
Magnesium and Vascular Calcification in Chronic Kidney Disease: Current Insights

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
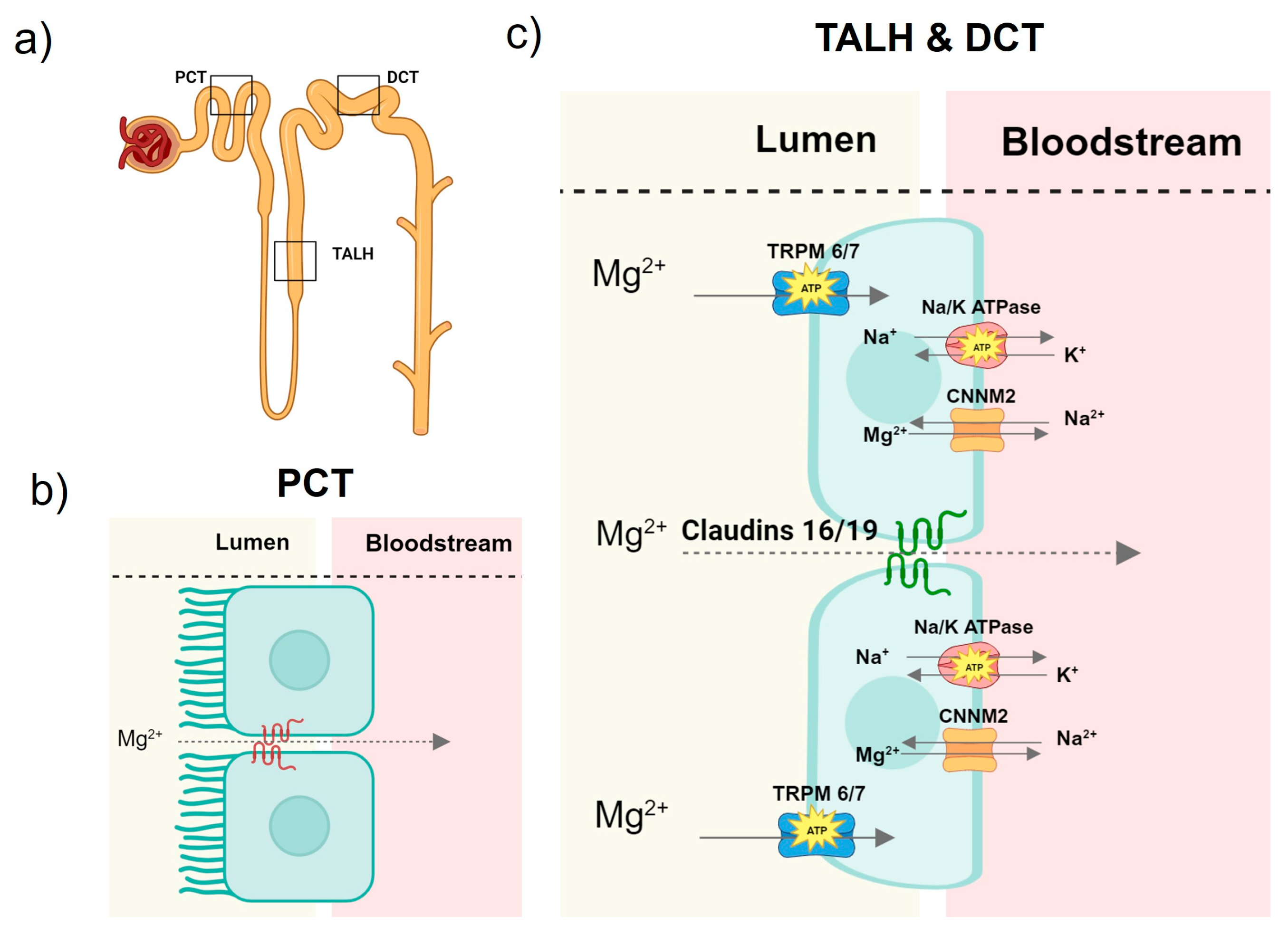
2. Mg Homeostasis
2.1. Magnesium Homeostasis in Health
2.2. Mg Disturbances in CKD
3. Vascular Calcification in CKD
4. Epidemiology for Mg and Vascular Calcification in CKD
5. Mg and Vascular Calcification: In Vitro and Animal Studies
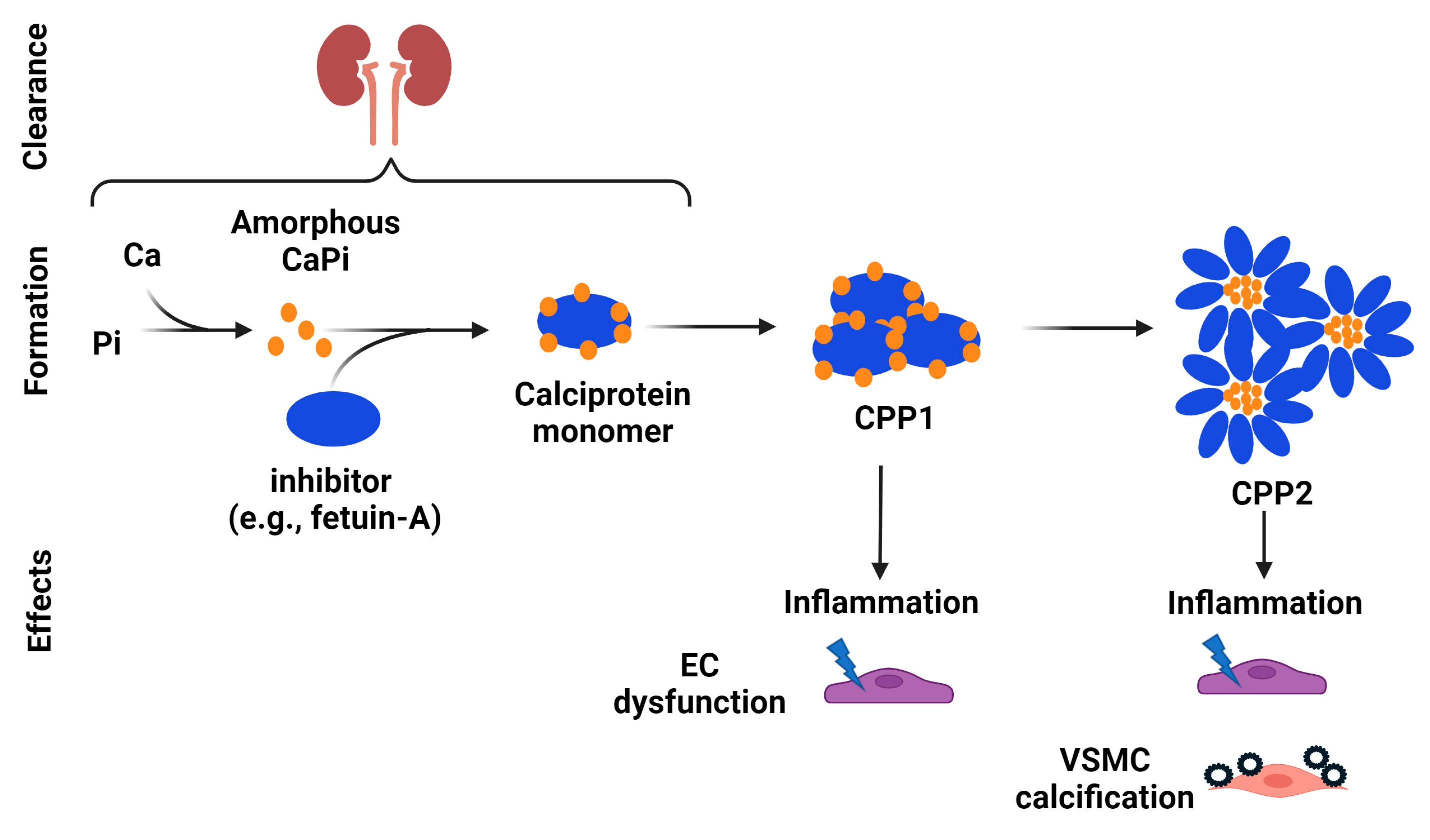
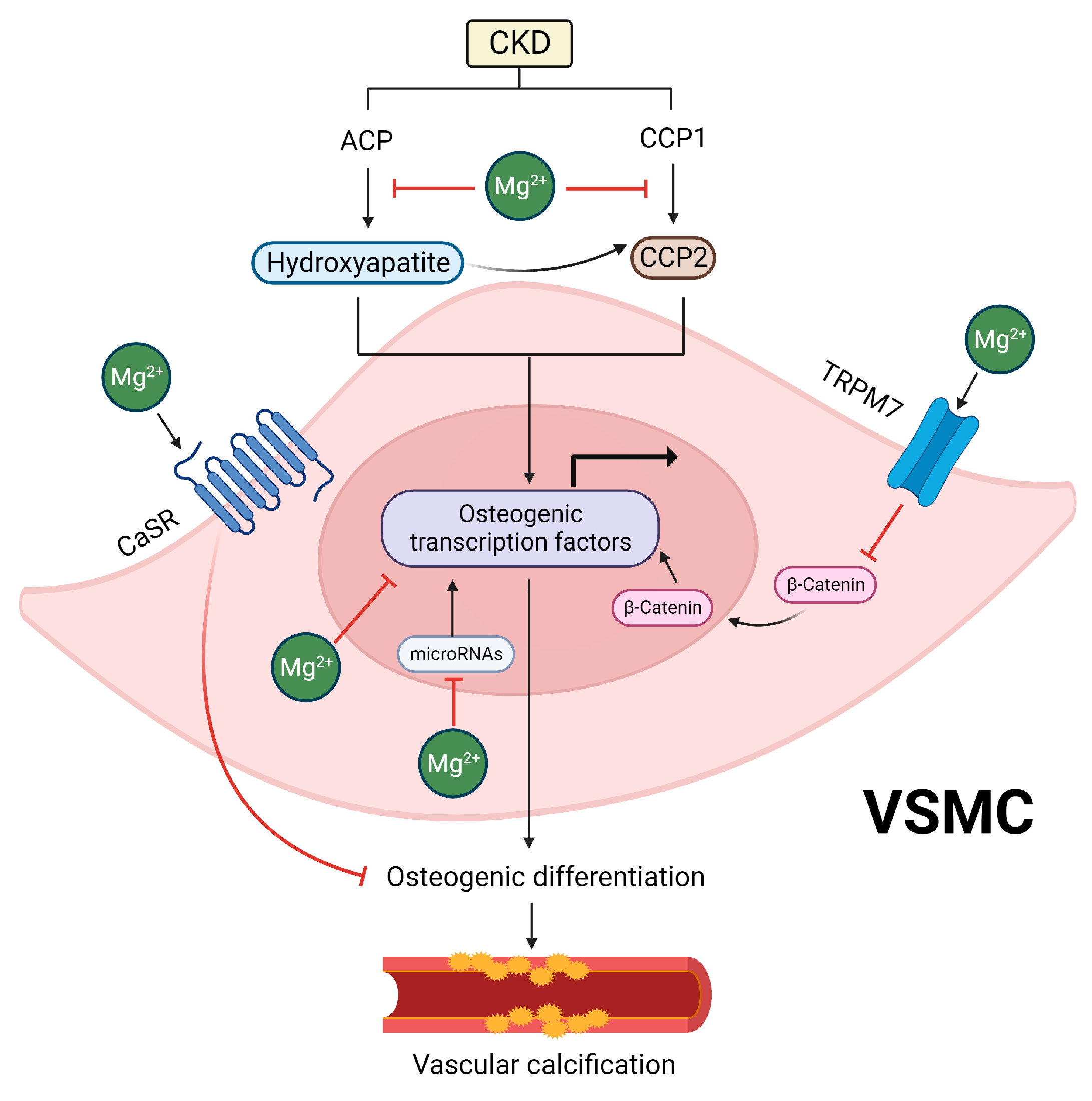
5.1. Inhibitory Effects of Mg on Hydroxyapatite and CPP Formation
5.2. Inhibitory Effects of Mg on the Osteogenic Differentiation of VSMCs
5.3. Indirect Effects of Mg Supplementation
5.4. Effects of Mg on Vascular Calcification In Vivo
6. Mg and Vascular Calcification: Clinical Studies
7. Perspectives
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Friedrich, H.E.; Mordike, B.L. Magnesium Technology; Springer: Berlin/Heidelberg, Germany, 2006; Volume 212. [Google Scholar]
- Weeks, M.E. The discovery of the elements. X. The alkaline earth metals and magnesium and cadmium. J. Chem. Educ. 1932, 9, 1046. [Google Scholar] [CrossRef]
- Multhaup, R.P. A history of magnesia alba. Ann. Sci. 1976, 33, 197. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.B.; Eklund, J.B. Magnesia Alba before Black. Pharm. Hist. 1972, 14, 139–146. [Google Scholar] [PubMed]
