2.1. Active and Inactive Classes Show Similar Key Chemical Properties
A diverse collection of scaffolds has been tested against ureases of different species, but the majority of studies have used jack bean urease as a surrogate due to its relatively low cost and commercial availability. Jack bean urease also shows generally good translation into the other species due to the highly conserved active site [
4]. Nevertheless, to avoid noise from aggregating data from different species when training our model, we chose to restrict our data to jack bean urease exclusively due to this organism having the largest set of activity data.
An initial dataset of 1614 active and inactive compounds (~1:1 ratio) tested against jack bean urease was assembled (
Table 1). Considering that compounds are assigned to either activity class according to their ratio of activity against the control in the corresponding assay, it is worth noting that the majority of the inactive compounds showed a maximum activity ratio of 2 (i.e., twice the control activity). This dataset was then split into two additional subsets: training and test set (75%:25%) (
Table 1) using clustered sampling (see methods) [
4], to avoid entire clusters being left out from training. An external validation set was also assembled in order to extend validation (external validation set). Additionally, while building the predictive model, newly reported compounds were continuously being collected, and these were used as a temporal dataset to assess the performance throughout time with newly reported scaffolds. All datasets showed a low Tanimoto mean value (0.13–0.15), and therefore, a high diversity in each dataset was identified (
Table 1). For all datasets, the majority of both active and inactive compounds had similar distributions of molecular weight and number of heavy atoms, lipophilicity (MolLogP) and topological polar surface area (TPSA) (
Figure S1).
2.2. Docking Score Is Predictive of Urease Inhibition
Having an understanding of the key structural underpinnings that drive ligand binding at the active site is an important step in the design of novel inhibitors. In this regard, docking is a very important tool, as it generally produces binding poses that can then be analyzed to determine key interactions that correlate with activity and can thus be used for screening. We carried out molecular docking calculations for all datasets using the X-ray structure of jack bean urease in the bound state (PDB 4H9M). We first validated the docking protocol with self-docking of acetohydroxamic acid and observed a high performance in predicting the crystallographic ligand pose (root mean square deviation < 1 Å). This protocol was then extended to the rest of the compounds. Since docking generates a number of energetically acceptable binding modes (poses), we compared the first docking pose and the top five poses and did not observe any significant differences in terms of docking scores between all poses, which varied mostly within 0.5 kcal/mol of each other (
Figure S2). However, since docking poses are determined by their docking score, which is often poorly correlated with the binding affinity [
10], we used all top five poses for subsequent analysis.
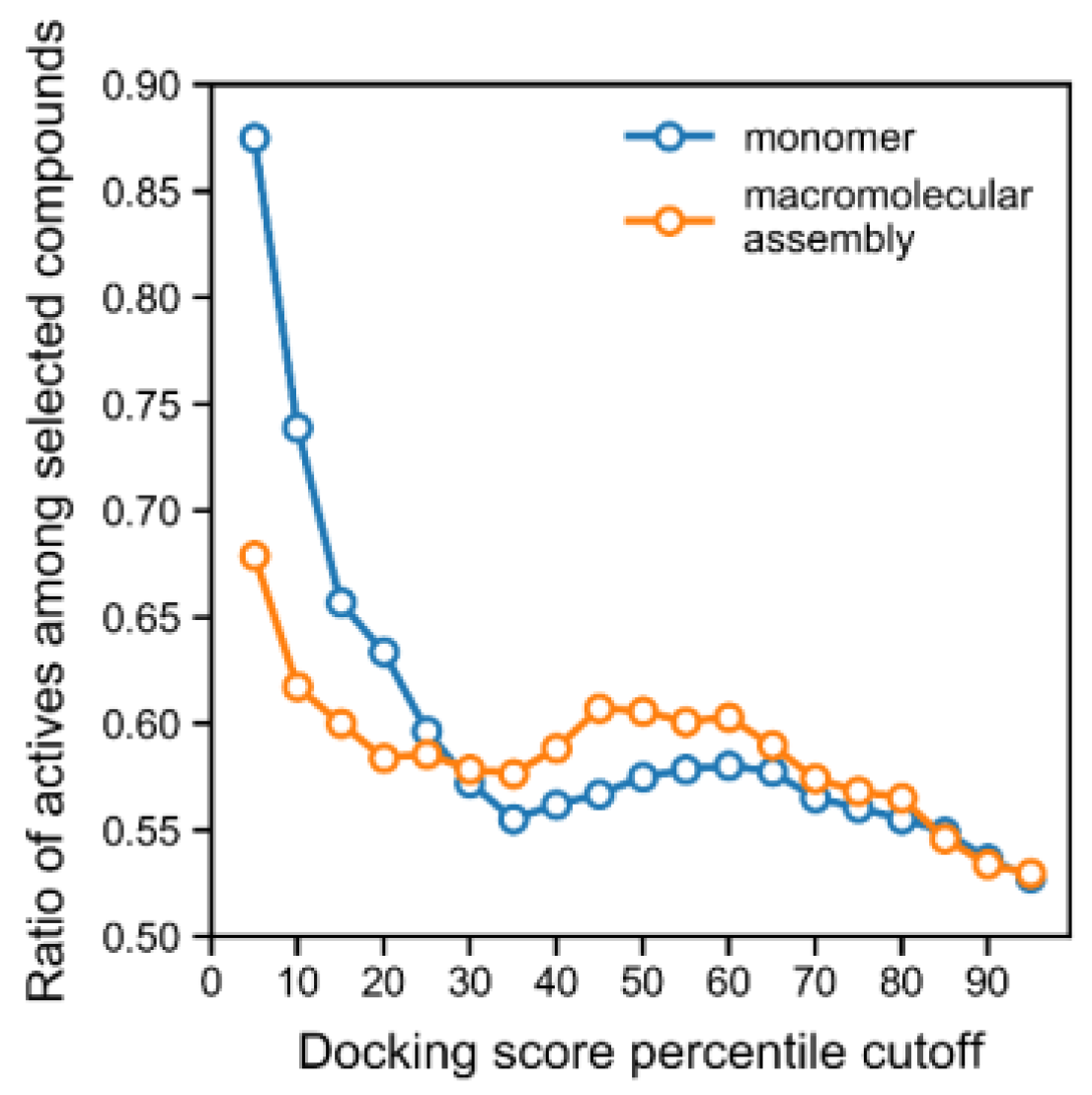
