
Application of Single-Cell Assay for Transposase-Accessible Chromatin with High Throughput Sequencing in Plant Science: Advances, Technical Challenges, and Prospects


 ,
,  and
and
Abstract
1. Introduction
2. scATAC-seq Unravels Epigenomic Regulatory Mechanisms Underlying Cellular Heterogeneity
2.1. Advances of Resolving Cellular Heterogeneity in Plants Using Traditional Bulk Assays
2.2. scATAC-seq Profiles Revolutionize Our Understanding of Cellular Heterogeneity
3. Challenges for Application of scATAC-seq in Plants
3.1. Preparation of Nuclei Suspensions Compatible with scATAC-seq
3.2. Analytical Tools Compatible with Plants
3.3. Challenges for Cell Type Annotation in scATAC-seq
3.4. The Annotation Quality of Reference Genomes
4. Current Strategies for Cell Type Annotation in scATAC-seq
4.1. Cell Type Annotation Based on scATAC-seq Data
4.2. Integration with scRNA-seq for Cell Type Annotation
4.3. Cell Type Annotation in Non-Model Organisms
5. Future Perspectives for Applying scATAC-seq in Plant Science
5.1. Refining Plant ENCODE Project at Single-Cell Level to Better Serve the Plant Science Community
5.2. Cross-Species Cell-Type Comparison Provides New Insights into Evolutionary Hierarchy
5.3. Toward Building a Cell Atlas through Multi-Omics Data Integration
5.4. scATAC-seq Intertwines with Lineage Tracing to Address Basic Questions in Plant Biology
5.5. Gene Regulatory Network Analyses Using scATAC-seq Data
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Preissl, S.; Gaulton, K.J.; Ren, B. Characterizing cis-regulatory elements using single-cell epigenomics. Nat. Rev. Genet. 2023, 24, 21–43. [Google Scholar] [CrossRef]
- Swinnen, G.; Goossens, A.; Pauwels, L. Lessons from Domestication: Targeting Cis-Regulatory Elements for Crop Improvement. Trends Plant Sci. 2019, 24, 1065. [Google Scholar] [CrossRef]
- Meyer, R.S.; Purugganan, M.D. Evolution of crop species: Genetics of domestication and diversification. Nat. Rev. Genet. 2013, 14, 840–852. [Google Scholar] [CrossRef]
- Springer, N.; de Leon, N.; Grotewold, E. Challenges of Translating Gene Regulatory Information into Agronomic Improvements. Trends Plant Sci. 2019, 24, 1075–1082. [Google Scholar] [CrossRef]
- Huang, C.; Sun, H.; Xu, D.; Chen, Q.; Liang, Y.; Wang, X.; Xu, G.; Tian, J.; Wang, C.; Li, D.; et al. ZmCCT9 enhances maize adaptation to higher latitudes. Proc. Natl. Acad. Sci. USA 2018, 115, E334–E341. [Google Scholar] [CrossRef]
- Studer, A.; Zhao, Q.; Ross-Ibarra, J.; Doebley, J. Identification of a functional transposon insertion in the maize domestication gene tb1. Nat. Genet. 2011, 43, U1160–U1164. [Google Scholar] [CrossRef]
- Jiang, S.; Huang, Z.; Li, Y.; Yu, C.; Yu, H.; Ke, Y.; Jiang, L.; Liu, J. Single-cell chromatin accessibility and transcriptome atlas of mouse embryos. Cell Rep. 2023, 42, 112210. [Google Scholar] [CrossRef] [PubMed]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Grandi, F.C.; Modi, H.; Kampman, L.; Corces, M.R. Chromatin accessibility profiling by ATAC-seq. Nat. Protoc. 2022, 17, 1518–1552. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Wu, B.; Litzenburger, U.M.; Ruff, D.; Gonzales, M.L.; Snyder, M.P.; Chang, H.Y.; Greenleaf, W.J. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 2015, 523, 486–490. [Google Scholar] [CrossRef]
- You, M.; Chen, L.; Zhang, D.; Zhao, P.; Chen, Z.; Qin, E.Q.; Gao, Y.; Davis, M.M.; Yang, P. Single-cell epigenomic landscape of peripheral immune cells reveals establishment of trained immunity in individuals convalescing from COVID-19. Nat. Cell Biol. 2021, 23, 620–630. [Google Scholar] [CrossRef]
- Wimmers, F.; Donato, M.; Kuo, A.; Ashuach, T.; Gupta, S.; Li, C.; Dvorak, M.; Foecke, M.H.; Chang, S.E.; Hagan, T.; et al. The single-cell epigenomic and transcriptional landscape of immunity to influenza vaccination. Cell 2021, 184, 3915–3935.e21. [Google Scholar] [CrossRef]