- Glasdam, S.-M.; Glasdam, S.; Peters, G.H. Chapter Six-The Importance of Magnesium in the Human Body: A Systematic Literature Review. In Advances in Clinical Chemistry; Makowski, G.S., Ed.; Elsevier: Amsterdam, The Netherlands, 2016; Volume 73, pp. 169–193. [Google Scholar]
- Bertini, I. Biological Inorganic Chemistry: Structure and Reactivity; University Science Books: Herndon, VA, USA, 2007; p. 794. [Google Scholar]
- Office of Dietary Supplements-Magnesium. Available online: https://ods.od.nih.gov/factsheets/Magnesium-HealthProfessional/ (accessed on 21 December 2023).
- Piovesan, D.; Profiti, G.; Martelli, P.L.; Casadio, R. The human “magnesome”: Detecting magnesium binding sites on human proteins. BMC Bioinform. 2012, 13, S10. [Google Scholar] [CrossRef]
- Nyabadza, A.; Shan, C.; Murphy, R.; Vazquez, M.; Brabazon, D. Laser-synthesised magnesium nanoparticles for amino acid and enzyme immobilisation. OpenNano 2023, 11, 100133. [Google Scholar] [CrossRef]
- Cockwell, P.; Fisher, L.-A. The global burden of chronic kidney disease. Lancet 2020, 395, 662–664. [Google Scholar] [CrossRef]
- Chronic Kidney Disease Basics|Chronic Kidney Disease Initiative|CDC. 2022. Available online: https://www.cdc.gov/kidneydisease/basics.html (accessed on 27 November 2023).