Typically, the correlation between the experimental inhibition and the docking energy is a poor surrogate for describing activity, and overall, we observed these limitations for a significant number of scaffolds produced from clustering the data. Indeed, docking scores failed to identify the majority of the most potent inhibitors (
Figure S3). Nevertheless, when considering the full training set, a high docking score was a good predictor of the active class (
p = 8.6 × 10
−19 Spearman’s test for the correlation between score and activity), and using an increasingly lower docking score cut-off led to the enrichment of compounds from the active class (
Figure 1). Indeed, all of the lowest docking scores (<−11), across all training and tests, corresponded entirely to active compounds (
Figure 1 and
Figure S3). Furthermore, we carried out docking on the macromolecular assembly made available in PDB (for the same PDB record, 4H9M), which consisted of a urease homodimer where the binding pocket is additionally shaped by a portion of the other monomer in the dimer. The calculations on this assembly structure interestingly did not improve the enrichment of the active class, but rather, degraded the predictive performance of the docking score, which could be observed with a smaller enrichment for the active class as a function of docking score cut-off (
Figure 1). Overall, the steadily increasing enrichment of actives for the “monomer” docking calculations shown in
Figure 1 shows that the docking score is a good predictor of activity and can be used as a filter in virtual screening.
The analysis of the 100 top-scoring compounds from docking revealed that these were predominantly substrate analogues and transition-state analogues as well as sulfonamides (known to be good binders of zinc). These top 100 compounds showed an average docking score of −10.86 and an average molecular weight of 527.20 g/mol (with 524.73 vs. 540.16 g/mol for active and inactive class, respectively). This small discrepancy between the averages for actives and inactives suggests that the relatively larger size of the inactive class may explain their apparently “improved” docking scores.
Even though the average activity ratio for the top 100 compounds was 2.414, the median was rather low, at 0.169, which means that the top-scoring compounds tend to be rather potent. Indeed, 84% of these compounds were actives (i.e., below a ratio of 1), 45% were under a ratio of 0.1, 13% under a ratio of 0.01 and 11% under 0.001. This means that the docking score would be theoretically able to find a compound with close to 2 μM of inhibitory activity with a 45% chance, and around 11% chance of finding one around with 200 nM of activity.
Since the docking score tends to penalize smaller molecules, there have been various approaches to cope with this bias and to allow finding more selective and potent inhibitors [
11]. One common approach is to calculate the ligand efficiency (i.e., docking score divided by number of heavy atoms). However, when we used ligand efficiency to sort the compounds in our training set, we observed that its predictive power was much lower (around 55%), with a Spearman’s test showing a non-significant correlation (
p = 0.21) even within categories of the same number of heavy atoms (
Figure S3).