- Ranzoni, A.M.; Tangherloni, A.; Berest, I.; Riva, S.G.; Myers, B.; Strzelecka, P.M.; Xu, J.; Panada, E.; Mohorianu, I.; Zaugg, J.B.; et al. Integrative Single-Cell RNA-Seq and ATAC-Seq Analysis of Human Developmental Hematopoiesis. Cell Stem Cell 2021, 28, 472–487.e7. [Google Scholar] [CrossRef]
- Takayama, N.; Murison, A.; Takayanagi, S.I.; Arlidge, C.; Zhou, S.; Garcia-Prat, L.; Chan-Seng-Yue, M.; Zandi, S.; Gan, O.I.; Boutzen, H.; et al. The Transition from Quiescent to Activated States in Human Hematopoietic Stem Cells Is Governed by Dynamic 3D Genome Reorganization. Cell Stem Cell 2021, 28, 488–501.e10. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Curbelo, D.; Ho, Y.J.; Burdziak, C.; Maag, J.L.V.; Morris, J.P.t.; Chandwani, R.; Chen, H.A.; Tsanov, K.M.; Barriga, F.M.; Luan, W.; et al. A gene-environment-induced epigenetic program initiates tumorigenesis. Nature 2021, 590, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Regner, M.J.; Wisniewska, K.; Garcia-Recio, S.; Thennavan, A.; Mendez-Giraldez, R.; Malladi, V.S.; Hawkins, G.; Parker, J.S.; Perou, C.M.; Bae-Jump, V.L.; et al. A multi-omic single-cell landscape of human gynecologic malignancies. Mol. Cell 2021, 81, 4924–4941.e10. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Liang, Z.; Wang, Y.; Yao, J.; Yuan, Z.; Hu, G.; Qu, R.; Xie, S.; Li, D.; Yang, L.; et al. Chromatin accessibility illuminates single-cell regulatory dynamics of rice root tips. BMC Biol. 2022, 20, 274. [Google Scholar] [CrossRef] [PubMed]
- Dorrity, M.W.; Alexandre, C.M.; Hamm, M.O.; Vigil, A.L.; Fields, S.; Queitsch, C.; Cuperus, J.T. The regulatory landscape of Arabidopsis thaliana roots at single-cell resolution. Nat. Commun. 2021, 12, 3334. [Google Scholar] [CrossRef] [PubMed]
- Marand, A.P.; Chen, Z.L.; Gallavotti, A.; Schmitz, R.J. A cis-regulatory atlas in maize at single-cell resolution. Cell 2021, 184, 3041–3055.e21. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.X.; Shang, G.D.; Wu, L.Y.; Xu, Z.G.; Zhao, X.Y.; Wang, J.W. Chromatin Accessibility Dynamics and a Hierarchical Transcriptional Regulatory Network Structure for Plant Somatic Embryogenesis. Dev. Cell 2020, 54, 742–757.e8. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Bie, X.M.; Lin, X.; Li, M.; Wang, H.; Zhang, X.; Yang, Y.; Zhang, C.; Zhang, X.S.; Xiao, J. Uncovering the transcriptional regulatory network involved in boosting wheat regeneration and transformation. Nat. Plants 2023, 9, 908–925. [Google Scholar] [CrossRef] [PubMed]
- Stepniak, K.; Machnicka, M.A.; Mieczkowski, J.; Macioszek, A.; Wojtas, B.; Gielniewski, B.; Poleszak, K.; Perycz, M.; Krol, S.K.; Guzik, R.; et al. Mapping chromatin accessibility and active regulatory elements reveals pathological mechanisms in human gliomas. Nat. Commun. 2021, 12, 3621. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.Y.; Yu, C.P.; Chang, C.K.; Chen, H.J.; Li, M.Y.; Chen, Y.H.; Shiu, S.H.; Ku, M.S.B.; Tu, S.L.; Lu, M.J.; et al. Regulators of early maize leaf development inferred from transcriptomes of laser capture microdissection (LCM)-isolated embryonic leaf cells. Proc. Natl. Acad. Sci. USA 2022, 119, e2208795119. [Google Scholar] [CrossRef]
- Pires, R.C.; Ferro, A.; Capote, T.; Usie, A.; Correia, B.; Pinto, G.; Menendez, E.; Marum, L. Laser Microdissection of Woody and Suberized Plant Tissues for RNA-Seq Analysis. Mol. Biotechnol. 2022, 65, 419–432. [Google Scholar] [CrossRef]
- Deal, R.B.; Henikoff, S. The INTACT method for cell type-specific gene expression and chromatin profiling in Arabidopsis thaliana. Nat. Protoc. 2011, 6, 56–68. [Google Scholar] [CrossRef]
