
Network Biology Analyses and Dynamic Modeling of Gene Regulatory Networks under Drought Stress Reveal Major Transcriptional Regulators in Arabidopsis
 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
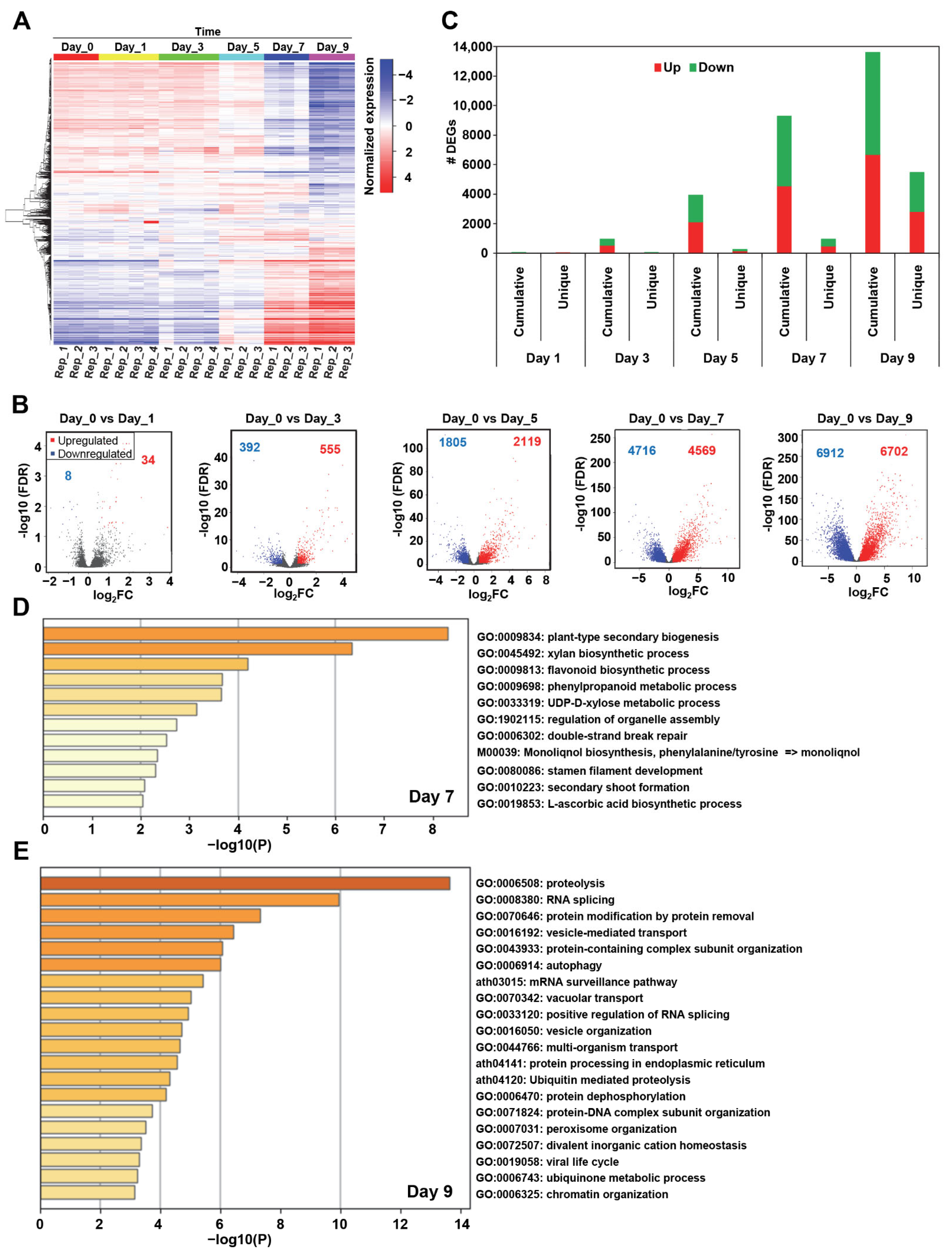
2.1. The Temporal Transcriptional Landscape of Arabidopsis during Drought Stress Identifies Unique Transcriptome Signatures and Specific Biological Pathways
2.2. Network Topology Analyses of Drought-Related Co-Expression Network Reveal 117 Major TFs
2.3. Dynamic Transcriptional Regulatory Modeling Discovered Major Bifurcated Events of the TFs, Which Participate in the “Water Deprivation Response” Pathway
2.4. Mathematical Transcriptional Simulation of Six Major Tfs Acquired by Integrating Drought-Related Co-Expression Data with Dynamic Transcriptional Modeling
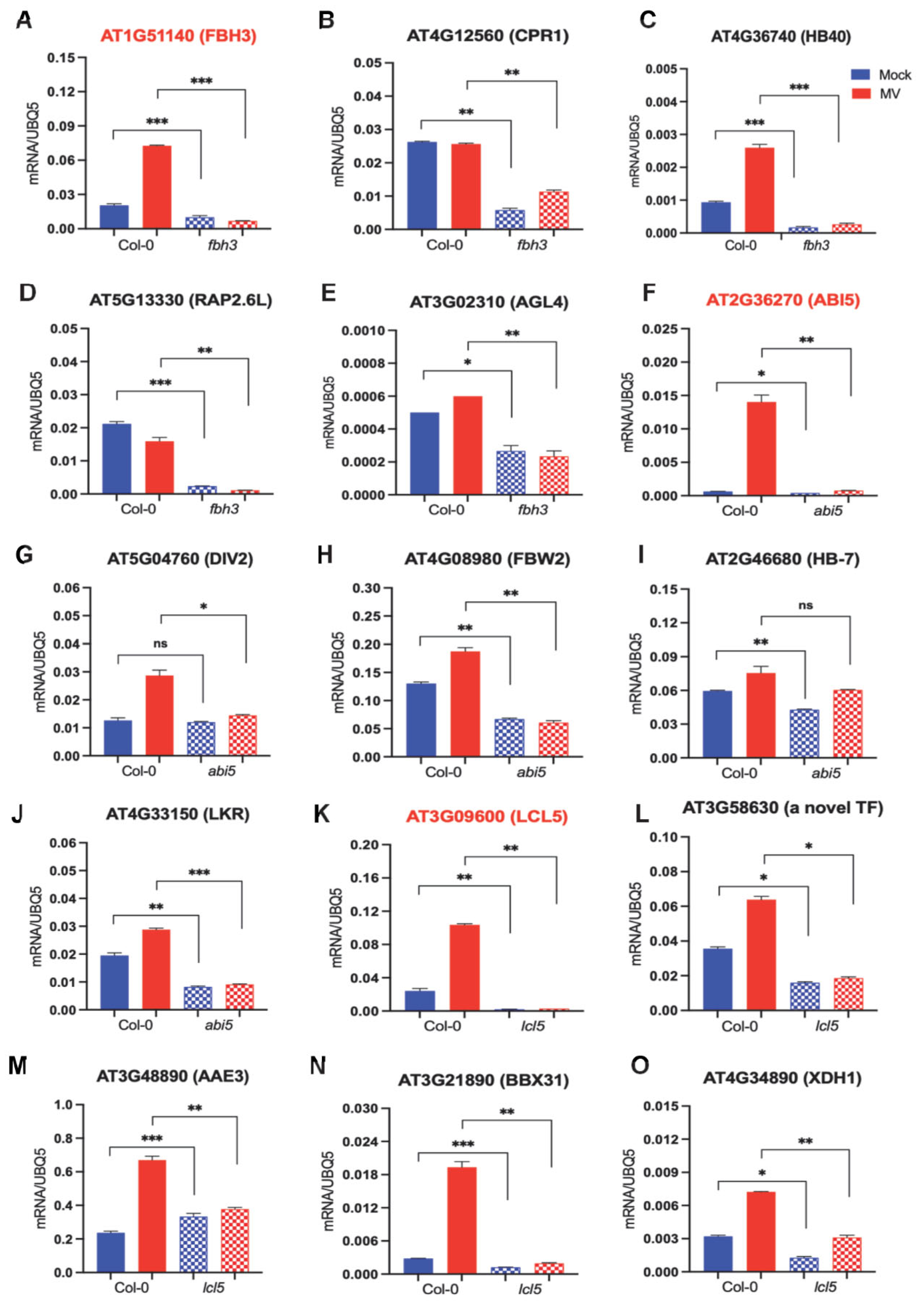
2.5. Experimental Evidence for the Regulatory Relationship between Major Drought-Related TFs and Their Target Genes
2.6. Experimental Evidence for the Regulatory Relationship between Significant Drought-Related TFs and Their Target Genes
2.7. Anthocyanin Production in the Mutants Corresponding to Selected Drought-Related TFs in Response to Reactive Oxygen Species (ROS) and Oxidative STRESS-Inducing Agents
3. Discussion
4. Materials and Methods
4.1. Plant Growth Conditions
4.2. Anthocyanin Quantification
4.3. RNA Extractions and Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
4.4. Drought Stress Analysis Data Acquisition
4.5. Differentially Expressed Genes (DEGs) Analysis and Functional Annotation Pathway Analyses
4.6. Weighted Gene Co-Expression Network Analysis (WGCNA) and Network Centrality Analyses
4.7. Dynamic Gene Regulatory Event Mining and Dynamic Simulations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rey, D.; Holman, I.P.; Knox, J.W. Developing drought resilience in irrigated agriculture in the face of increasing water scarcity. Reg. Environ. Chang. 2017, 17, 1527–1540. [Google Scholar] [CrossRef]
- Fahad, S.; Bajwa, A.A.; Nazir, U.; Anjum, S.A.; Farooq, A.; Zohaib, A.; Sadia, S.; Nasim, W.; Adkins, S.; Saud, S.; et al. Crop Production under Drought and Heat Stress: Plant Responses and Management Options. Front. Plant Sci. 2017, 8, 1147. [Google Scholar] [CrossRef]
- Leng, G.; Hall, J. Crop yield sensitivity of global major agricultural countries to droughts and the projected changes in the future. Sci. Total Environ. 2019, 654, 811–821. [Google Scholar] [CrossRef] [PubMed]