Considering that the docking score allowed for a good enrichment profile for the active compounds (
Figure 1, monomer curve), we used this metric to predict actives. Whenever the docking score was used to filter compounds, we considered a cut-off of −9.43 kcal/mol, which corresponded to the 10th percentile of all docking scores. This cut-off was selected for being associated with an acceptable precision (0.74) when predicting actives (see
Figure 1). The predictive performance of this docking model was also validated on the external set and temporal set, and the previously selected cut-off yielded a precision for the active class of 0.76 and 0.87, respectively. On the other hand, docking scores ranged between −1.98 and −11.45 kcal/mol.
Azizian et al. used docking to screen 737,685 compounds from ZINC8 which led to the selection of three compounds of different classes, one of which showed inhibitory activity and was further modified to generate barbituric acid analogues with moderate activity [
12]. Similarly, docking of an in-house dataset of approximately 10,000 compounds led to the discovery of compounds with moderate activity [
13]. Even though these studies proposed moderate activities, the predictive power of docking was not validated with a set of known actives, and the docking score was used with sole reliance on a good self-docking performance.
2.3. Protein–Ligand Interaction Fingerprints (PLIF) Identify Key Residues of Interactions
A useful way of drawing information that helps guide drug design is to analyze the protein–ligand interactions in the docking poses. Even though the docking score itself showed good predictive performance, it lacked information regarding what structural elements were contributing to the activity. This can be addressed with PLIFs, as they provide additional three-dimensional information on the types of interaction observed. If many inhibitors interact with a protein via a similar type of interaction, potentially they have similar activity. Therefore, this allows “rescuing” compounds that may have been penalized by both the scoring function of docking or missed by the machine learning model [
14].