- Kim, E.D.; Dorrity, M.W.; Fitzgerald, B.A.; Seo, H.; Sepuru, K.M.; Queitsch, C.; Mitsuda, N.; Han, S.K.; Torii, K.U. Dynamic chromatin accessibility deploys heterotypic cis/trans-acting factors driving stomatal cell-fate commitment. Nat. Plants 2022, 8, 1453–1466. [Google Scholar] [CrossRef]
- Maher, K.A.; Bajic, M.; Kajala, K.; Reynoso, M.; Pauluzzi, G.; West, D.A.; Zumstein, K.; Woodhouse, M.; Bubb, K.; Dorrity, M.W.; et al. Profiling of Accessible Chromatin Regions across Multiple Plant Species and Cell Types Reveals Common Gene Regulatory Principles and New Control Modules. Plant Cell 2018, 30, 15–36. [Google Scholar] [CrossRef]
- Sijacic, P.; Bajic, M.; McKinney, E.C.; Meagher, R.B.; Deal, R.B. Changes in chromatin accessibility between Arabidopsis stem cells and mesophyll cells illuminate cell type-specific transcription factor networks. Plant J. 2018, 94, 215–231. [Google Scholar] [CrossRef]
- Tannenbaum, M.; Sarusi-Portuguez, A.; Krispil, R.; Schwartz, M.; Loza, O.; Benichou, J.I.C.; Mosquna, A.; Hakim, O. Regulatory chromatin landscape in Arabidopsis thaliana roots uncovered by coupling INTACT and ATAC-seq. Plant Methods 2018, 14, 113. [Google Scholar] [CrossRef]
- Frerichs, A.; Engelhorn, J.; Altmuller, J.; Gutierrez-Marcos, J.; Werr, W. Specific chromatin changes mark lateral organ founder cells in the Arabidopsis inflorescence meristem. J. Exp. Bot. 2019, 70, 3867–3879. [Google Scholar] [CrossRef]
- Sullivan, A.M.; Arsovski, A.A.; Thompson, A.; Sandstrom, R.; Thurman, R.E.; Neph, S.; Johnson, A.K.; Sullivan, S.T.; Sabo, P.J.; Neri, F.V., 3rd; et al. Mapping and Dynamics of Regulatory DNA in Maturing Arabidopsis thaliana Siliques. Front. Plant Sci. 2019, 10, 1434. [Google Scholar] [CrossRef] [PubMed]
- Pott, S.; Lieb, J.D. Single-cell ATAC-seq: Strength in numbers. Genome Biol. 2015, 16, 172. [Google Scholar] [CrossRef]
- Satpathy, A.T.; Granja, J.M.; Yost, K.E.; Qi, Y.Y.; Meschi, F.; McDermott, G.P.; Olsen, B.N.; Mumbach, M.R.; Pierce, S.E.; Corces, M.R.; et al. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat. Biotechnol. 2019, 37, 925–936. [Google Scholar] [CrossRef] [PubMed]
- Cusanovich, D.A.; Reddington, J.P.; Garfield, D.A.; Daza, R.M.; Aghamirzaie, D.; Marco-Ferreres, R.; Pliner, H.A.; Christiansen, L.; Qiu, X.; Steemers, F.J.; et al. The cis-regulatory dynamics of embryonic development at single-cell resolution. Nature 2018, 555, 538–542. [Google Scholar] [CrossRef]
- Cusanovich, D.A.; Hill, A.J.; Aghamirzaie, D.; Daza, R.M.; Pliner, H.A.; Berletch, J.B.; Filippova, G.N.; Huang, X.; Christiansen, L.; DeWitt, W.S.; et al. A Single-Cell Atlas of In Vivo Mammalian Chromatin Accessibility. Cell 2018, 174, 1309–1324.e18. [Google Scholar] [CrossRef]
- Lu, Z.; Ricci, W.A.; Schmitz, R.J.; Zhang, X. Identification of cis-regulatory elements by chromatin structure. Curr. Opin. Plant Biol. 2018, 42, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Louwers, M.; Bader, R.; Haring, M.; van Driel, R.; de Laat, W.; Stam, M. Tissue- and Expression Level-Specific Chromatin Looping at Maize b1 Epialleles. Plant Cell 2009, 21, 832–842. [Google Scholar] [CrossRef]
- Salvi, S.; Sponza, G.; Morgante, M.; Tomes, D.; Niu, X.; Fengler, K.A.; Meeley, R.; Ananiev, E.V.; Svitashev, S.; Bruggemann, E.; et al. Conserved noncoding genomic sequences associated with a flowering-time quantitative trait locus in maize. Proc. Natl. Acad. Sci. USA 2007, 104, 11376–11381. [Google Scholar] [CrossRef]
- Xu, G.H.; Wang, X.F.; Huang, C.; Xu, D.Y.; Li, D.; Tian, J.G.; Chen, Q.Y.; Wang, C.L.; Liang, Y.M.; Wu, Y.Y.; et al. Complex genetic architecture underlies maize tassel domestication. New Phytol. 2017, 214, 852–864. [Google Scholar] [CrossRef]