- United States Renal Data System. 2023 USRDS Annual Data Report: Epidemiology of Kidney Disease in the United States; National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2023. [Google Scholar]
- Jayalath, R.W.; Mangan, S.H.; Golledge, J. Aortic Calcification. Eur. J. Vasc. Endovasc. Surg. 2005, 30, 476–488. [Google Scholar] [CrossRef]
- Wilson, P.W.; Kauppila, L.I.; O’Donnell, C.J.; Kiel, D.P.; Hannan, M.; Polak, J.M.; Cupples, L.A. Abdominal aortic calcific deposits are an important predictor of vascular morbidity and mortality. Circulation 2001, 103, 1529–1534. [Google Scholar] [CrossRef]
- Gerke, O.; Lindholt, J.S.; Abdo, B.H.; Lambrechtsen, J.; Frost, L.; Steffensen, F.H.; Karon, M.; Egstrup, K.; Urbonaviciene, G.; Busk, M.; et al. Prevalence and extent of coronary artery calcification in the middle-aged and elderly population. Eur. J. Prev. Cardiol. 2021, 28, 2048–2055. [Google Scholar] [CrossRef]
- Raggi, P.; Boulay, A.; Chasan, T.S.; Amin, N.; Dillon, M.; Burke, S.K.; Chertow, G.M. Cardiac calcification in adult hemodialysis patients. J. Am. Coll. Cardiol. 2002, 39, 695–701. [Google Scholar] [CrossRef]
- Ohtake, T.; Ishioka, K.; Honda, K.; Oka, M.; Maesato, K.; Mano, T.; Ikee, R.; Moriya, H.; Hidaka, S.; Kobayashi, S. Impact of coronary artery calcification in hemodialysis patients: Risk factors and associations with prognosis. Hemodial. Int. 2010, 14, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Jahnen-Dechent, W.; Ketteler, M. Magnesium basics. Clin. Kidney J. 2012, 5, i3–i14. [Google Scholar] [CrossRef] [PubMed]
- Rondanelli, M.; Faliva, M.A.; Tartara, A.; Gasparri, C.; Perna, S.; Infantino, V.; Riva, A.; Petrangolini, G.; Peroni, G. An update on magnesium and bone health. Biometals 2021, 34, 715–736. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.T.; Soman, S.S.; Yee, J. Magnesium Balance and Measurement. Adv. Chronic Kidney Dis. 2018, 25, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.J.; Sharma, S. Magnesium. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Volpe, S.L. Magnesium in Disease Prevention and Overall Health12. Adv. Nutr. 2013, 4, 378S–383S. [Google Scholar] [CrossRef] [PubMed]
- Pennington, J.A.T.; Youngt, B. Sodium, potassium, calcium, phosphorus, and magnesium in foods from the United States total diet study. J. Food Compos. Anal. 1990, 3, 145–165. [Google Scholar] [CrossRef]
- Blaine, J.; Chonchol, M.; Levi, M. Renal Control of Calcium, Phosphate, and Magnesium Homeostasis. Clin. J. Am. Soc. Nephrol. CJASN 2015, 10, 1257–1272. [Google Scholar] [CrossRef]
- Chubanov, V.; Gudermann, T. TRPM6. In Mammalian Transient Receptor Potential (TRP) Cation Channels: Volume I; Nilius, B., Flockerzi, V., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 503–520. [Google Scholar]
- Ryazanova, L.V.; Pavur, K.S.; Petrov, A.N.; Dorovkov, M.V.; Ryazanov, A.G. Novel Type of Signaling Molecules: Protein Kinases Covalently Linked with Ion Channels. Mol. Biol. 2001, 35, 271–283. [Google Scholar] [CrossRef]
- Schlingmann, K.P.; Waldegger, S.; Konrad, M.; Chubanov, V.; Gudermann, T. TRPM6 and TRPM7—Gatekeepers of human magnesium metabolism. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2007, 1772, 813–821. [Google Scholar] [CrossRef]
- Romani, A.M.P. Cellular magnesium homeostasis. Arch. Biochem. Biophys. 2011, 512, 1–23. [Google Scholar] [CrossRef]
- TRPM6 Gene: MedlinePlus Genetics. Available online: https://medlineplus.gov/ (accessed on 21 December 2023).
- TRPM6 Transient Receptor Potential Cation Channel Subfamily M Member 6 [Homo Sapiens (Human)]-Gene-NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene/140803 (accessed on 20 November 2023).
- Yamazaki, D.; Funato, Y.; Miura, J.; Sato, S.; Toyosawa, S.; Furutani, K.; Kurachi, Y.; Omori, Y.; Furukawa, T.; Tsuda, T.; et al. Basolateral Mg2+ Extrusion via CNNM4 Mediates Transcellular Mg2+ Transport across Epithelia: A Mouse Model. PLoS Genet. 2013, 9, e1003983. [Google Scholar] [CrossRef] [PubMed]
- Philipp Schuchardt, J.; Hahn, A. Intestinal Absorption and Factors Influencing Bioavailability of Magnesium—An Update. Curr. Nutr. Food Sci. 2017, 13, 260–278. [Google Scholar] [CrossRef]
- Paxton, S.; Peckham, M.; Knibbs, A. The Leeds Histology Guide. Available online: www.histology.leeds.ac.uk (accessed on 21 December 2023).
- Curry, J.; Yu, A. Magnesium Handling in the Kidney. Adv. Chronic Kidney Dis. 2018, 25, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Whang, R.; Hampton, E.M.; Whang, D.D. Magnesium Homeostasis and Clinical Disorders of Magnesium Deficiency. Ann. Pharmacother. 1994, 28, 220–226. [Google Scholar] [CrossRef] [PubMed]
- de Baaij, J.H.F. Magnesium reabsorption in the kidney. Am. J. Physiol.-Ren. Physiol. 2023, 324, F227–F244. [Google Scholar] [CrossRef] [PubMed]
- Prot-Bertoye, C.; Houillier, P. Claudins in Renal Physiology and Pathology. Genes 2020, 11, 290. [Google Scholar] [CrossRef] [PubMed]
- Funato, Y.; Miki, H. Molecular function and biological importance of CNNM family Mg2+ transporters. J. Biochem. 2019, 165, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Favus, M.J.; Bushinsky, D.A.; Lemann, J., Jr. Chapter 13. Regulation of Calcium, Magnesium, and Phosphate Metabolism; American Society for Bone and Mineral Research: Washington, DC, USA, 2006. [Google Scholar]
- Schmitz, C.; Perraud, A.-L.; Johnson, C.O.; Inabe, K.; Smith, M.K.; Penner, R.; Kurosaki, T.; Fleig, A.; Scharenberg, A.M. Regulation of Vertebrate Cellular Mg2+ Homeostasis by TRPM7. Cell 2003, 114, 191–200. [Google Scholar] [CrossRef]
- Seo, J.W.; Park, T.J. Magnesium Metabolism. Electrolytes Blood Press. E BP 2008, 6, 86–95. [Google Scholar] [CrossRef]
- Felsenfeld, A.J.; Levine, B.S.; Rodriguez, M. Pathophysiology of Calcium, Phosphorus, and Magnesium Dysregulation in Chronic Kidney Disease. Semin. Dial. 2015, 28, 564–577. [Google Scholar] [CrossRef]
- Uwitonze, A.M.; Razzaque, M.S. Role of Magnesium in Vitamin D Activation and Function. J. Osteopath. Med. 2018, 118, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Ikari, A.; Okude, C.; Sawada, H.; Takahashi, T.; Sugatani, J.; Miwa, M. Down-regulation of TRPM6-mediated magnesium influx by cyclosporin A. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2008, 377, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Ikari, A.; Sanada, A.; Sawada, H.; Okude, C.; Tonegawa, C.; Sugatani, J. Decrease in transient receptor potential melastatin 6 mRNA stability caused by rapamycin in renal tubular epithelial cells. Biochim. Et Biophys. Acta (BBA)-Biomembr. 2011, 1808, 1502–1508. [Google Scholar] [CrossRef]