Both active and inactive classes of compounds on average interacted with a similar number of residues (5.85 ± 1.99 residues for actives vs. 5.97 ± 2.09 residues for the inactives), and similar amounts of interactions for each interaction type were observed for both classes (
Table S1). Surprisingly, despite several families of inhibitors being designed to bind to the metal center, slightly more metal interactions per compound were observed for the inactive class (0.740 and 0.925, respectively).
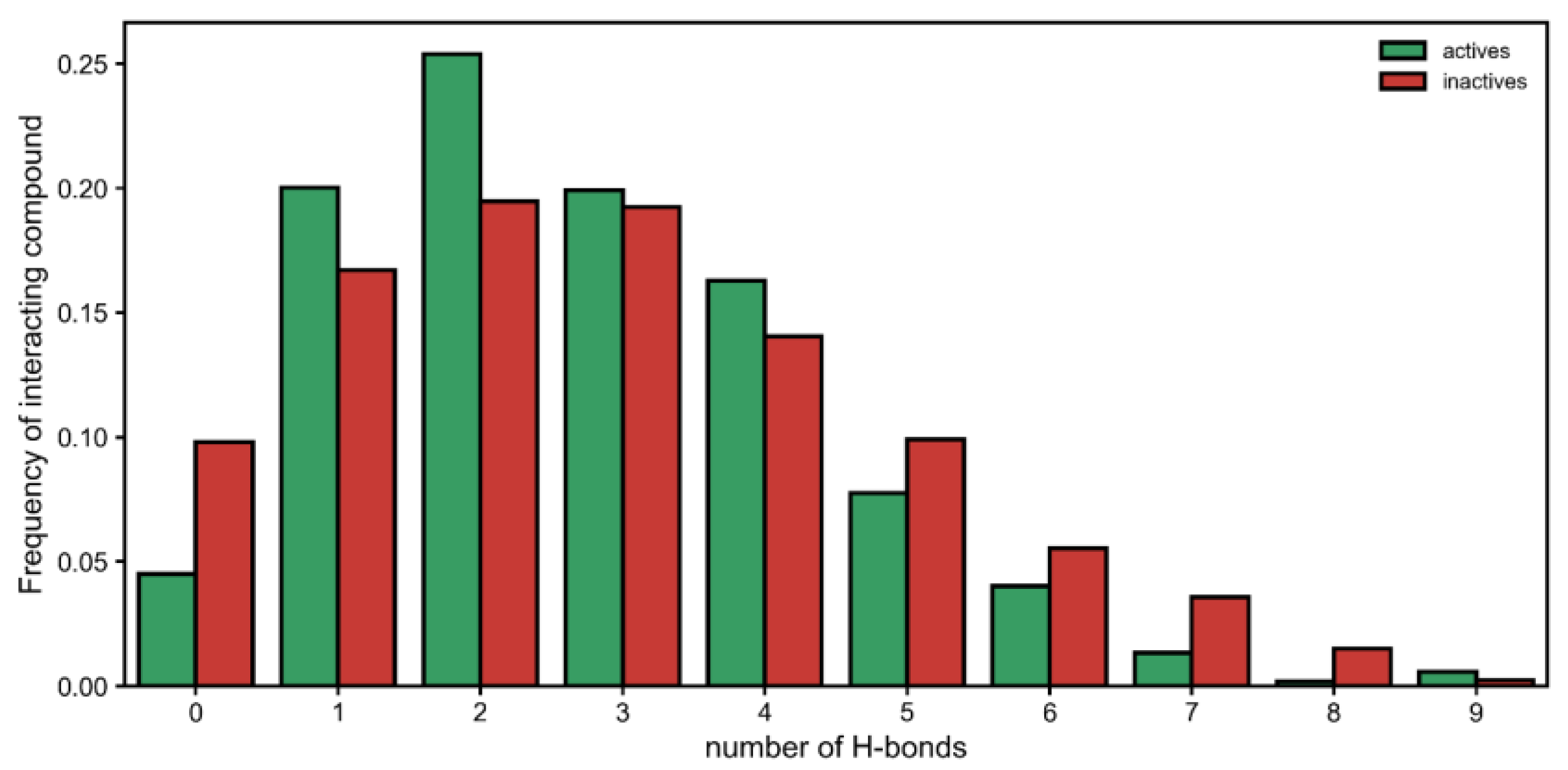
Moreover, by looking at each interaction type, we observed that there was a significant difference in hydrogen bonds established by active and inactive classes (
Figure 2). On one hand, the inactive compounds were more likely to have no hydrogen bonds with urease. Then, having one or two interactions seemed to be a better predictor of the active class. However, when five or more hydrogen bonds were established, this seemed to either (1) deteriorate activity (occurring more in inactives) or (2) become increasingly more irrelevant toward generating activity (
Figure 2). Therefore, having many interactions via hydrogen boding might be either detrimental or at least unhelpful.
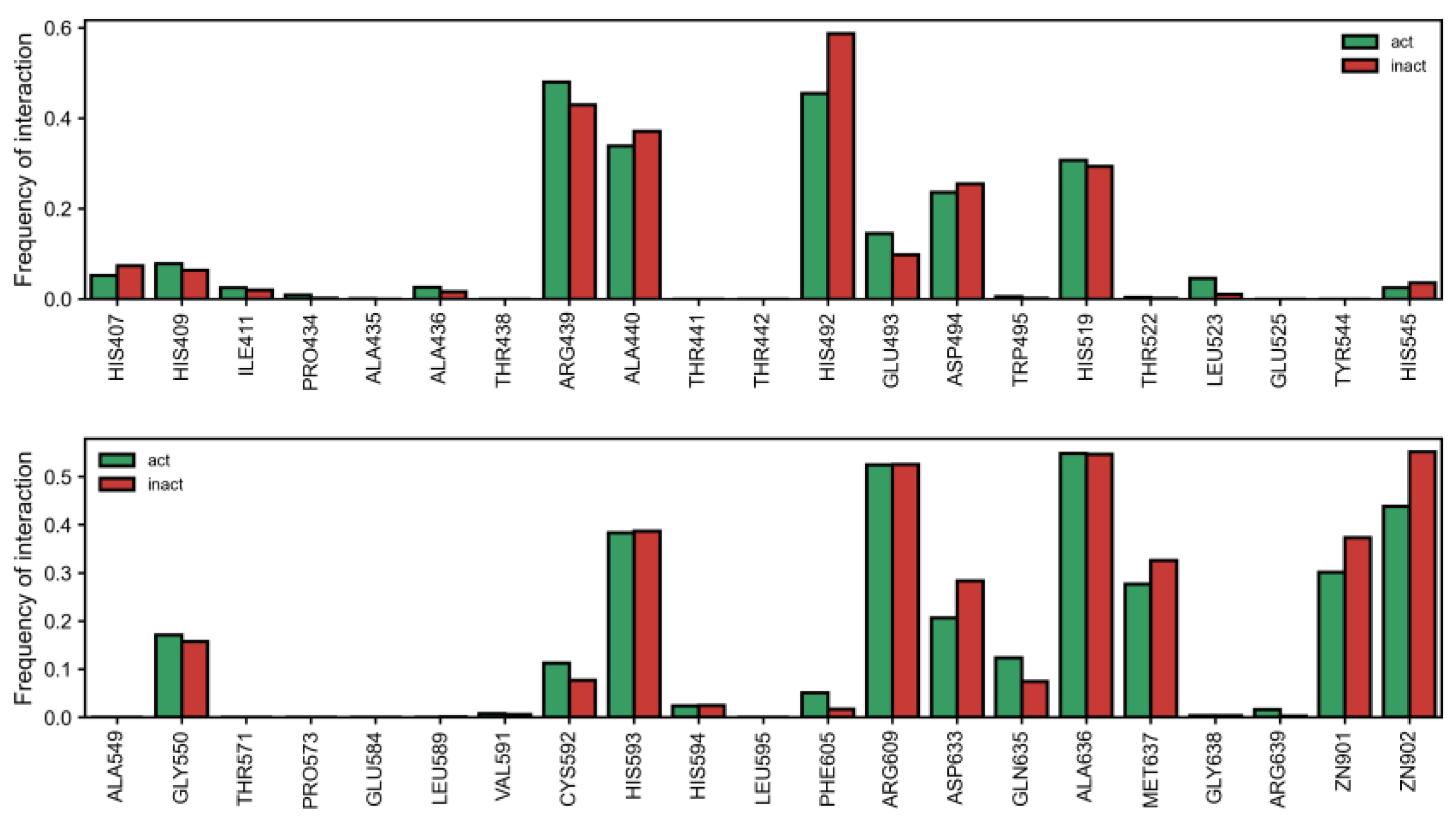
Together with the type of interaction, the types of residues that interact with compounds also appears to dictate activity in general. Surprisingly, we observed that more inactives interacted with the Zn atom (replacing Ni in the docking calculation) (
Figure 3). Furthermore, we also noticed a general trend where the residues most associated with the active class were those farther from the metal center (
Figure S4). However, given that a relatively large percentage of actives bind to the metal ions (~30% and ~40% to Zn901 and Zn902, respectively (
Figure 3), this suggests that lengthier compounds that are able to extend from the metal center outward and establish interactions away from the Zn (Ni) atoms may be desirable. The pattern of inhibitory interaction was also not solely due to a single residue, as compounds showed a complex interaction pattern with many residues. Nevertheless, it should be noted that there were clearly residues that interacted more than others with compounds of either class (