- Obidiegwu, J.E.; Bryan, G.J.; Jones, H.G.; Prashar, A. Coping with drought: Stress and adaptive responses in potato and perspectives for improvement. Front. Plant Sci. 2015, 6, 542. [Google Scholar] [CrossRef]
- Lv, L.; Zhang, W.; Sun, L.; Zhao, A.; Zhang, Y.; Wang, L.; Liu, Y.; Li, Z.; Li, H.; Chen, X. Gene co-expression network analysis to identify critical modules and candidate genes of drought-resistance in wheat. PLoS ONE 2020, 15, e0236186. [Google Scholar] [CrossRef]
- Tolosa, L.N.; Zhang, Z. The Role of Major Transcription Factors in Solanaceous Food Crops under Different Stress Conditions: Current and Future Perspectives. Plants 2020, 9, 56. [Google Scholar] [CrossRef]
- Ximenez-Embun, M.G.; Gonzalez-Guzman, M.; Arbona, V.; Gomez-Cadenas, A.; Ortego, F.; Castanera, P. Plant-Mediated Effects of Water Deficit on the Performance of Tetranychus evansi on Tomato Drought-Adapted Accessions. Front. Plant Sci. 2018, 9, 1490. [Google Scholar] [CrossRef]
- Bista, D.R.; Heckathorn, S.A.; Jayawardena, D.M.; Mishra, S.; Boldt, J.K. Effects of Drought on Nutrient Uptake and the Levels of Nutrient-Uptake Proteins in Roots of Drought-Sensitive and -Tolerant Grasses. Plants 2018, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Bouzid, M.; He, F.; Schmitz, G.; Hausler, R.E.; Weber, A.P.M.; Mettler-Altmann, T.; De Meaux, J. Arabidopsis species deploy distinct strategies to cope with drought stress. Ann. Bot. 2019, 124, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Gamboa-Tuz, S.D.; Pereira-Santana, A.; Zamora-Briseno, J.A.; Castano, E.; Espadas-Gil, F.; Ayala-Sumuano, J.T.; Keb-Llanes, M.A.; Sanchez-Teyer, F.; Rodriguez-Zapata, L.C. Transcriptomics and co-expression networks reveal tissue-specific responses and regulatory hubs under mild and severe drought in papaya (Carica papaya L.). Sci. Rep. 2018, 8, 14539. [Google Scholar] [CrossRef]
- Chen, M.; Zhu, X.; Zhang, Y.; Du, Z.; Chen, X.; Kong, X.; Sun, W.; Chen, C. Drought stress modify cuticle of tender tea leaf and mature leaf for transpiration barrier enhancement through common and distinct modes. Sci. Rep. 2020, 10, 6696. [Google Scholar] [CrossRef]
- Khadka, V.S.; Vaughn, K.; Xie, J.; Swaminathan, P.; Ma, Q.; Cramer, G.R.; Fennell, A.Y. Transcriptomic response is more sensitive to water deficit in shoots than roots of Vitis riparia (Michx.). BMC Plant Biol. 2019, 19, 72. [Google Scholar] [CrossRef]