- Adrian, J.; Farrona, S.; Reimer, J.J.; Albani, M.C.; Coupland, G.; Turck, F. cis-Regulatory elements and chromatin state coordinately control temporal and spatial expression of FLOWERING LOCUS T in Arabidopsis. Plant Cell 2010, 22, 1425–1440. [Google Scholar] [CrossRef]
- Liu, L.Y.; Adrian, J.; Pankin, A.; Hu, J.Y.; Dong, X.; von Korff, M.; Turck, F. Induced and natural variation of promoter length modulates the photoperiodic response of FLOWERING LOCUS T. Nat. Commun. 2014, 5, 4558. [Google Scholar] [CrossRef] [PubMed]
- McGarry, R.C.; Ayre, B.G. A DNA element between At4g28630 and At4g28640 confers companion-cell specific expression following the sink-to-source transition in mature minor vein phloem. Planta 2008, 228, 839–849. [Google Scholar] [CrossRef]
- Yang, W.; Jefferson, R.A.; Huttner, E.; Moore, J.M.; Gagliano, W.B.; Grossniklaus, U. An egg apparatus-specific enhancer of Arabidopsis, identified by enhancer detection. Plant Physiol. 2005, 139, 1421–1432. [Google Scholar] [CrossRef]
- Rodgers-Melnick, E.; Vera, D.L.; Bass, H.W.; Buckler, E.S. Open chromatin reveals the functional maize genome. Proc. Natl. Acad. Sci. USA 2016, 113, E3177–E3184. [Google Scholar] [CrossRef]
- Heintzman, N.D.; Hon, G.C.; Hawkins, R.D.; Kheradpour, P.; Stark, A.; Harp, L.F.; Ye, Z.; Lee, L.K.; Stuart, R.K.; Ching, C.W.; et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009, 459, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Shu, X.; Zhu, P.; Pei, D. Chromatin accessibility dynamics during cell fate reprogramming. EMBO Rep. 2021, 22, e51644. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.J.; Schultz, M.D.; Urich, M.A.; Nery, J.R.; Pelizzola, M.; Libiger, O.; Alix, A.; McCosh, R.B.; Chen, H.; Schork, N.J.; et al. Patterns of population epigenomic diversity. Nature 2013, 495, 193–198. [Google Scholar] [CrossRef]
- Schmitz, R.J.; Grotewold, E.; Stam, M. Cis-regulatory sequences in plants: Their importance, discovery, and future challenges. Plant Cell 2022, 34, 718–741. [Google Scholar] [CrossRef]
- Zhong, Z.; Feng, S.; Duttke, S.H.; Potok, M.E.; Zhang, Y.; Gallego-Bartolome, J.; Liu, W.; Jacobsen, S.E. DNA methylation-linked chromatin accessibility affects genomic architecture in Arabidopsis. Proc. Natl. Acad. Sci. USA 2021, 118, e2023347118. [Google Scholar] [CrossRef]
- Jores, T.; Tonnies, J.; Wrightsman, T.; Buckler, E.S.; Cuperus, J.T.; Fields, S.; Queitsch, C. Synthetic promoter designs enabled by a comprehensive analysis of plant core promoters. Nat. Plants 2021, 7, 842–855. [Google Scholar] [CrossRef]
- Peng, T.; Zhai, Y.; Atlasi, Y.; Ter Huurne, M.; Marks, H.; Stunnenberg, H.G.; Megchelenbrink, W. STARR-seq identifies active, chromatin-masked, and dormant enhancers in pluripotent mouse embryonic stem cells. Genome Biol. 2020, 21, 243. [Google Scholar] [CrossRef] [PubMed]
- Muerdter, F.; Boryn, L.M.; Arnold, C.D. STARR-seq-principles and applications. Genomics 2015, 106, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Feng, Q.; Snouffer, A.; Zhang, B.Y.; Rodriguez, G.R.; van der Knaap, E. Increasing Fruit Weight by Editing a Cis-Regulatory Element in Tomato KLUH Promoter Using CRISPR/Cas9. Front. Plant Sci. 2022, 13, 879642. [Google Scholar] [CrossRef] [PubMed]
- Thibivilliers, S.B.; Anderson, D.K.; Libault, M.Y. Isolation of Plant Nuclei Compatible with Microfluidic Single-nucleus ATAC-sequencing. Bio-Protocol 2021, 11, e4240. [Google Scholar] [CrossRef] [PubMed]
- Farmer, A.; Thibivilliers, S.; Ryu, K.H.; Schiefelbein, J.; Libault, M. Single-nucleus RNA and ATAC sequencing reveals the impact of chromatin accessibility on gene expression in Arabidopsis roots at the single-cell level. Mol. Plant 2021, 14, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Xu, X.C.; Smaczniak, C.; Schumacher, J.; Yan, W.H.; Bluthgen, N.; Greb, T.; Jonsson, H.; Traas, J.; Kaufmann, K.; et al. A 3D gene expression atlas of the floral meristem based on spatial reconstruction of single nucleus RNA sequencing data. Nat. Commun. 2022, 13, 2838. [Google Scholar] [CrossRef] [PubMed]