Figure 3). Besides Zn, several residues that interacted more frequently with inactive compounds were HIS492 (OR of 0.58), and to a lesser degree, ALA440 (OR = 0.87), ASP494 (OR = 0.90), ASP633 (OR = 0.65) and MET637 (OR = 0.79). Contrarily, interactions with ARG439 (OR = 1.22) were more frequently observed in the active class, which, alongside the higher percentage of pi-cation interactions, may explain their favorable profile (
Table S1). Indeed, pi-cations with ARG439 were almost double in active over inactive classes of compounds. Other important residues for activity were CYS592 (OR = 1.52), GLN635 (OR = 1.74) and GLU493 (OR = 1.55).
Next, we exhaustively mined interaction patterns that were predictive of activity by evaluating all combinations of urease residues (three, four or five simultaneous residues) where at least a minimum of 70% of 40 compounds were shown to be from the active class (see
Table 2). Starting with combinations of three residues, we observed the combinations of the same residues to be the most relevant to make predictions. In particular, it seems that ARG-609 is a very important residue and appears in many of these combinations to contribute to the enrichment of compounds belonging to the active class. Curiously, these combinations also pointed to the importance of ASP494. After mapping these residues in the crystal structure, we observed that ASP494 was right in front of the ARG609 and, therefore, they probably interact with one another via salt bridges, and it is likely that compounds that can insert into that site may disrupt this interaction.
Increasing the length of combinations to four residues resulted in different combinations of nearby residues but generally evidenced the positive role of ARG609, HIS519 and ASP633 in combinations associated with activity.
Further increasing the length of combinations to five revealed that the combination ALA636/ARG439/HIS492/MET637/ZN902 (combination 10) had a hit rate of 70% for 54 compounds (
Table 2). Again, ARG609 was found to be important for activity, and another interesting combination found was ASP633/GLY550/HIS519 with both metal ions of the active site (combination 14). In combination with these residues, HIS593 was relevant but only in combination with ASP494 and not GLU493. This was expected since in the pocket in this residue is positioned next to ARG609 and in front of ASP494, while relatively far from GLU493.