- Bian, Z.; Wang, Y.; Zhang, X.; Grundy, S.; Hardy, K.; Yang, Q.; Lu, C. A Transcriptome Analysis Revealing the New Insight of Green Light on Tomato Plant Growth and Drought Stress Tolerance. Front. Plant Sci. 2021, 12, 649283. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.H.; Agarwal, S.; Rai, A.; Memaya, M.B.; Mehrotra, S.; Mehrotra, R. Co-expression network analysis of protein phosphatase 2A (PP2A) genes with stress-responsive genes in Arabidopsis thaliana reveals 13 key regulators. Sci. Rep. 2020, 10, 21480. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Yamaguchi-Shinozaki, K.; Shinozaki, K. The transcriptional regulatory network in the drought response and its crosstalk in abiotic stress responses including drought, cold, and heat. Front. Plant Sci. 2014, 5, 170. [Google Scholar] [CrossRef]
- Baldoni, E.; Genga, A.; Cominelli, E. Plant MYB Transcription Factors: Their Role in Drought Response Mechanisms. Int. J. Mol. Sci. 2015, 16, 15811–15851. [Google Scholar] [CrossRef]
- Singh, D.; Laxmi, A. Transcriptional regulation of drought response: A tortuous network of transcriptional factors. Front. Plant Sci. 2015, 6, 895. [Google Scholar] [CrossRef]
- Tully, J.P.; Hill, A.E.; Ahmed, H.M.; Whitley, R.; Skjellum, A.; Mukhtar, M.S. Expression-based network biology identifies immune-related functional modules involved in plant defense. BMC Genomics 2014, 15, 421. [Google Scholar] [CrossRef]
- Mishra, B.; Sun, Y.; Ahmed, H.; Liu, X.; Mukhtar, M.S. Global temporal dynamic landscape of pathogen-mediated subversion of Arabidopsis innate immunity. Sci. Rep. 2017, 7, 7849. [Google Scholar] [CrossRef]
- Alvarez, J.M.; Brooks, M.D.; Swift, J.; Coruzzi, G.M. Time-Based Systems Biology Approaches to Capture and Model Dynamic Gene Regulatory Networks. Annu. Rev. Plant Biol. 2021, 72, 105–131. [Google Scholar] [CrossRef]
- Mishra, B.; Sun, Y.; Howton, T.C.; Kumar, N.; Mukhtar, M.S. Dynamic modeling of transcriptional gene regulatory network uncovers distinct pathways during the onset of Arabidopsis leaf senescence. NPJ Syst. Biol. Appl. 2018, 4, 35. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.; Howton, T.C.; Sun, Y.; Weinberger, N.; Belkhadir, Y.; Mukhtar, M.S. Network biology discovers pathogen contact points in host protein-protein interactomes. Nat. Commun. 2018, 9, 2312. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Kumar, N.; Mukhtar, M.S. Network biology to uncover functional and structural properties of the plant immune system. Curr. Opin. Plant Biol. 2021, 62, 102057. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Kumar, N.; Mukhtar, M.S. Systems Biology and Machine Learning in Plant-Pathogen Interactions. Mol. Plant Microbe Interact. 2019, 32, 45–55. [Google Scholar] [CrossRef]