- Conde, D.; Triozzi, P.M.; Pereira, W.J.; Schmidt, H.W.; Balmant, K.M.; Knaack, S.A.; Redondo-Lopez, A.; Roy, S.; Dervinis, C.; Kirst, M. Single-nuclei transcriptome analysis of the shoot apex vascular system differentiation in Populus. Development 2022, 149, dev200632. [Google Scholar] [CrossRef]
- Pliner, H.A.; Packer, J.S.; McFaline-Figueroa, J.L.; Cusanovich, D.A.; Daza, R.M.; Aghamirzaie, D.; Srivatsan, S.; Qiu, X.; Jackson, D.; Minkina, A.; et al. Cicero Predicts cis-Regulatory DNA Interactions from Single-Cell Chromatin Accessibility Data. Mol. Cell 2018, 71, 858–871.e8. [Google Scholar] [CrossRef]
- Stuart, T.; Srivastava, A.; Madad, S.; Lareau, C.A.; Satija, R. Single-cell chromatin state analysis with Signac. Nat. Methods 2021, 18, 1333–1341. [Google Scholar] [CrossRef]
- Yan, F.; Powell, D.R.; Curtis, D.J.; Wong, N.C. From reads to insight: A hitchhiker’s guide to ATAC-seq data analysis. Genome Biol. 2020, 21, 22. [Google Scholar] [CrossRef]
- Nair, V.D.; Vasoya, M.; Nair, V.; Smith, G.R.; Pincas, H.; Ge, Y.C.; Douglas, C.M.; Esser, K.A.; Sealfon, S.C. Optimization of the Omni-ATAC protocol to chromatin accessibility profiling in snap-frozen rat adipose and muscle tissues. Methodsx 2022, 9, 101681. [Google Scholar] [CrossRef] [PubMed]
- Nadelmann, E.R.; Gorham, J.M.; Reichart, D.; Delaughter, D.M.; Wakimoto, H.; Lindberg, E.L.; Litvinukova, M.; Maatz, H.; Curran, J.J.; Ischiu Gutierrez, D.; et al. Isolation of Nuclei from Mammalian Cells and Tissues for Single-Nucleus Molecular Profiling. Curr. Protoc. 2021, 1, e132. [Google Scholar] [CrossRef] [PubMed]
- Wiegleb, G.; Reinhardt, S.; Dahl, A.; Posnien, N. Tissue dissociation for single-cell and single-nuclei RNA sequencing for low amounts of input material. Front. Zool. 2022, 19, 27. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, A.; Blanco-Carmona, E.; Demirdizen, E.; Sun, X.Y.; Herold-Mende, C.; Schlesner, M.; Turcan, S. Nuclei Isolation from Fresh Frozen Brain Tumors for Single-Nucleus RNA-seq and ATAC-seq. Jove-J. Vis. Exp. 2020, 162, e61542. [Google Scholar] [CrossRef]
- Corces, M.R.; Trevino, A.E.; Hamilton, E.G.; Greenside, P.G.; Sinnott-Armstrong, N.A.; Vesuna, S.; Satpathy, A.T.; Rubin, A.J.; Montine, K.S.; Wu, B.; et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 2017, 14, 959–962. [Google Scholar] [CrossRef] [PubMed]
- Sikorskaite, S.; Rajamaki, M.L.; Baniulis, D.; Stanys, V.; Valkonen, J.P.T. Protocol: Optimised methodology for isolation of nuclei from leaves of species in the Solanaceae and Rosaceae families. Plant Methods 2013, 9, 31. [Google Scholar] [CrossRef]
- Loureiro, J.; Kron, P.; Temsch, E.M.; Koutecky, P.; Lopes, S.; Castro, M.; Castro, S. Isolation of plant nuclei for estimation of nuclear DNA content: Overview and best practices. Cytom. Part A 2021, 99, 318–327. [Google Scholar] [CrossRef]
- Thibivilliers, S.; Anderson, D.; Libault, M. Isolation of Plant Root Nuclei for Single Cell RNA Sequencing. Curr. Protoc. Plant Biol. 2020, 5, e20120. [Google Scholar] [CrossRef]
- Wang, K.; Zhao, C.; Xiang, S.; Duan, K.; Chen, X.; Guo, X.; Sahu, S.K. An optimized FACS-free single-nucleus RNA sequencing (snRNA-seq) method for plant science research. Plant Sci. 2023, 326, 111535. [Google Scholar] [CrossRef]
- Conde, D.; Triozzi, P.M.; Balmant, K.M.; Doty, A.L.; Miranda, M.; Boullosa, A.; Schmidt, H.W.; Pereira, W.J.; Dervinis, C.; Kirst, M. A robust method of nuclei isolation for single-cell RNA sequencing of solid tissues from the plant genus Populus. PLoS ONE 2021, 16, e0251149. [Google Scholar] [CrossRef]