Building a decision tree with PLIFs revealed interesting patterns that differentiated the active from inactive class. For example, interaction with HIS492, MET637, and HIS593, but not with ALA636 and ZN901, was able to enrich the number of active over inactive compounds (126:51 ratio). This rule was even more interesting when we considered that the residues at the top of the corresponding branch individually interacted more frequently with inactive compounds (
Figure 3). Similarly, interaction with HIS492, MET637, not with HIS593, and with ARG-439 and GLY-638, enriched actives over inactive compounds (110:17). This highlights that even if interactions with ASP494 and MET637 are unfavorable, the interaction with ARG439 is overwhelmingly correlated with activity. For compounds that did not interact with HIS492 but interact with GLY550 and PHE605, if they then interacted with HIS593, we found a small enrichment of 102/78 for the active over inactive class, but if they did not interact with HIS593 but rather with MET637, the rule enriched with the inactive class (61 active to 122 inactive). Despite the observations that interactions with the metal centers seemed unfavorable, the decision tree algorithm found that compounds not interacting with HIS492 and GLY550 but interacting with ASP494, MET637 and ZN901 were two times more likely to be active (68/33). Even though decision trees provided interesting patterns of interaction, they performed the worst in separating active from inactive compounds compared to the combinations described above.
Overall, the PLIF analysis suggested a complex network of interactions between compounds and the jack bean urease active site. Many different combinations of residues seem to “fine tune” the interaction between a ligand and the protein and dictate inhibitory activity. In particular, a small set of residues appeared to be dominant in defining the type of interaction and activity.
2.4. Machine Learning Can Accurately Predict Activity of Urease Inhibitors
Many QSAR models have been developed for different classes of urease inhibitors [
15,
16,
17]. However, achieving a general and accurate prediction model that can be applied in structure-based virtual screening is still a very demanding task. For this purpose, we built a machine learning to produce a classification model that predicts whether a compound is active or not (activity defined by the ratio against a control) using aggregated chemical information extracted from docking and general chemical features.
We used three tree-based machine learning algorithms to build models that the predict active and inactive compounds: random forest (RF), extreme gradient boosting (XGBoost) and decision tree. The three candidate models were optimized through hyperparameter tuning using the grid search function (GridSearchCV), and optimized models were then tested against the test set (performance results are summarized in
Table 3). The RF model (built with 200 trees) showed the highest F1-score values for both active and inactive (0.80 vs. 0.77) and was selected as the best model. This model also outperformed the other two in terms of precision and recall, with the only exception being the recall of the inactive class, which showed the highest value in the XGBoost model.
Regarding the features that composed our model, the majority of the top features included physicochemical descriptors, and interestingly, many of these were 3D in nature: SpherocityIndex, RadiusOfGyration, Eccentricity and Asphericity (see details in RDKit’s documentation
https://www.rdkit.org/docs/source/rdkit.Chem.Descriptors3D.html, accessed on 2 April 2023). Furthermore, among the top 50 most important features, we also found Morgan Fringerprint bits and PLIF bits (such as HIS492, HIS593 or ALA636) to be relevant. Both the high importance of 3D physicochemical features as well as PLIFs suggested that addressing the three-dimensionality of the binding is important and provides useful information when modelling urease inhibition. This is something that has not been accounted for by other models previously published.
The optimal modelling conditions for the RF model were then selected to build an ensemble of 100 RF models where varying random seeds were used for the sampling of data and features during training. The resulting 100 predictions for each compound were then aggregated using majority voting to produce the final classifications. The trained model was subsequently tested on the test validation set (
Figure 4A) and the external set (
Figure 4B) and showed precision similar to that of the base model for the active class. However, enhancing the prediction with an applicability domain (AD) method, this model was able to effectively filter out unreliable predictions (i.e., mispredictions). As the AD cut-off became more stringent (lower value), the precision of predictions in both active and inactive classes steadily increased. In fact, using the most stringent cut-off, it was possible to achieve 100% precision when predicting the active class. The drawback of using such a stringent cut-off is that data coverage becomes very low (i.e., only 5.2% and 7.38% of predictions were accepted to be reliable). Based on these results, in order to use the model for screening purposes, we used the reliability-density neighborhood method to established an AD cut-off that corresponded to the maximum value at which we accepted predictions with sufficient reliability. This value was selected from the AD performance curve measured in the test set (
Figure 4A), where a cut-off of 0.258 was the highest value associated with ~90% precision for both active and inactive classes. This result not only validated the reliability of the predictions produced by the machine learning model but also validated the usefulness of the AD cut-off selected. Additionally, it is worth noting that both the AD curves for the test set and the external set showed an overall indirect correlation between the AD cut-off and precision of predictions. This meant that the method used to define the model’s AD has the desired relationship with prediction performance (i.e., as the cut-off becomes more stringent, prediction performance tends to increase steadily).