- Garbutt, C.C.; Bangalore, P.V.; Kannar, P.; Mukhtar, M.S. Getting to the edge: Protein dynamical networks as a new frontier in plant-microbe interactions. Front. Plant Sci. 2014, 5, 312. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, R.Z.; Zaidi, S.S.; Akhtar, K.P.; Strickler, S.; Woldemariam, M.; Mishra, B.; Mukhtar, M.S.; Scheffler, B.E.; Scheffler, J.A.; Jander, G.; et al. Transcriptomics reveals multiple resistance mechanisms against cotton leaf curl disease in a naturally immune cotton species, Gossypium arboreum. Sci. Rep. 2017, 7, 15880. [Google Scholar] [CrossRef]
- Zaidi, S.S.; Naqvi, R.Z.; Asif, M.; Strickler, S.; Shakir, S.; Shafiq, M.; Khan, A.M.; Amin, I.; Mishra, B.; Mukhtar, M.S.; et al. Molecular insight into cotton leaf curl geminivirus disease resistance in cultivated cotton (Gossypium hirsutum). Plant Biotechnol. J. 2020, 18, 691–706. [Google Scholar] [CrossRef]
- Naqvi, R.Z.; Zaidi, S.S.; Mukhtar, M.S.; Amin, I.; Mishra, B.; Strickler, S.; Mueller, L.A.; Asif, M.; Mansoor, S. Transcriptomic analysis of cultivated cotton Gossypium hirsutum provides insights into host responses upon whitefly-mediated transmission of cotton leaf curl disease. PLoS ONE 2019, 14, e0210011. [Google Scholar] [CrossRef]
- Mishra, B.; Athar, M.; Mukhtar, M.S. Transcriptional circuitry atlas of genetic diverse unstimulated murine and human macrophages define disparity in population-wide innate immunity. Sci. Rep. 2021, 11, 7373. [Google Scholar] [CrossRef]
- Bizouerne, E.; Buitink, J.; Vu, B.L.; Vu, J.L.; Esteban, E.; Pasha, A.; Provart, N.; Verdier, J.; Leprince, O. Gene co-expression analysis of tomato seed maturation reveals tissue-specific regulatory networks and hubs associated with the acquisition of desiccation tolerance and seed vigour. BMC Plant Biol. 2021, 21, 124. [Google Scholar] [CrossRef]
- Sakuraba, Y.; Kim, Y.S.; Han, S.H.; Lee, B.D.; Paek, N.C. The Arabidopsis Transcription Factor NAC016 Promotes Drought Stress Responses by Repressing AREB1 Transcription through a Trifurcate Feed-Forward Regulatory Loop Involving NAP. Plant Cell 2015, 27, 1771–1787. [Google Scholar] [CrossRef]