- Groenestege, W.M.T.; Hoenderop, J.G.; van den Heuvel, L.; Knoers, N.; Bindels, R.J. The epithelial Mg2+ channel transient receptor potential melastatin 6 is regulated by dietary Mg2+ content and estrogens. J. Am. Soc. Nephrol. JASN 2006, 17, 1035–1043. [Google Scholar] [CrossRef]
- Rössig, A.; Hill, K.; Nörenberg, W.; Weidenbach, S.; Zierler, S.; Schaefer, M.; Gudermann, T.; Chubanov, V. Pharmacological agents selectively acting on the channel moieties of TRPM6 and TRPM7. Cell Calcium 2022, 106, 102640. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Hamano, T.; Sakaguchi, Y.; Yamaguchi, S.; Kubota, K.; Senda, M.; Yonemoto, S.; Shimada, K.; Matsumoto, A.; Hashimoto, N.; et al. Proteinuria-associated renal magnesium wasting leads to hypomagnesemia: A common electrolyte abnormality in chronic kidney disease. Nephrol. Dial. Transplant. 2019, 34, 1154–1162. [Google Scholar] [CrossRef]
- Mitsopoulos, E.; Griveas, I.; Zanos, S.; Anagnostopoulos, K.; Giannakou, A.; Pavlitou, A.; Sakellariou, G. Increase in Serum Magnesium Level in Haemodialysis Patients Receiving Sevelamer Hydrochloride. Int. Urol. Nephrol. 2005, 37, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Liabeuf, S.; Lambert, O.; Metzger, M.; Hamroun, A.; Laville, M.; Laville, S.M.; Frimat, L.; Pecoits-Filho, R.; Fouque, D.; Massy, Z.A.; et al. Adverse outcomes of proton pump inhibitors in patients with chronic kidney disease: The CKD-REIN cohort study. Br. J. Clin. Pharmacol. 2021, 87, 2967–2976. [Google Scholar] [CrossRef]
- Gommers, L.M.M.; Hoenderop, J.G.J.; de Baaij, J.H.F. Mechanisms of proton pump inhibitor-induced hypomagnesemia. Acta Physiol. 2022, 235, e13846. [Google Scholar] [CrossRef]
- Srinutta, T.; Chewcharat, A.; Takkavatakarn, K.; Praditpornsilpa, K.; Eiam-Ong, S.; Jaber, B.L.; Susantitaphong, P. Proton pump inhibitors and hypomagnesemia: A meta-analysis of observational studies. Medicine 2019, 98, e17788. [Google Scholar] [CrossRef]
- Alexander, R.T.; Dimke, H. Effect of diuretics on renal tubular transport of calcium and magnesium. Am. J. Physiol.-Ren. Physiol. 2017, 312, F998–F1015. [Google Scholar] [CrossRef] [PubMed]
- Martin, B.J.; Milligan, K. Diuretic-Associated Hypomagnesemia in the Elderly. Arch. Intern. Med. 1987, 147, 1768–1771. [Google Scholar] [CrossRef] [PubMed]
- Nijenhuis, T.; Vallon, V.; Kemp, A.W.C.M.v.d.; Loffing, J.; Hoenderop, J.G.J.; Bindels, R.J.M. Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J. Clin. Investig. 2005, 115, 1651–1658. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Harrison, S.D.; Cope, M.J.; Park, C.; Lee, L.; Salaymeh, F.; Madsen, D.; Benton, W.W.; Berman, L.; Buysse, J. Mechanism of Action and Pharmacology of Patiromer, a Nonabsorbed Cross-Linked Polymer That Lowers Serum Potassium Concentration in Patients With Hyperkalemia. J. Cardiovasc. Pharmacol. Ther. 2016, 21, 456–465. [Google Scholar] [CrossRef] [PubMed]
- FDA. VELTASSA (Patiromer) for Oral Suspension. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/205739s016lbl.pdf (accessed on 21 December 2023).
- Bakris, G.L.; Pitt, B.; Weir, M.R.; Freeman, M.W.; Mayo, M.R.; Garza, D.; Stasiv, Y.; Zawadzki, R.; Berman, L.; Bushinsky, D.A.; et al. Effect of Patiromer on Serum Potassium Level in Patients With Hyperkalemia and Diabetic Kidney Disease: The AMETHYST-DN Randomized Clinical Trial. JAMA 2015, 314, 151–161. [Google Scholar] [CrossRef]
- Gonella, M.; Ballanti, P.; Della Rocca, C.; Calabrese, G.; Pratesi, G.; Vagelli, G.; Mazzotta, A.; Bonucci, E. Improved bone morphology by normalizing serum magnesium in chronically hemodialyzed patients. Miner. Electrolyte Metab. 1988, 14, 240–245. [Google Scholar] [PubMed]
- Alhosaini, M.; Leehey, D.J. Magnesium and Dialysis: The Neglected Cation. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2015, 66, 523–531. [Google Scholar] [CrossRef]
- Cascella, M.; Vaqar, S. Hypermagnesemia. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Gragossian, A.; Bashir, K.; Friede, R. Hypomagnesemia; StatPearls Publishing: Treasure Island, FL, USA, 2018. [Google Scholar]
- Lamprea-Montealegre, J.A.; McClelland, R.L.; Astor, B.C.; Matsushita, K.; Shlipak, M.; de Boer, I.H.; Szklo, M. Chronic kidney disease, plasma lipoproteins, and coronary artery calcium incidence: The Multi-Ethnic Study of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 652–658. [Google Scholar] [CrossRef]
- Chen, W.; Fitzpatrick, J.; Monroy-Trujillo, J.M.; Sozio, S.M.; Jaar, B.G.; Estrella, M.M.; Wu, T.T.; Melamed, M.L.; Parekh, R.S.; Bushinsky, D.A. Diabetes Mellitus Modifies the Associations of Serum Magnesium Concentration With Arterial Calcification and Stiffness in Incident Hemodialysis Patients. Kidney Int. Rep. 2019, 4, 806–813. [Google Scholar] [CrossRef]
- Demer, L.L.; Tintut, Y. Vascular calcification: Pathobiology of a multifaceted disease. Circulation 2008, 117, 2938–2948. [Google Scholar] [CrossRef]
- London, G.M.; Marchais, S.J.; Guerin, A.P. Arterial stiffness and function in end-stage renal disease. Adv. Chronic Kidney Dis. 2004, 11, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Blacher, J.; Guerin, A.P.; Pannier, B.; Marchais, S.J.; Safar, M.E.; London, G.M. Impact of aortic stiffness on survival in end-stage renal disease. Circulation 1999, 99, 2434–2439. [Google Scholar] [CrossRef] [PubMed]
- Giachelli, C.M. The emerging role of phosphate in vascular calcification. Kidney Int. 2009, 75, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Jono, S.; McKee, M.D.; Murry, C.E.; Shioi, A.; Nishizawa, Y.; Mori, K.; Morii, H.; Giachelli, C.M. Phosphate regulation of vascular smooth muscle cell calcification. Circ. Res. 2000, 87, E10–E17. [Google Scholar] [CrossRef]
- Steitz, S.A.; Speer, M.Y.; Curinga, G.; Yang, H.Y.; Haynes, P.; Aebersold, R.; Schinke, T.; Karsenty, G.; Giachelli, C.M. Smooth muscle cell phenotypic transition associated with calcification: Upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ. Res. 2001, 89, 1147–1154. [Google Scholar] [CrossRef]
- Byon, C.H.; Javed, A.; Dai, Q.; Kappes, J.C.; Clemens, T.L.; Darley-Usmar, V.M.; McDonald, J.M.; Chen, Y. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J. Biol. Chem. 2008, 283, 15319–15327. [Google Scholar] [CrossRef]