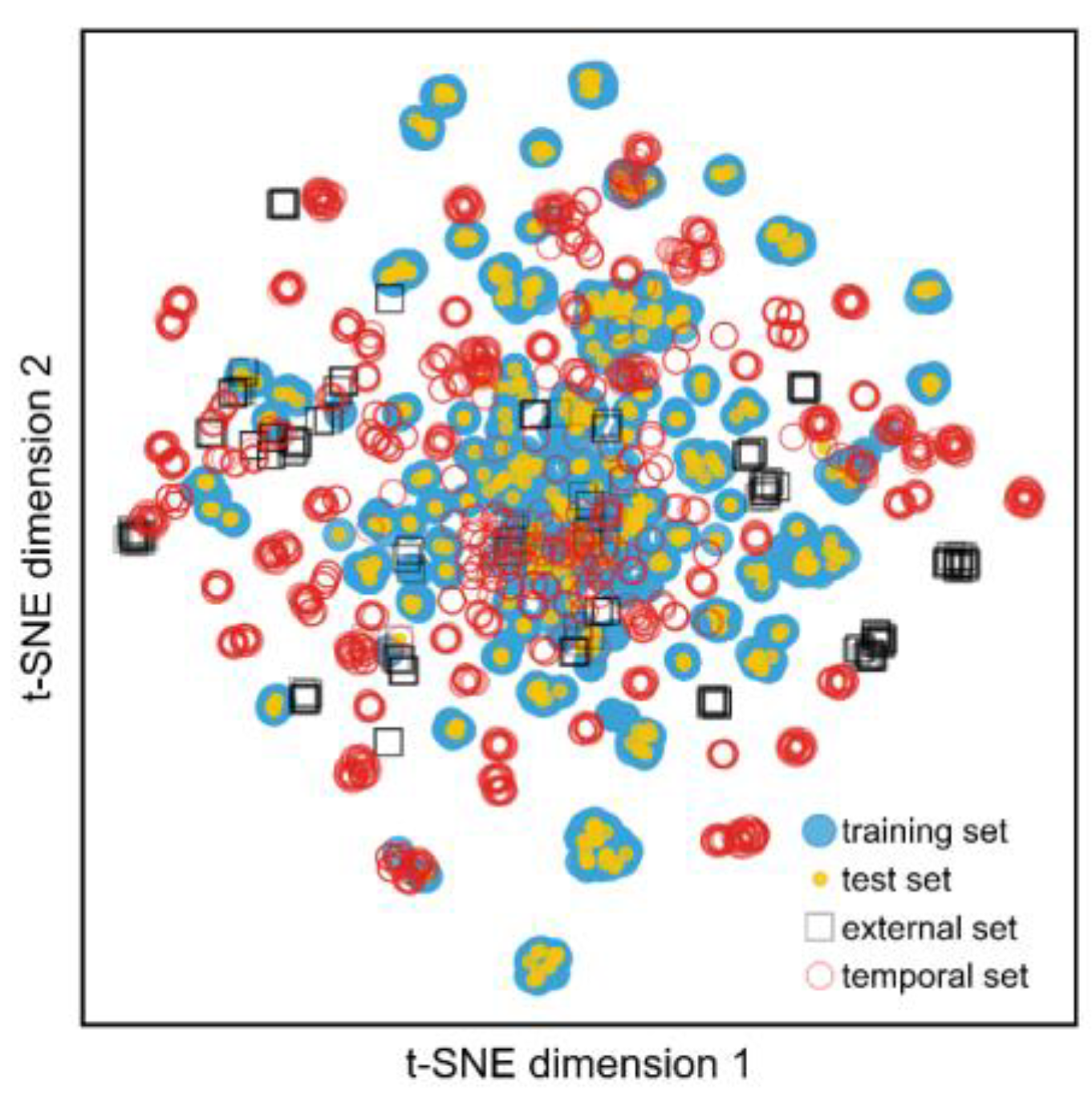
To further validate our model’s predictive performance, we tested it on data published after the publications in the training set (i.e., from 2021 to 2023). After curating this dataset, we were left with 728 compounds with activity data against jack bean urease. Contrary to previous tested datasets, we observed a precision of 0.67 and recall of 0.82 for the active class. Given the passage of time and its inherent chemical space drift, a portion of these predictions were extrapolated from the chemical space covered by the model (
Figure 5). Therefore, employing an AD filter was even more important in this scenario and, indeed, doing so yielded a significant increase in the precision and recall of the active class (i.e., 0.67 and 0.71, respectively). However, we should note that, in this case, an even more stringent AD (0.25) cut-off was used compared to the test set, since the original cut-off produced significantly poorer precision (0.67). This indicates that the model performance should be “recalibrated” periodically as new scaffolds are being published.