- Ding, J.; Hagood, J.S.; Ambalavanan, N.; Kaminski, N.; Bar-Joseph, Z. iDREM: Interactive visualization of dynamic regulatory networks. PLoS Comput. Biol. 2018, 14, e1006019. [Google Scholar] [CrossRef]
- Mishra, B.; Kumar, N.; Liu, J.; Pajerowska-Mukhtar, K.M. Dynamic Regulatory Event Mining by iDREM in Large-Scale Multi-omics Datasets During Biotic and Abiotic Stress in Plants. Methods Mol. Biol. 2021, 2328, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Di Cara, A.; Garg, A.; De Micheli, G.; Xenarios, I.; Mendoza, L. Dynamic simulation of regulatory networks using SQUAD. BMC Bioinformatics 2007, 8, 462. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, S.; Bashir, K.; Matsui, A.; Tanaka, M.; Seki, M. Transcriptomic Analysis of Soil-Grown Arabidopsis thaliana Roots and Shoots in Response to a Drought Stress. Front. Plant Sci. 2016, 7, 180. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Clark, N.M.; Buckner, E.; Fisher, A.P.; Nelson, E.C.; Nguyen, T.T.; Simmons, A.R.; de Luis Balaguer, M.A.; Butler-Smith, T.; Sheldon, P.J.; Bergmann, D.C.; et al. Stem-cell-ubiquitous genes spatiotemporally coordinate division through regulation of stem-cell-specific gene networks. Nat. Commun. 2019, 10, 5574. [Google Scholar] [CrossRef]
- Takahashi, Y.; Ebisu, Y.; Shimazaki, K.I. Reconstitution of Abscisic Acid Signaling from the Receptor to DNA via bHLH Transcription Factors. Plant Physiol. 2017, 174, 815–822. [Google Scholar] [CrossRef]
- Skubacz, A.; Daszkowska-Golec, A.; Szarejko, I. The Role and Regulation of ABI5 (ABA-Insensitive 5) in Plant Development, Abiotic Stress Responses and Phytohormone Crosstalk. Front. Plant Sci. 2016, 7, 1884. [Google Scholar] [CrossRef]
- Yang, P.; Wang, J.; Huang, F.Y.; Yang, S.; Wu, K. The Plant Circadian Clock and Chromatin Modifications. Genes 2018, 9, 561. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Mahmood, K.; Rothstein, S.J. ROS Induces Anthocyanin Production Via Late Biosynthetic Genes and Anthocyanin Deficiency Confers the Hypersensitivity to ROS-Generating Stresses in Arabidopsis. Plant Cell Physiol. 2017, 58, 1364–1377. [Google Scholar] [CrossRef]
- Cha, J.Y.; Kim, W.Y.; Kang, S.B.; Kim, J.I.; Baek, D.; Jung, I.J.; Kim, M.R.; Li, N.; Kim, H.J.; Nakajima, M.; et al. A novel thiol-reductase activity of Arabidopsis YUC6 confers drought tolerance independently of auxin biosynthesis. Nat. Commun. 2015, 6, 8041. [Google Scholar] [CrossRef]