- Tu, X.Y.; Marand, A.P.; Schmitz, R.J.; Zhong, S.L. A combinatorial indexing strategy for low-cost epigenomic profiling of plant single cells. Plant Commun. 2022, 3, 100308. [Google Scholar] [CrossRef]
- Zhang, K.; Zemke, N.R.; Armand, E.J.; Ren, B. A fast, scalable and versatile tool for analysis of single-cell omics data. Nat. Methods 2024. [Google Scholar] [CrossRef]
- Wang, D.; Hu, X.; Ye, H.; Wang, Y.; Yang, Q.; Liang, X.; Wang, Z.; Zhou, Y.; Wen, M.; Yuan, X.; et al. Cell-specific clock-controlled gene expression program regulates rhythmic fiber cell growth in cotton. Genome Biol. 2023, 24, 49. [Google Scholar] [CrossRef]
- Bravo Gonzalez-Blas, C.; Minnoye, L.; Papasokrati, D.; Aibar, S.; Hulselmans, G.; Christiaens, V.; Davie, K.; Wouters, J.; Aerts, S. cisTopic: Cis-regulatory topic modeling on single-cell ATAC-seq data. Nat. Methods 2019, 16, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Danese, A.; Richter, M.L.; Chaichoompu, K.; Fischer, D.S.; Theis, F.J.; Colome-Tatche, M. EpiScanpy: Integrated single-cell epigenomic analysis. Nat. Commun. 2021, 12, 5228. [Google Scholar] [CrossRef]
- Wang, C.; Sun, D.; Huang, X.; Wan, C.; Li, Z.; Han, Y.; Qin, Q.; Fan, J.; Qiu, X.; Xie, Y.; et al. Integrative analyses of single-cell transcriptome and regulome using MAESTRO. Genome Biol. 2020, 21, 198. [Google Scholar] [CrossRef]
- Granja, J.M.; Corces, M.R.; Pierce, S.E.; Bagdatli, S.T.; Choudhry, H.; Chang, H.Y.; Greenleaf, W.J. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 2021, 53, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.; Preissl, S.; Li, Y.; Hou, X.; Lucero, J.; Wang, X.; Motamedi, A.; Shiau, A.K.; Zhou, X.; Xie, F.; et al. Comprehensive analysis of single cell ATAC-seq data with SnapATAC. Nat. Commun. 2021, 12, 1337. [Google Scholar] [CrossRef] [PubMed]
- Vlot, A.H.C.; Maghsudi, S.; Ohler, U. Cluster-independent marker feature identification from single-cell omics data using SEMITONES. Nucleic Acids Res. 2022, 50, e107. [Google Scholar] [CrossRef]
- Shahan, R.; Hsu, C.W.; Nolan, T.M.; Cole, B.J.; Taylor, I.W.; Greenstreet, L.; Zhang, S.; Afanassiev, A.; Vlot, A.H.C.; Schiebinger, G.; et al. A single-cell Arabidopsis root atlas reveals developmental trajectories in wild-type and cell identity mutants. Dev. Cell 2022, 57, 543–560.e9. [Google Scholar] [CrossRef]
- Dai, M.; Pei, X.; Wang, X.J. Accurate and fast cell marker gene identification with COSG. Brief. Bioinform. 2022, 23, bbab579. [Google Scholar] [CrossRef]
- Sun, S.; Shen, X.; Li, Y.; Li, Y.; Wang, S.; Li, R.; Zhang, H.; Shen, G.; Guo, B.; Wei, J.; et al. Single-cell RNA sequencing provides a high-resolution roadmap for understanding the multicellular compartmentation of specialized metabolism. Nat. Plants 2023, 9, 179–190. [Google Scholar] [CrossRef]
- Deyneko, I.V.; Mustafaev, O.N.; Tyurin Acapital, A.C.; Zhukova, K.V.; Varzari, A.; Goldenkova-Pavlova, I.V. Modeling and cleaning RNA-seq data significantly improve detection of differentially expressed genes. BMC Bioinform. 2022, 23, 488. [Google Scholar] [CrossRef]
- Chen, X.Y.; Chen, S.Q.; Song, S.; Gao, Z.J.; Hou, L.; Zhang, X.G.; Lv, H.R.; Jiang, R. Cell type annotation of single-cell chromatin accessibility data via supervised Bayesian embedding. Nat. Mach. Intell. 2022, 4, 116–126. [Google Scholar] [CrossRef]
- Ma, W.; Lu, J.; Wu, H. Cellcano: Supervised cell type identification for single cell ATAC-seq data. Nat. Commun. 2022, 14, 1864. [Google Scholar] [CrossRef]
