer of the insecticidal polyether antibiotic nanchangmycin and the antiparasitic macrolide meilingmycin, contains multiple polyketide gene clusters. Microbiology 2002, 148, 361–371. [Google Scholar] [CrossRef]
- Bierman, M.; Logan, R.; O’Brien, K.; Seno, E.T.; Rao, R.N.; Schoner, B.E. Plasmid cloning vectors for the conjugal transfer of DNA from Escherichia coli to Streptomyces spp. Gene 1992, 116, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Cano-Prieto, C.; García-Salcedo, R.; Sánchez-Hidalgo, M.; Braña, A.F.; Fiedler, H.P.; Méndez, C.; Salas, J.A.; Olano, C. Genome Mining of Streptomyces sp. Tü 6176: Characterization of the Nataxazole Biosynthesis Pathway. ChemBioChem 2015, 16, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.G.; Young, L.; Chuang, R.Y.; Venter, J.C.; Hutchison, C.A. 3rd, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Martín, J.; Crespo, G.; González-Menéndez, V.; Pérez-Moreno, G.; Sánchez-Carrasco, P.; Pérez-Victoria, I.; Ruiz-Pérez, L.M.; González-Pacanowska, D.; Vicente, F.; Genilloud, O.; et al. MDN-0104, an antiplasmodial betaine lipid from Heterospora chenopodii. J. Nat. Prod. 2014, 77, 2118–2123. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutant Strain | Plasmid | Deleted Genes |
|---|---|---|
| CS113R-ΔC1orf2 | pHZ-CS113-C1orf2 | dpnZ |
| CS113R-ΔC1orf15 | pHZ-CS113-C1orf15 | dpnO2 |
| CS113R-ΔC1orf18 | pHZ-CS113-C1orf18 | dpnS2 |
| Recombinant Strain | Plasmid | Expressed Genes |
| CS113R-indC1 | pOJ260ind-C1 | indC |
| ΔC1orf2 (pSETETc-C1orf2) | pSETETc-C1orf2 | dpnZ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Gutiérrez, C.; Pérez-Victoria, I.; Montero, I.; Fernández-De la Hoz, J.; Malmierca, M.G.; Martín, J.; Salas, J.A.; Olano, C.; Reyes, F.; Méndez, C. Unearthing a Cryptic Biosynthetic Gene Cluster for the Piperazic Acid-Bearing Depsipeptide Diperamycin in the Ant-Dweller Streptomyces sp. CS113. Int. J. Mol. Sci. 2024, 25, 2347. https://doi.org/10.3390/ijms25042347
García-Gutiérrez C, Pérez-Victoria I, Montero I, Fernández-De la Hoz J, Malmierca MG, Martín J, Salas JA, Olano C, Reyes F, Méndez C. Unearthing a Cryptic Biosynthetic Gene Cluster for the Piperazic Acid-Bearing Depsipeptide Diperamycin in the Ant-Dweller Streptomyces sp. CS113. International Journal of Molecular Sciences. 2024; 25(4):2347. https://doi.org/10.3390/ijms25042347
Chicago/Turabian StyleGarcía-Gutiérrez, Coral, Ignacio Pérez-Victoria, Ignacio Montero, Jorge Fernández-De la Hoz, Mónica G. Malmierca, Jesús Martín, José A. Salas, Carlos Olano, Fernando Reyes, and Carmen Méndez. 2024. "Unearthing a Cryptic Biosynthetic Gene Cluster for the Piperazic Acid-Bearing Depsipeptide Diperamycin in the Ant-Dweller Streptomyces sp. CS113" International Journal of Molecular Sciences 25, no. 4: 2347. https://doi.org/10.3390/ijms25042347
APA StyleGarcía-Gutiérrez, C., Pérez-Victoria, I., Montero, I., Fernández-De la Hoz, J., Malmierca, M. G., Martín, J., Salas, J. A., Olano, C., Reyes, F., & Méndez, C. (2024). Unearthing a Cryptic Biosynthetic Gene Cluster for the Piperazic Acid-Bearing Depsipeptide Diperamycin in the Ant-Dweller Streptomyces sp. CS113. International Journal of Molecular Sciences, 25(4), 2347. https://doi.org/10.3390/ijms25042347