- Farago, D.; Zsigmond, L.; Benyo, D.; Alcazar, R.; Rigo, G.; Ayaydin, F.; Rabilu, S.A.; Hunyadi-Gulyas, E.; Szabados, L. Small paraquat resistance proteins modulate paraquat and ABA responses and confer drought tolerance to overexpressing Arabidopsis plants. Plant Cell Environ. 2022, 45, 1985–2003. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Tian, Y.S.; Xing, X.J.; Peng, R.H.; Zhu, B.; Gao, J.J.; Yao, Q.H. Over-expression of AtGSTU19 provides tolerance to salt, drought and methyl viologen stresses in Arabidopsis. Physiol. Plant 2016, 156, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Chun, H.J.; Lim, L.H.; Cheong, M.S.; Baek, D.; Park, M.S.; Cho, H.M.; Lee, S.H.; Jin, B.J.; No, D.H.; Cha, Y.J.; et al. Arabidopsis CCoAOMT1 Plays a Role in Drought Stress Response via ROS- and ABA-Dependent Manners. Plants 2021, 10, 831. [Google Scholar] [CrossRef]
- Cruz de Carvalho, M.H. Drought stress and reactive oxygen species: Production, scavenging and signaling. Plant Signal. Behav. 2008, 3, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Salehi, M. Global water shortage and potable water safety; Today’s concern and tomorrow’s crisis. Environ. Int. 2022, 158, 106936. [Google Scholar] [CrossRef]
- Kumar, N.; Mishra, B.; Mehmood, A.; Mohammad, A.; Mukhtar, M.S. Integrative Network Biology Framework Elucidates Molecular Mechanisms of SARS-CoV-2 Pathogenesis. iScience 2020, 23, 101526. [Google Scholar] [CrossRef] [PubMed]
- McCormack, M.E.; Lopez, J.A.; Crocker, T.H.; Mukhtar, M.S. Making the right connections: Network biology and plant immune system dynamics. Curr. Plant Biol. 2016, 5, 2–12. [Google Scholar] [CrossRef]
- Liu, X.; Afrin, T.; Pajerowska-Mukhtar, K.M. Arabidopsis GCN2 kinase contributes to ABA homeostasis and stomatal immunity. Commun. Biol. 2019, 2, 302. [Google Scholar] [CrossRef]
- Miao, Z.; Han, Z.; Zhang, T.; Chen, S.; Ma, C. A systems approach to a spatio-temporal understanding of the drought stress response in maize. Sci. Rep. 2017, 7, 6590. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Xu, L.; Dossa, K.; Zhou, K.; Zhu, M.; Xie, H.; Tang, S.; Yu, Y.; Guo, X.; Zhou, B. Identification of putative drought-responsive genes in rice using gene co-expression analysis. Bioinformation 2019, 15, 480–489. [Google Scholar] [CrossRef]