- Mody, N.; Parhami, F.; Sarafian, T.A.; Demer, L.L. Oxidative stress modulates osteoblastic differentiation of vascular and bone cells. Free. Radic. Biol. Med. 2001, 31, 509–519. [Google Scholar] [CrossRef]
- Fleisch, H.; Bisaz, S. Mechanism of calcification: Inhibitory role of pyrophosphate. Nature 1962, 195, 911. [Google Scholar] [CrossRef]
- Paloian, N.J.; Giachelli, C.M. A current understanding of vascular calcification in CKD. Am. J. Physiol. Renal. Physiol. 2014, 307, F891–F900. [Google Scholar] [CrossRef]
- Shroff, R.C.; McNair, R.; Skepper, J.N.; Figg, N.; Schurgers, L.J.; Deanfield, J.; Rees, L.; Shanahan, C.M. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J. Am. Soc. Nephrol. 2010, 21, 103–112. [Google Scholar] [CrossRef]
- Sanchez-Duffhues, G.; Garcia de Vinuesa, A.; van de Pol, V.; Geerts, M.E.; de Vries, M.R.; Janson, S.G.; van Dam, H.; Lindeman, J.H.; Goumans, M.J.; Ten Dijke, P. Inflammation induces endothelial-to-mesenchymal transition and promotes vascular calcification through downregulation of BMPR2. J. Pathol. 2019, 247, 333–346. [Google Scholar] [CrossRef]
- Mikhaylova, L.; Malmquist, J.; Nurminskaya, M. Regulation of in vitro vascular calcification by BMP4, VEGF and Wnt3a. Calcif. Tissue Int. 2007, 81, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Kanno, Y.; Into, T.; Lowenstein, C.J.; Matsushita, K. Nitric oxide regulates vascular calcification by interfering with TGF- signalling. Cardiovasc. Res. 2008, 77, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Alique, M.; Bodega, G.; Corchete, E.; Garcia-Menendez, E.; de Sequera, P.; Luque, R.; Rodriguez-Padron, D.; Marques, M.; Portoles, J.; Carracedo, J.; et al. Microvesicles from indoxyl sulfate-treated endothelial cells induce vascular calcification in vitro. Comput. Struct Biotechnol. J. 2020, 18, 953–966. [Google Scholar] [CrossRef]
- Alique, M.; Ramirez-Carracedo, R.; Bodega, G.; Carracedo, J.; Ramirez, R. Senescent Microvesicles: A Novel Advance in Molecular Mechanisms of Atherosclerotic Calcification. Int. J. Mol. Sci. 2018, 19, 2003. [Google Scholar] [CrossRef]
- Chen, W.; Chen, Y. Cardiometabolic Syndrome and Vascular Calcification. Korean Soc. CardioMetabolic Syndr. 2022, 2, 1–21. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, X.; Wu, H. Metabolic Stress and Cardiovascular Disease in Diabetes Mellitus: The Role of Protein O-GlcNAc Modification. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1911–1924. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lim, J.; Lu, J.; Pedego, T.M.; Demer, L.; Tintut, Y. Protective Role of Smad6 in Inflammation-Induced Valvular Cell Calcification. J. Cell. Biochem. 2015, 116, 2354–2364. [Google Scholar] [CrossRef]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced glycation end products: Sparking the development of diabetic vascular injury. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef]
- Du, X.L.; Edelstein, D.; Rossetti, L.; Fantus, I.G.; Goldberg, H.; Ziyadeh, F.; Wu, J.; Brownlee, M. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc. Natl. Acad. Sci. USA 2000, 97, 12222–12226. [Google Scholar] [CrossRef]
- Andres-Bergos, J.; Tardio, L.; Larranaga-Vera, A.; Gomez, R.; Herrero-Beaumont, G.; Largo, R. The increase in O-linked N-acetylglucosamine protein modification stimulates chondrogenic differentiation both in vitro and in vivo. J. Biol. Chem. 2012, 287, 33615–33628. [Google Scholar] [CrossRef]
- El-Abbadi, M.M.; Pai, A.S.; Leaf, E.M.; Yang, H.Y.; Bartley, B.A.; Quan, K.K.; Ingalls, C.M.; Liao, H.W.; Giachelli, C.M. Phosphate feeding induces arterial medial calcification in uremic mice: Role of serum phosphorus, fibroblast growth factor-23, and osteopontin. Kidney Int. 2009, 75, 1297–1307. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Curinga, G.; Giachelli, C.M. Elevated extracellular calcium levels induce smooth muscle cell matrix mineralization in vitro. Kidney Int. 2004, 66, 2293–2299. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.X.; Kircelli, F.; O’Neill, K.D.; Chen, X.; Moe, S.M. Verapamil inhibits calcification and matrix vesicle activity of bovine vascular smooth muscle cells. Kidney Int. 2010, 77, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Pasch, A.; Farese, S.; Graber, S.; Wald, J.; Richtering, W.; Floege, J.; Jahnen-Dechent, W. Nanoparticle-based test measures overall propensity for calcification in serum. J. Am. Soc. Nephrol. JASN 2012, 23, 1744–1752. [Google Scholar] [CrossRef]
- Heiss, A.; Pipich, V.; Jahnen-Dechent, W.; Schwahn, D. Fetuin-A is a mineral carrier protein: Small angle neutron scattering provides new insight on Fetuin-A controlled calcification inhibition. Biophys. J. 2010, 99, 3986–3995. [Google Scholar] [CrossRef] [PubMed]
- Miura, Y.; Iwazu, Y.; Shiizaki, K.; Akimoto, T.; Kotani, K.; Kurabayashi, M.; Kurosu, H.; Kuro, O.M. Identification and quantification of plasma calciprotein particles with distinct physical properties in patients with chronic kidney disease. Sci. Rep. 2018, 8, 1256. [Google Scholar] [CrossRef] [PubMed]
- Koeppert, S.; Ghallab, A.; Peglow, S.; Winkler, C.F.; Graeber, S.; Buscher, A.; Hengstler, J.G.; Jahnen-Dechent, W. Live Imaging of Calciprotein Particle Clearance and Receptor Mediated Uptake: Role of Calciprotein Monomers. Front. Cell Dev. Biol. 2021, 9, 633925. [Google Scholar] [CrossRef]
- Koppert, S.; Buscher, A.; Babler, A.; Ghallab, A.; Buhl, E.M.; Latz, E.; Hengstler, J.G.; Smith, E.R.; Jahnen-Dechent, W. Cellular Clearance and Biological Activity of Calciprotein Particles Depend on Their Maturation State and Crystallinity. Front. Immunol. 2018, 9, 1991. [Google Scholar] [CrossRef]
- Wald, J.; Wiese, S.; Eckert, T.; Jahnen-Dechent, W.; Richtering, W.; Heiss, A. Formation and stability kinetics of calcium phosphate-fetuin-A colloidal particles probed by time-resolved dynamic light scattering. Soft Matter. 2011, 7, 2869–2874. [Google Scholar] [CrossRef]
- Heiss, A.; DuChesne, A.; Denecke, B.; Grotzinger, J.; Yamamoto, K.; Renne, T.; Jahnen-Dechent, W. Structural basis of calcification inhibition by alpha 2-HS glycoprotein/fetuin-A. Formation of colloidal calciprotein particles. J. Biol. Chem. 2003, 278, 13333–13341. [Google Scholar] [CrossRef] [PubMed]
- Shishkova, D.; Velikanova, E.; Sinitsky, M.; Tsepokina, A.; Gruzdeva, O.; Bogdanov, L.; Kutikhin, A. Calcium Phosphate Bions Cause Intimal Hyperplasia in Intact Aortas of Normolipidemic Rats through Endothelial Injury. Int. J. Mol. Sci. 2019, 20, 5728. [Google Scholar] [CrossRef] [PubMed]