- Hao, Y.H.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M.; Zheng, S.W.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.X.; Wu, T.Y.; Wan, S.; Yang, J.Y.H.; Wong, W.H.; Wang, Y.X.R. scJoint integrates atlas-scale single-cell RNA-seq and ATAC-seq data with transfer learning. Nat. Biotechnol. 2022, 40, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yang, C.; Zhang, X. scDART: Integrating unmatched scRNA-seq and scATAC-seq data and learning cross-modality relationship simultaneously. Genome Biol. 2022, 23, 139. [Google Scholar] [CrossRef]
- Clarke, Z.A.; Andrews, T.S.; Atif, J.; Pouyabahar, D.; Innes, B.T.; MacParland, S.A.; Bader, G.D. Tutorial: Guidelines for annotating single-cell transcriptomic maps using automated and manual methods. Nat. Protoc. 2021, 16, 2749–2764. [Google Scholar] [CrossRef] [PubMed]
- MacParland, S.A.; Liu, J.C.; Ma, X.Z.; Innes, B.T.; Bartczak, A.M.; Gage, B.K.; Manuel, J.; Khuu, N.; Echeverri, J.; Linares, I.; et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun. 2018, 9, 4383. [Google Scholar] [CrossRef]
- Hodge, R.D.; Bakken, T.E.; Miller, J.A.; Smith, K.A.; Barkan, E.R.; Graybuck, L.T.; Close, J.L.; Long, B.; Johansen, N.; Penn, O.; et al. Conserved cell types with divergent features in human versus mouse cortex. Nature 2019, 573, 61–68. [Google Scholar] [CrossRef]
- Pliner, H.A.; Shendure, J.; Trapnell, C. Supervised classification enables rapid annotation of cell atlases. Nat. Methods 2019, 16, 983–986. [Google Scholar] [CrossRef]
- Lane, A.K.; Niederhuth, C.E.; Ji, L.; Schmitz, R.J. pENCODE: A plant encyclopedia of DNA elements. Annu. Rev. Genet. 2014, 48, 49–70. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.Y.; Zhu, T.; Zhou, X.; Yu, R.; He, Z.; Zhang, P.; Wu, Z.; Chen, M.; Kaufmann, K.; Chen, D. ChIP-Hub provides an integrative platform for exploring plant regulome. Nat. Commun. 2022, 13, 3413. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Xie, L.; Zhang, Q.; Ouyang, W.; Deng, L.; Guan, P.; Ma, M.; Li, Y.; Zhang, Y.; Xiao, Q.; et al. Integrative analysis of reference epigenomes in 20 rice varieties. Nat. Commun. 2020, 11, 2658. [Google Scholar] [CrossRef]
- Xie, L.; Liu, M.; Zhao, L.; Cao, K.; Wang, P.; Xu, W.; Sung, W.K.; Li, X.; Li, G. RiceENCODE: A comprehensive epigenomic database as a rice Encyclopedia of DNA Elements. Mol. Plant 2021, 14, 1604–1606. [Google Scholar] [CrossRef]
- Shafer, M.E.R. Cross-Species Analysis of Single-Cell Transcriptomic Data. Front. Cell Dev. Biol. 2019, 7, 175. [Google Scholar] [CrossRef]
- Liu, T.B.; Li, J.; Yu, L.Q.; Sun, H.X.; Li, J.; Dong, G.Y.; Hu, Y.Y.; Li, Y.; Shen, Y.; Wu, J.; et al. Cross-species single-cell transcriptomic analysis reveals pre-gastrulation developmental differences among pigs, monkeys, and humans. Cell Discov. 2021, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.Q.; Chen, Y.; Liu, Y.; Lin, W.H.; Wang, J.W. Single-cell transcriptome atlas and chromatin accessibility landscape reveal differentiation trajectories in the rice root. Nat. Commun. 2021, 12, 2053. [Google Scholar] [CrossRef]
- Kajala, K.; Gouran, M.; Shaar-Moshe, L.; Mason, G.A.; Rodriguez-Medina, J.; Kawa, D.; Pauluzzi, G.; Reynoso, M.; Canto-Pastor, A.; Manzano, C.; et al. Innovation, conservation, and repurposing of gene function in root cell type development. Cell 2021, 184, 5070. [Google Scholar] [CrossRef]
- Xie, J.; Qian, K.; Si, J.; Xiao, L.; Ci, D.; Zhang, D. Conserved noncoding sequences conserve biological networks and influence genome evolution. Heredity 2018, 120, 437–451. [Google Scholar] [CrossRef]
- Rozenblatt-Rosen, O.; Shin, J.W.; Rood, J.E.; Hupalowska, A.; Regev, A.; Heyn, H.; Technology, H.C.A.S. Building a high-quality Human Cell Atlas. Nat. Biotechnol. 2021, 39, 149–153. [Google Scholar] [CrossRef]
- Wagner, D.E.; Klein, A.M. Lineage tracing meets single-cell omics: Opportunities and challenges. Nat. Rev. Genet. 2020, 21, 410–427. [Google Scholar] [CrossRef]