- Pajerowska-Mukhtar, K.M.; Wang, W.; Tada, Y.; Oka, N.; Tucker, C.L.; Fonseca, J.P.; Dong, X. The HSF-like transcription factor TBF1 is a major molecular switch for plant growth-to-defense transition. Curr. Biol. 2012, 22, 103–112. [Google Scholar] [CrossRef]
- Ma, S.; Bohnert, H.J.; Dinesh-Kumar, S.P. AtGGM2014, an Arabidopsis gene co-expression network for functional studies. Sci. China Life Sci. 2015, 58, 276–286. [Google Scholar] [CrossRef]
- Shaik, R.; Ramakrishna, W. Genes and co-expression modules common to drought and bacterial stress responses in Arabidopsis and rice. PLoS ONE 2013, 8, e77261. [Google Scholar] [CrossRef]
- Chen, P.; Wei, F.; Jian, H.; Hu, T.; Wang, B.; Lv, X.; Wang, H.; Fu, X.; Yu, S.; Wei, H.; et al. A Comprehensive Gene Co-Expression Network Analysis Reveals a Role of GhWRKY46 in Responding to Drought and Salt Stresses. Int. J. Mol. Sci. 2022, 23, 2181. [Google Scholar] [CrossRef]
- Liu, S.; Zenda, T.; Dong, A.; Yang, Y.; Wang, N.; Duan, H. Global Transcriptome and Weighted Gene Co-expression Network Analyses of Growth-Stage-Specific Drought Stress Responses in Maize. Front. Genet. 2021, 12, 645443. [Google Scholar] [CrossRef]
- Desvoyes, B.; Gutierrez, C. Roles of plant retinoblastoma protein: Cell cycle and beyond. EMBO J. 2020, 39, e105802. [Google Scholar] [CrossRef] [PubMed]
- Simmons, A.R.; Davies, K.A.; Wang, W.; Liu, Z.; Bergmann, D.C. SOL1 and SOL2 regulate fate transition and cell divisions in the Arabidopsis stomatal lineage. Development 2019, 14, dev171066. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, T.; Sari, N.; Misaki, R.; Fujiyama, K. Biochemical characterization of Arabidopsis clade F polygalacturonase shows a substrate preference toward oligogalacturonic acids. J. Biosci. Bioeng. 2022, 133, 1–7. [Google Scholar] [CrossRef]
- Jing, Y.; Shi, L.; Li, X.; Zheng, H.; Gao, J.; Wang, M.; He, L.; Zhang, W. OXS2 is Required for Salt Tolerance Mainly through Associating with Salt Inducible Genes, CA1 and Araport11, in Arabidopsis. Sci. Rep. 2019, 9, 20341. [Google Scholar] [CrossRef] [PubMed]
- Song, J.T.; Lu, H.; Greenberg, J.T. Divergent roles in Arabidopsis thaliana development and defense of two homologous genes, aberrant growth and death2 and AGD2-LIKE DEFENSE RESPONSE PROTEIN1, encoding novel aminotransferases. Plant Cell 2004, 16, 353–366. [Google Scholar] [CrossRef] [PubMed]
- Mukhtar, M.S.; McCormack, M.E.; Argueso, C.T.; Pajerowska-Mukhtar, K.M. Pathogen Tactics to Manipulate Plant Cell Death. Curr. Biol. 2016, 26, R608–R619. [Google Scholar] [CrossRef]