- Shishkova, D.K.; Velikanova, E.A.; Bogdanov, L.A.; Sinitsky, M.Y.; Kostyunin, A.E.; Tsepokina, A.V.; Gruzdeva, O.V.; Mironov, A.V.; Mukhamadiyarov, R.A.; Glushkova, T.V.; et al. Calciprotein Particles Link Disturbed Mineral Homeostasis with Cardiovascular Disease by Causing Endothelial Dysfunction and Vascular Inflammation. Int. J. Mol. Sci. 2021, 22, 2458. [Google Scholar] [CrossRef] [PubMed]
- Shishkova, D.; Markova, V.; Sinitsky, M.; Tsepokina, A.; Velikanova, E.; Bogdanov, L.; Glushkova, T.; Kutikhin, A. Calciprotein Particles Cause Endothelial Dysfunction under Flow. Int. J. Mol. Sci. 2020, 21, 8802. [Google Scholar] [CrossRef] [PubMed]
- Aghagolzadeh, P.; Bachtler, M.; Bijarnia, R.; Jackson, C.; Smith, E.R.; Odermatt, A.; Radpour, R.; Pasch, A. Calcification of vascular smooth muscle cells is induced by secondary calciprotein particles and enhanced by tumor necrosis factor-alpha. Atherosclerosis 2016, 251, 404–414. [Google Scholar] [CrossRef]
- Viegas, C.S.B.; Santos, L.; Macedo, A.L.; Matos, A.A.; Silva, A.P.; Neves, P.L.; Staes, A.; Gevaert, K.; Morais, R.; Vermeer, C.; et al. Chronic Kidney Disease Circulating Calciprotein Particles and Extracellular Vesicles Promote Vascular Calcification: A Role for GRP (Gla-Rich Protein). Arter. Thromb. Vasc. Biol. 2018, 38, 575–587. [Google Scholar] [CrossRef]
- Ter Braake, A.D.; Eelderink, C.; Zeper, L.W.; Pasch, A.; Bakker, S.J.L.; de Borst, M.H.; Hoenderop, J.G.J.; de Baaij, J.H.F. Calciprotein particle inhibition explains magnesium-mediated protection against vascular calcification. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.-Eur. Ren. Assoc. 2020, 35, 765–773. [Google Scholar] [CrossRef]
- Feenstra, L.; Kutikhin, A.G.; Shishkova, D.K.; Buikema, H.; Zeper, L.W.; Bourgonje, A.R.; Krenning, G.; Hillebrands, J.L. Calciprotein Particles Induce Endothelial Dysfunction by Impairing Endothelial Nitric Oxide Metabolism. Arter. Thromb. Vasc. Biol. 2023, 43, 443–455. [Google Scholar] [CrossRef]
- Patel, L.; Bernard, L.M.; Elder, G.J. Sevelamer Versus Calcium-Based Binders for Treatment of Hyperphosphatemia in CKD: A Meta-Analysis of Randomized Controlled Trials. Clin. J. Am. Soc. Nephrol. CJASN 2016, 11, 232–244. [Google Scholar] [CrossRef]
- Block, G.A.; Wheeler, D.C.; Persky, M.S.; Kestenbaum, B.; Ketteler, M.; Spiegel, D.M.; Allison, M.A.; Asplin, J.; Smits, G.; Hoofnagle, A.N.; et al. Effects of phosphate binders in moderate CKD. J. Am. Soc. Nephrol. JASN 2012, 23, 1407–1415. [Google Scholar] [CrossRef]
- Toussaint, N.D.; Pedagogos, E.; Lioufas, N.M.; Elder, G.J.; Pascoe, E.M.; Badve, S.V.; Valks, A.; Block, G.A.; Boudville, N.; Cameron, J.D.; et al. A Randomized Trial on the Effect of Phosphate Reduction on Vascular End Points in CKD (IMPROVE-CKD). J. Am. Soc. Nephrol. JASN 2020, 31, 2653–2666. [Google Scholar] [CrossRef] [PubMed]
- Lioufas, N.; Pascoe, E.; Hawley, C.; Elder, G.; Badve, S.; Block, G.; Johnson, D.; Toussaint, N. Systematic Review and Meta-analyses of Effects of Phosphate-lowering Agents in Non-dialysis Chronic Kidney Disease. J. Am. Soc. Nephrol. JASN 2021, 33, 59–76. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, Y.; Fujii, N.; Shoji, T.; Hayashi, T.; Rakugi, H.; Isaka, Y. Hypomagnesemia is a significant predictor of cardiovascular and non-cardiovascular mortality in patients undergoing hemodialysis. Kidney Int. 2014, 85, 174–181. [Google Scholar] [CrossRef]
- Xiong, J.; He, T.; Wang, M.; Nie, L.; Zhang, Y.; Wang, Y.; Huang, Y.; Feng, B.; Zhang, J.; Zhao, J. Serum magnesium, mortality, and cardiovascular disease in chronic kidney disease and end-stage renal disease patients: A systematic review and meta-analysis. J. Nephrol. 2019, 32, 791–802. [Google Scholar] [CrossRef]
- Molnar, A.O.; Biyani, M.; Hammond, I.; Harmon, J.P.; Lavoie, S.; McCormick, B.; Sood, M.M.; Wagner, J.; Pena, E.; Zimmerman, D.L. Lower serum magnesium is associated with vascular calcification in peritoneal dialysis patients: A cross sectional study. BMC Nephrol. 2017, 18, 129. [Google Scholar] [CrossRef]
- Hruby, A.; O’Donnell, C.J.; Jacques, P.F.; Meigs, J.B.; Hoffmann, U.; McKeown, N.M. Magnesium intake is inversely associated with coronary artery calcification: The Framingham Heart Study. JACC Cardiovasc. Imaging 2014, 7, 59–69. [Google Scholar] [CrossRef]
- Leenders, N.H.J.; Bos, C.; Hoekstra, T.; Schurgers, L.J.; Vervloet, M.G.; Hoenderop, J.G.J. Dietary magnesium supplementation inhibits abdominal vascular calcification in an experimental animal model of chronic kidney disease. Nephrol. Dial. Transpl. 2022, 37, 1049–1058. [Google Scholar] [CrossRef]
- Yao, Z.; Xu, Y.; Ma, W.; Sun, X.Y.; Jia, S.; Zheng, Y.; Liu, X.; Fan, Y.; Wang, C. Magnesium Citrate Protects Against Vascular Calcification in an Adenine-induced Chronic Renal Failure Rat Model. J. Cardiovasc. Pharmacol. 2018, 72, 270–276. [Google Scholar] [CrossRef]
- Diaz-Tocados, J.M.; Peralta-Ramirez, A.; Rodriguez-Ortiz, M.E.; Raya, A.I.; Lopez, I.; Pineda, C.; Herencia, C.; Montes de Oca, A.; Vergara, N.; Steppan, S.; et al. Dietary magnesium supplementation prevents and reverses vascular and soft tissue calcifications in uremic rats. Kidney Int. 2017, 92, 1084–1099. [Google Scholar] [CrossRef]
- Zelt, J.G.; McCabe, K.M.; Svajger, B.; Barron, H.; Laverty, K.; Holden, R.M.; Adams, M.A. Magnesium Modifies the Impact of Calcitriol Treatment on Vascular Calcification in Experimental Chronic Kidney Disease. J. Pharmacol. Exp. Ther. 2015, 355, 451–462. [Google Scholar] [CrossRef]
- Salem, S.; Bruck, H.; Bahlmann, F.H.; Peter, M.; Passlick-Deetjen, J.; Kretschmer, A.; Steppan, S.; Volsek, M.; Kribben, A.; Nierhaus, M.; et al. Relationship between magnesium and clinical biomarkers on inhibition of vascular calcification. Am. J. Nephrol. 2012, 35, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Zhang, J.; Xu, J.; Cui, L.; Zhang, H.; Zhang, S.; Feng, X. Magnesium prevents beta-glycerophosphate-induced calcification in rat aortic vascular smooth muscle cells. Biomed Rep. 2015, 3, 593–597. [Google Scholar] [CrossRef] [PubMed]