- Baron, C.S.; van Oudenaarden, A. Unravelling cellular relationships during development and regeneration using genetic lineage tracing. Nat. Rev. Mol. Cell Biol. 2019, 20, 753–765. [Google Scholar] [CrossRef]
- Smetana, O.; Makila, R.; Lyu, M.; Amiryousefi, A.; Sanchez Rodriguez, F.; Wu, M.F.; Sole-Gil, A.; Leal Gavarron, M.; Siligato, R.; Miyashima, S.; et al. High levels of auxin signalling define the stem-cell organizer of the vascular cambium. Nature 2019, 565, 485–489. [Google Scholar] [CrossRef]
- Zhai, N.; Xu, L. Pluripotency acquisition in the middle cell layer of callus is required for organ regeneration. Nat. Plants 2021, 7, 1453–1460. [Google Scholar] [CrossRef]
- Badia, I.M.P.; Wessels, L.; Muller-Dott, S.; Trimbour, R.; Ramirez Flores, R.O.; Argelaguet, R.; Saez-Rodriguez, J. Gene regulatory network inference in the era of single-cell multi-omics. Nat. Rev. Genet. 2023, 24, 739–754. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Tang, K.; Xu, Y.; Wei, H.; Han, T.; Wang, C. Single-cell gene regulation network inference by large-scale data integration. Nucleic Acids Res. 2022, 50, e126. [Google Scholar] [CrossRef] [PubMed]
- Kartha, V.K.; Duarte, F.M.; Hu, Y.; Ma, S.; Chew, J.G.; Lareau, C.A.; Earl, A.; Burkett, Z.D.; Kohlway, A.S.; Lebofsky, R.; et al. Functional inference of gene regulation using single-cell multi-omics. Cell Genom. 2022, 2, 100166. [Google Scholar] [CrossRef] [PubMed]
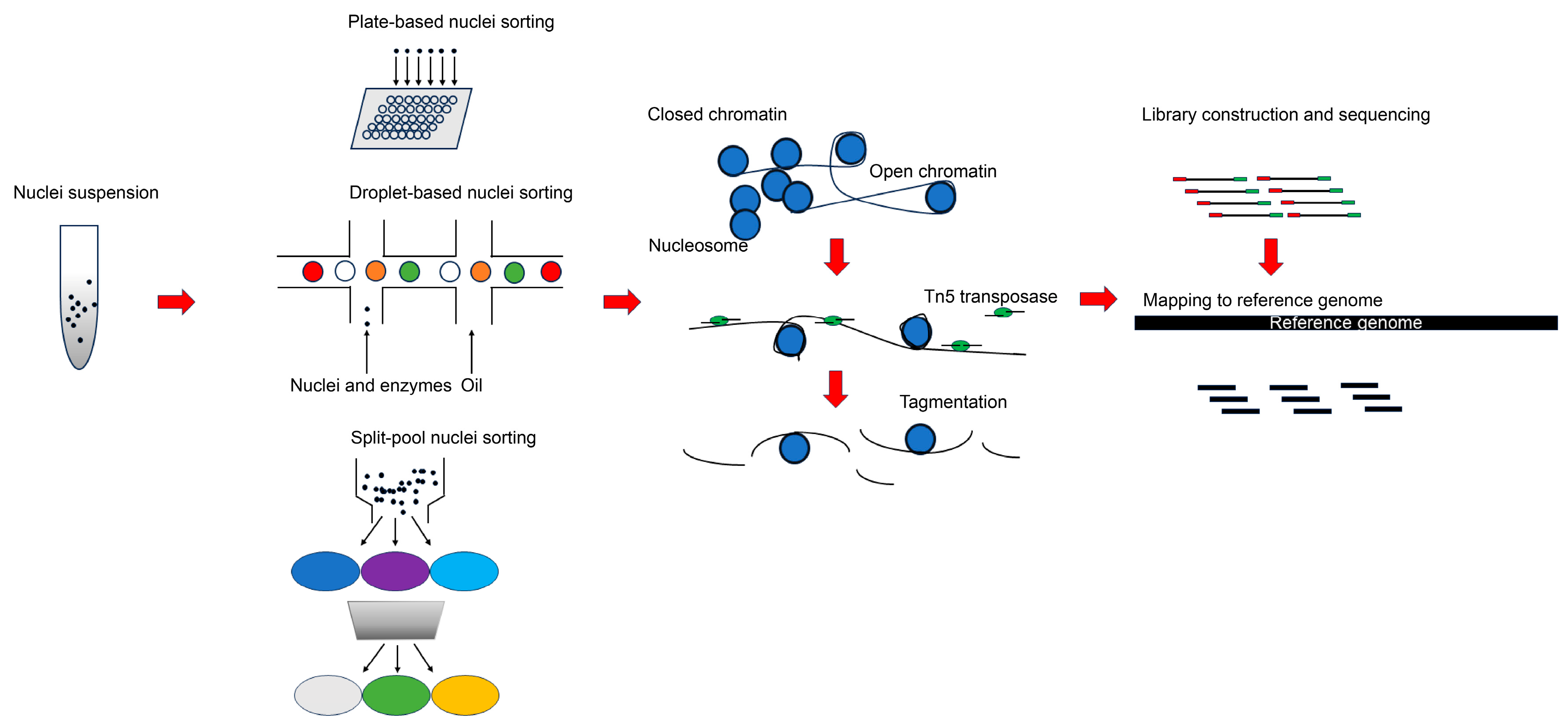
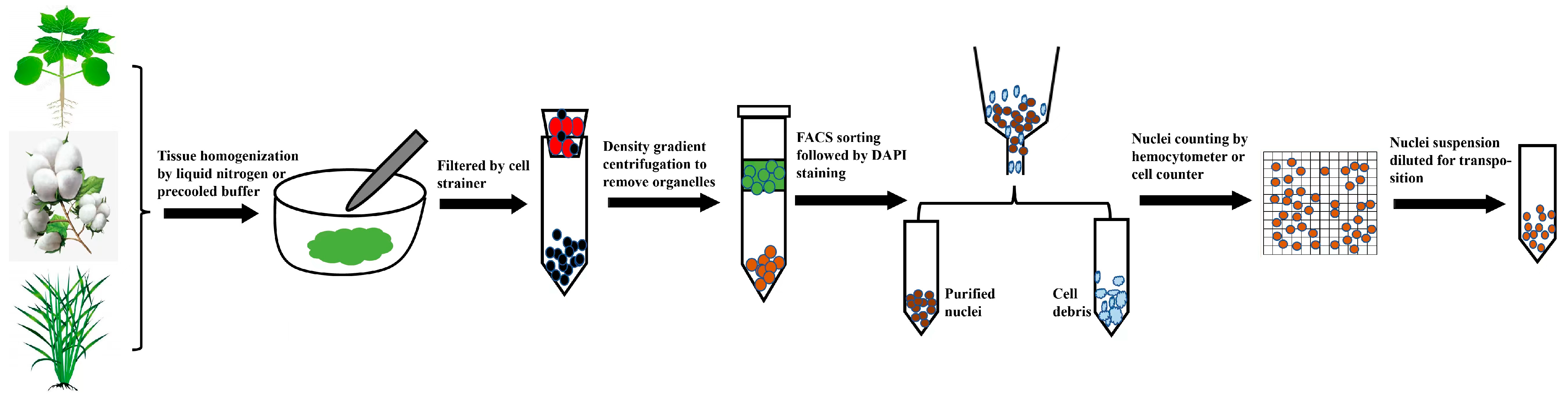
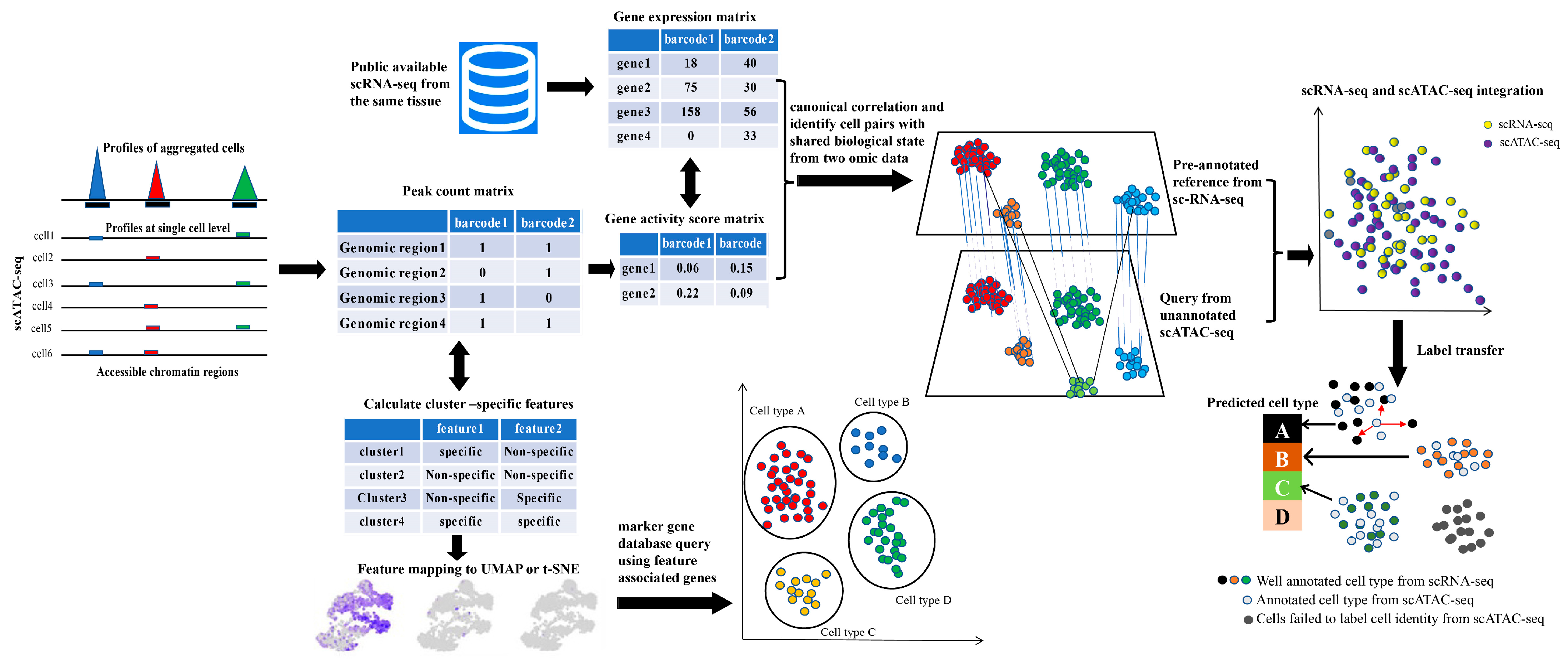


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analytical Tools | Platform | Motif Enrichment | Doublet Removal | Pseudotime Analysis | Batch Correction | Upstream Analysis | Applicable to Other Omics Data | Multi-Omics Integration | Reported in Plants |
|---|---|---|---|---|---|---|---|---|---|
| Cicero | R | No | No | Yes | Yes | No | No | No | No [59] |
| snapATAC2 | Python/R | Yes | No | No | Yes | Yes | Yes | Yes | No [73] |
| Signac | R | Yes | No | Yes | No | No | No | Yes | Yes [19] |
| ArchR | R | Yes | Yes | Yes | Yes | No | No | Yes | Yes [18] |
| SCALE | Python | No | No | No | Yes | No | No | No | Yes [74] |
| cisTopic | R | Yes | No | No | Yes | No | Yes | No | No [75] |
| epiScanpy | Python | No | No | Yes | Yes | No | Yes | No | No [76] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, C.; Wei, Y.; Abbas, M.; Agula, H.; Wang, E.; Meng, Z.; Zhang, R. Application of Single-Cell Assay for Transposase-Accessible Chromatin with High Throughput Sequencing in Plant Science: Advances, Technical Challenges, and Prospects. Int. J. Mol. Sci. 2024, 25, 1479. https://doi.org/10.3390/ijms25031479
Lu C, Wei Y, Abbas M, Agula H, Wang E, Meng Z, Zhang R. Application of Single-Cell Assay for Transposase-Accessible Chromatin with High Throughput Sequencing in Plant Science: Advances, Technical Challenges, and Prospects. International Journal of Molecular Sciences. 2024; 25(3):1479. https://doi.org/10.3390/ijms25031479
Chicago/Turabian StyleLu, Chao, Yunxiao Wei, Mubashir Abbas, Hasi Agula, Edwin Wang, Zhigang Meng, and Rui Zhang. 2024. "Application of Single-Cell Assay for Transposase-Accessible Chromatin with High Throughput Sequencing in Plant Science: Advances, Technical Challenges, and Prospects" International Journal of Molecular Sciences 25, no. 3: 1479. https://doi.org/10.3390/ijms25031479
APA StyleLu, C., Wei, Y., Abbas, M., Agula, H., Wang, E., Meng, Z., & Zhang, R. (2024). Application of Single-Cell Assay for Transposase-Accessible Chromatin with High Throughput Sequencing in Plant Science: Advances, Technical Challenges, and Prospects. International Journal of Molecular Sciences, 25(3), 1479. https://doi.org/10.3390/ijms25031479