Previous in vitro screening efforts for sets of 84 and 3904 compounds were able to discover new urease inhibitors with different mechanisms of action [
6,
7]. Screening a large number of compounds is, however, usually costly, and for this reason, there has been an interest in predicting urease inhibition for virtual screening. Recently, using machine learning classification and regression models such as associative neural networks, k-nearest neighbors, XGBOOST, and WEKA-RF (RF model in WEKA), with cross-validation on a small dataset containing 647 compounds (518/129 split) showed the potential of predicting activity [
8]. Even though the experimental compounds (thiazole based scaffold) were all weaker than thiourea (around 50 μM and, therefore, classified as inactive by our model), the predicted activity was accurate [
8]. Using the combination of docking with a Monte Carlo method-based QSAR model using simplified molecular-input line-entry system (SMILES) and GRAPH descriptors on a dataset of 436 urease inhibitors from BindDB similarly achieved high prediction performance for the test sets but was not tested against an experimental validation or any external dataset [
18]. Alternatively, virtual screening with the 3D shape-based Rapid Overlay of Chemical Structures Tanimoto score based on the compound
o-chloro-hippurohydroxamic acid was used to screen the enamine library of 1.83 million compounds, of which 1700 were then docked into urease. Afterwards, eight compounds of different classes from the top 100 ranked compounds were tested in vitro, and all showed activity lower than thiourea (0.32–12.53 μM vs. 22.61 μM respectively) and were shown to be competitive and mixed-type inhibitors [
9]. Even results from the virtual screening described above are promising; these models were still very limited in the number of compounds used. Based on our results, this is very important, as even after using a relatively large dataset, its accuracy dropped significantly for new compounds with distinctive scaffolds. For instance, the loss in prediction power in the temporal dataset can be attributed to the drift from the original chemical space known by the model, given that the collected data for this dataset were published after the data that made up the training and test sets. On the other hand, a limitation of using docking-based methods for urease is the assumption of “competitive” binding. However, from the reported literature that was used to assemble this dataset, compounds tested against urease were mostly mixed-type inhibitors and several uncompetitive and non-competitive inhibitors as well as covalent inhibitors that were also present. Their inclusion thus may skew the results, as their binding to the active site is not realistic, and current models do not discriminate the type of mechanism by which compounds may act.
2.5. Proof-of-Concept Shows the Usefulness of Our Machine Learning Model: In Vitro Urease Inhibition Assay of an in-House Library
We then screened an in-house library of 106 compounds. Molecular docking yielded no actives, but the machine learning model predicted eight actives (albeit outside of the applicability domain), from which five were visually selected for in vitro testing. The compounds predicted to be active included some sulfonamide and sulfinamide analogues (
Figure 6), and these were further selected to test in vitro against jack bean urease (
Table 4). Benzene sulfonamides are a well-established class of urease inhibitors, with very potent compounds having been reported [
19], whereas the sulfinamide group has never been tested against urease, to the best of our knowledge. The selected compounds were, however, very closely similar to sulfonamides previously reported to have weak activity [
20].
As described in
Table 4, all compounds predicted by the ML model were outside the applicability domain and, therefore, the predictions should not be accepted. However, these were still tested, as they belong to an often-active scaffold. Indeed, using the AD filter would have prevented us from accepting two mispredictions. All active compounds showed moderate-to-strong activity, and compound
2 and
3 with the lowest IC
50 were further investigated for their mechanism of inhibition by a preliminary enzyme kinetics assay using different concentrations of compounds and substrate. A Lineweaver–Burk curve was then fitted to these data, being used to provide an initial indication of the mode of inhibition. For compound
2, the decrease in
Vmax and increase in
KM indicated a mixed-type inhibition (
Figure S5). On the other hand, for compound
3, it produced parallel lines in the double reciprocal plot, and both
Vmax and
KM decreased with increasing inhibitor concentration, suggesting an uncompetitive inhibition pattern. Even though our docking protocol is specifically meant to find competitive inhibitors, the fact that compound
3 was shown to be uncompetitive further highlights the difficulty in predictions based on this approach.
From the limited number of compounds, it appeared that the methyl group (compound 1) was detrimental for the activity of this class of compounds. On the other hand, the sulfinamide group seemed to be significantly more potent but led to an uncompetitive type of inhibition (compound 3). However, restricting the degrees of freedom of the compound with the introduction of a carbonyl group led to complete loss of activity (compound 4), even though the fragment of the molecule (compound 5) seemed to have activity similar to that of thiourea.


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}