- Louvet, L.; Buchel, J.; Steppan, S.; Passlick-Deetjen, J.; Massy, Z.A. Magnesium prevents phosphate-induced calcification in human aortic vascular smooth muscle cells. Nephrol. Dial. Transpl. 2013, 28, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Kircelli, F.; Peter, M.E.; Sevinc Ok, E.; Celenk, F.G.; Yilmaz, M.; Steppan, S.; Asci, G.; Ok, E.; Passlick-Deetjen, J. Magnesium reduces calcification in bovine vascular smooth muscle cells in a dose-dependent manner. Nephrol. Dial. Transpl. 2012, 27, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Henaut, L.; Massy, Z.A. Magnesium as a Calcification Inhibitor. Adv. Chronic. Kidney Dis. 2018, 25, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Ter Braake, A.D.; Shanahan, C.M.; de Baaij, J.H.F. Magnesium Counteracts Vascular Calcification: Passive Interference or Active Modulation? Arter. Thromb. Vasc. Biol. 2017, 37, 1431–1445. [Google Scholar] [CrossRef] [PubMed]
- Ter Braake, A.D.; Tinnemans, P.T.; Shanahan, C.M.; Hoenderop, J.G.J.; de Baaij, J.H.F. Magnesium prevents vascular calcification in vitro by inhibition of hydroxyapatite crystal formation. Sci. Rep. 2018, 8, 2069. [Google Scholar] [CrossRef]
- Ter Braake, A.D.; Vervloet, M.G.; de Baaij, J.H.F.; Hoenderop, J.G.J. Magnesium to prevent kidney disease-associated vascular calcification: Crystal clear? Nephrol. Dial. Transpl. 2022, 37, 421–429. [Google Scholar] [CrossRef]
- Lagier, R.; Baud, C.A. Magnesium whitlockite, a calcium phosphate crystal of special interest in pathology. Pathol. Res. Pr. 2003, 199, 329–335. [Google Scholar] [CrossRef]
- TenHuisen, K.S.; Brown, P.W. Effects of magnesium on the formation of calcium-deficient hydroxyapatite from CaHPO4.2H2O and Ca4(PO4)2O. J. Biomed. Mater. Res. 1997, 36, 306–314. [Google Scholar] [CrossRef]
- Ryan, L.M.; Cheung, H.S.; LeGeros, R.Z.; Kurup, I.V.; Toth, J.; Westfall, P.R.; McCarthy, G.M. Cellular responses to whitlockite. Calcif. Tissue Int. 1999, 65, 374–377. [Google Scholar] [CrossRef] [PubMed]
- LeGeros, R.Z.; Contiguglia, S.R.; Alfrey, A.C. Pathological calcifications associated with uremia: Two types of calcium phosphate deposits. Calcif. Tissue Res. 1973, 13, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Verberckmoes, S.C.; Persy, V.; Behets, G.J.; Neven, E.; Hufkens, A.; Zebger-Gong, H.; Muller, D.; Haffner, D.; Querfeld, U.; Bohic, S.; et al. Uremia-related vascular calcification: More than apatite deposition. Kidney Int. 2007, 71, 298–303. [Google Scholar] [CrossRef][Green Version]
- Schlieper, G.; Aretz, A.; Verberckmoes, S.C.; Kruger, T.; Behets, G.J.; Ghadimi, R.; Weirich, T.E.; Rohrmann, D.; Langer, S.; Tordoir, J.H.; et al. Ultrastructural analysis of vascular calcifications in uremia. J. Am. Soc. Nephrol. 2010, 21, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Blumenthal, N.C.; Betts, F.; Posner, A.S. Stabilization of amorphous calcium phosphate by Mg and ATP. Calcif. Tissue Res. 1977, 23, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Gelli, R.; Pucci, V.; Ridi, F.; Baglioni, P. A study on biorelevant calciprotein particles: Effect of stabilizing agents on the formation and crystallization mechanisms. J. Colloid Interface Sci. 2022, 620, 431–441. [Google Scholar] [CrossRef]
- Tangvoraphonkchai, K.; Davenport, A. Magnesium and Cardiovascular Disease. Adv. Chronic. Kidney Dis. 2018, 25, 251–260. [Google Scholar] [CrossRef]
- Sakaguchi, Y. The emerging role of magnesium in CKD. Clin. Exp. Nephrol. 2022, 26, 379–384. [Google Scholar] [CrossRef]
- Peride, I.; Tiglis, M.; Neagu, T.P.; Niculae, A.; Checherita, I.A. Magnesium-A More Important Role in CKD-MBD than We Thought. Diagnostics 2022, 12, 880. [Google Scholar] [CrossRef]
- Disthabanchong, S.; Srisuwarn, P. Mechanisms of Vascular Calcification in Kidney Disease. Adv. Chronic. Kidney Dis. 2019, 26, 417–426. [Google Scholar] [CrossRef]
- Alesutan, I.; Tuffaha, R.; Auer, T.; Feger, M.; Pieske, B.; Lang, F.; Voelkl, J. Inhibition of osteo/chondrogenic transformation of vascular smooth muscle cells by MgCl2 via calcium-sensing receptor. J. Hypertens. 2017, 35, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Montes de Oca, A.; Guerrero, F.; Martinez-Moreno, J.M.; Madueno, J.A.; Herencia, C.; Peralta, A.; Almaden, Y.; Lopez, I.; Aguilera-Tejero, E.; Gundlach, K.; et al. Magnesium inhibits Wnt/beta-catenin activity and reverses the osteogenic transformation of vascular smooth muscle cells. PLoS ONE 2014, 9, e89525. [Google Scholar] [CrossRef] [PubMed]
- Montezano, A.C.; Zimmerman, D.; Yusuf, H.; Burger, D.; Chignalia, A.Z.; Wadhera, V.; van Leeuwen, F.N.; Touyz, R.M. Vascular smooth muscle cell differentiation to an osteogenic phenotype involves TRPM7 modulation by magnesium. Hypertension 2010, 56, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Louvet, L.; Metzinger, L.; Buchel, J.; Steppan, S.; Massy, Z.A. Magnesium Attenuates Phosphate-Induced Deregulation of a MicroRNA Signature and Prevents Modulation of Smad1 and Osterix during the Course of Vascular Calcification. BioMed Res. Int. 2016, 2016, 7419524. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Yao, G.; Savoia, C.; Touyz, R.M. Transient receptor potential melastatin 7 ion channels regulate magnesium homeostasis in vascular smooth muscle cells: Role of angiotensin II. Circ. Res. 2005, 96, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.U.; Kirton, J.P.; Wilkinson, F.L.; Towers, E.; Sinha, S.; Rouhi, M.; Vizard, T.N.; Sage, A.P.; Martin, D.; Ward, D.T.; et al. Calcification is associated with loss of functional calcium-sensing receptor in vascular smooth muscle cells. Cardiovasc. Res. 2009, 81, 260–268. [Google Scholar] [CrossRef]
- Brown, E.M.; Gamba, G.; Riccardi, D.; Lombardi, M.; Butters, R.; Kifor, O.; Sun, A.; Hediger, M.A.; Lytton, J.; Hebert, S.C. Cloning and characterization of an extracellular Ca(2+)-sensing receptor from bovine parathyroid. Nature 1993, 366, 575–580. [Google Scholar] [CrossRef]
- Maier, J.A. Endothelial cells and magnesium: Implications in atherosclerosis. Clin. Sci. 2012, 122, 397–407. [Google Scholar] [CrossRef]
- Maier, J.A.; Bernardini, D.; Rayssiguier, Y.; Mazur, A. High concentrations of magnesium modulate vascular endothelial cell behaviour in vitro. Biochim. Biophys. Acta 2004, 1689, 6–12. [Google Scholar] [CrossRef]
- Rochelson, B.; Dowling, O.; Schwartz, N.; Metz, C.N. Magnesium sulfate suppresses inflammatory responses by human umbilical vein endothelial cells (HuVECs) through the NFkappaB pathway. J. Reprod. Immunol. 2007, 73, 101–107. [Google Scholar] [CrossRef]
- Lin, X.; Shan, S.K.; Xu, F.; Zhong, J.Y.; Wu, F.; Duan, J.Y.; Guo, B.; Li, F.X.; Wang, Y.; Zheng, M.H.; et al. The crosstalk between endothelial cells and vascular smooth muscle cells aggravates high phosphorus-induced arterial calcification. Cell Death Dis. 2022, 13, 650. [Google Scholar] [CrossRef] [PubMed]
- Ter Braake, A.D.; Smit, A.E.; Bos, C.; van Herwaarden, A.E.; Alkema, W.; van Essen, H.W.; Bravenboer, N.; Vervloet, M.G.; Hoenderop, J.G.J.; de Baaij, J.H.F. Magnesium prevents vascular calcification in Klotho deficiency. Kidney Int. 2020, 97, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Leibrock, C.; Pelzl, L.; Gawaz, M.; Pieske, B.; Alesutan, I.; Voelkl, J. Therapeutic Interference With Vascular Calcification-Lessons From Klotho-Hypomorphic Mice and Beyond. Front. Endocrinol. 2018, 9, 207. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, Y.; Hamano, T.; Obi, Y.; Monden, C.; Oka, T.; Yamaguchi, S.; Matsui, I.; Hashimoto, N.; Matsumoto, A.; Shimada, K.; et al. A Randomized Trial of Magnesium Oxide and Oral Carbon Adsorbent for Coronary Artery Calcification in Predialysis CKD. J. Am. Soc. Nephrol. JASN 2019, 30, 1073–1085. [Google Scholar] [CrossRef] [PubMed]
- Bressendorff, I.; Hansen, D.; Schou, M.; Kragelund, C.; Svensson, M.; Hashemi, B.; Kristensen, T.; Vrist, M.H.; Borg, R.; Tougaard, B.; et al. The Effect of Magnesium Supplementation on Vascular Calcification in CKD: A Randomized Clinical Trial (MAGiCAL-CKD). J. Am. Soc. Nephrol. JASN 2023, 34, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Goodkin, D.A.; Bragg-Gresham, J.L.; Koenig, K.G.; Wolfe, R.A.; Akiba, T.; Andreucci, V.E.; Saito, A.; Rayner, H.C.; Kurokawa, K.; Port, F.K.; et al. Association of comorbid conditions and mortality in hemodialysis patients in Europe, Japan, and the United States: The Dialysis Outcomes and Practice Patterns Study (DOPPS). J. Am. Soc. Nephrol. JASN 2003, 14, 3270–3277. [Google Scholar] [CrossRef]
- Pham, P.C.; Pham, P.M.; Pham, S.V.; Miller, J.M.; Pham, P.T. Hypomagnesemia in patients with type 2 diabetes. Clin. J. Am. Soc. Nephrol. CJASN 2007, 2, 366–373. [Google Scholar] [CrossRef]
- Katz, R.; Budoff, M.J.; O’Brien, K.D.; Wong, N.D.; Nasir, K. The metabolic syndrome and diabetes mellitus as predictors of thoracic aortic calcification as detected by non-contrast computed tomography in the Multi-Ethnic Study of Atherosclerosis. Diabet. Med. 2016, 33, 912–919. [Google Scholar] [CrossRef]
- Schmaderer, C.; Braunisch, M.C.; Suttmann, Y.; Lorenz, G.; Pham, D.; Haller, B.; Angermann, S.; Matschkal, J.; Renders, L.; Baumann, M.; et al. Reduced Mortality in Maintenance Haemodialysis Patients on High versus Low Dialysate Magnesium: A Pilot Study. Nutrients 2017, 9, 926. [Google Scholar] [CrossRef]
- Bressendorff, I.; Hansen, D.; Schou, M.; Pasch, A.; Brandi, L. The Effect of Increasing Dialysate Magnesium on Serum Calcification Propensity in Subjects with End Stage Kidney Disease: A Randomized, Controlled Clinical Trial. Clin. J. Am. Soc. Nephrol. 2018, 13, 1373–1380. [Google Scholar] [CrossRef]
- Bressendorff, I.; Hansen, D.; Pasch, A.; Holt, S.G.; Schou, M.; Brandi, L.; Smith, E.R. The effect of increasing dialysate magnesium on calciprotein particles, inflammation and bone markers: Post hoc analysis from a randomized controlled clinical trial. Nephrol. Dial. Transplant. 2021, 36, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Ritman, E.L. Small-animal CT-Its Difference from, and Impact on, Clinical CT. Nucl. Instrum. Methods Phys. Res. A 2007, 580, 968–970. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Gao, J.; Lv, Q.; Cai, H.; Wang, F.; Ye, R.; Liu, X. Calcification in Atherosclerotic Plaque Vulnerability: Friend or Foe? Front. Physiol. 2020, 11, 56. [Google Scholar] [CrossRef]
- Dweck, M.R.; Jones, C.; Joshi, N.V.; Fletcher, A.M.; Richardson, H.; White, A.; Marsden, M.; Pessotto, R.; Clark, J.C.; Wallace, W.A.; et al. Assessment of valvular calcification and inflammation by positron emission tomography in patients with aortic stenosis. Circulation 2012, 125, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Derlin, T.; Wisotzki, C.; Richter, U.; Apostolova, I.; Bannas, P.; Weber, C.; Mester, J.; Klutmann, S. In vivo imaging of mineral deposition in carotid plaque using 18F-sodium fluoride PET/CT: Correlation with atherogenic risk factors. J. Nucl. Med. 2011, 52, 362–368. [Google Scholar] [CrossRef]
- Dweck, M.R.; Jenkins, W.S.; Vesey, A.T.; Pringle, M.A.; Chin, C.W.; Malley, T.S.; Cowie, W.J.; Tsampasian, V.; Richardson, H.; Fletcher, A.; et al. 18F-sodium fluoride uptake is a marker of active calcification and disease progression in patients with aortic stenosis. Circ. Cardiovasc. Imaging 2014, 7, 371–378. [Google Scholar] [CrossRef]
- Dweck, M.R.; Chow, M.W.; Joshi, N.V.; Williams, M.C.; Jones, C.; Fletcher, A.M.; Richardson, H.; White, A.; McKillop, G.; van Beek, E.J.; et al. Coronary arterial 18F-sodium fluoride uptake: A novel marker of plaque biology. J. Am. Coll Cardiol. 2012, 59, 1539–1548. [Google Scholar] [CrossRef]
- Irkle, A.; Vesey, A.T.; Lewis, D.Y.; Skepper, J.N.; Bird, J.L.; Dweck, M.R.; Joshi, F.R.; Gallagher, F.A.; Warburton, E.A.; Bennett, M.R.; et al. Identifying active vascular microcalcification by (18)F-sodium fluoride positron emission tomography. Nat. Commun. 2015, 6, 7495. [Google Scholar] [CrossRef]
- Outcomes of a Higher vs. Lower Hemodialysate Magnesium Concentration (Dial-Mag Canada). Available online: https://clinicaltrials.gov/study/NCT04079582 (accessed on 21 December 2023).



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaslow, S.J.; Oliveira-Paula, G.H.; Chen, W. Magnesium and Vascular Calcification in Chronic Kidney Disease: Current Insights. Int. J. Mol. Sci. 2024, 25, 1155. https://doi.org/10.3390/ijms25021155
Zaslow SJ, Oliveira-Paula GH, Chen W. Magnesium and Vascular Calcification in Chronic Kidney Disease: Current Insights. International Journal of Molecular Sciences. 2024; 25(2):1155. https://doi.org/10.3390/ijms25021155
Chicago/Turabian StyleZaslow, Shari J., Gustavo H. Oliveira-Paula, and Wei Chen. 2024. "Magnesium and Vascular Calcification in Chronic Kidney Disease: Current Insights" International Journal of Molecular Sciences 25, no. 2: 1155. https://doi.org/10.3390/ijms25021155
APA StyleZaslow, S. J., Oliveira-Paula, G. H., & Chen, W. (2024). Magnesium and Vascular Calcification in Chronic Kidney Disease: Current Insights. International Journal of Molecular Sciences, 25(2), 1155. https://doi.org/10.3390/ijms25021155