- Gonzalez-Fuente, M.; Carrere, S.; Monachello, D.; Marsella, B.G.; Cazale, A.C.; Zischek, C.; Mitra, R.M.; Reze, N.; Cottret, L.; Mukhtar, M.S.; et al. EffectorK, a comprehensive resource to mine for Ralstonia, Xanthomonas, and other published effector interactors in the Arabidopsis proteome. Mol. Plant Pathol. 2020, 21, 1257–1270. [Google Scholar] [CrossRef]
- Sun, Y.; Detchemendy, T.W.; Pajerowska-Mukhtar, K.M.; Mukhtar, M.S. NPR1 in JazzSet with Pathogen Effectors. Trends Plant Sci. 2018, 23, 469–472. [Google Scholar] [CrossRef]
- Sun, F.; Liu, P.; Xu, J.; Dong, H. Mutation in RAP2.6L, a transactivator of the ERF transcription factor family, enhances Arabidopsis resistance to Pseudomonas syringae. Physiol. Mol. Plant Pathol. 2010, 74, 295–302. [Google Scholar] [CrossRef]
- Earley, K.; Smith, M.; Weber, R.; Gregory, B.; Poethig, R. An endogenous F-box protein regulates ARGONAUTE1 in Arabidopsis thaliana. Silence 2010, 1, 15. [Google Scholar] [CrossRef] [PubMed]
- Fang, Q.; Wang, Q.; Mao, H.; Xu, J.; Wang, Y.; Hu, H.; He, S.; Tu, J.; Cheng, C.; Tian, G.; et al. AtDIV2, an R-R-type MYB transcription factor of Arabidopsis, negatively regulates salt stress by modulating ABA signaling. Plant Cell Rep. 2018, 37, 1499–1511. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Wang, H.; Jia, X.; Gao, H.; Mao, K.; Ma, F. The HD-Zip I transcription factor MdHB7-like confers tolerance to salinity in transgenic apple (Malus domestica). Physiol. Plant 2021, 172, 1452–1464. [Google Scholar] [CrossRef]
- Zhao, S.; Gao, H.; Jia, X.; Wei, J.; Mao, K.; Ma, F. MdHB-7 Regulates Water Use Efficiency in Transgenic Apple (Malus domestica) Under Long-Term Moderate Water Deficit. Front. Plant Sci. 2021, 12, 740492. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Liu, G.; Wei, Y.; Chan, Z. The zinc-finger transcription factor ZAT6 is essential for hydrogen peroxide induction of anthocyanin synthesis in Arabidopsis. Plant Mol. Biol. 2018, 97, 165–176. [Google Scholar] [CrossRef]
- McCormack, M.E.; Liu, X.; Jordan, M.R.; Pajerowska-Mukhtar, K.M. An improved high-throughput screening assay for tunicamycin sensitivity in Arabidopsis seedlings. Front. Plant Sci. 2015, 6, 663. [Google Scholar] [CrossRef]
- Barrett, T.; Troup, D.B.; Wilhite, S.E.; Ledoux, P.; Rudnev, D.; Evangelista, C.; Kim, I.F.; Soboleva, A.; Tomashevsky, M.; Marshall, K.A.; et al. NCBI GEO: Archive for high-throughput functional genomic data. Nucleic Acids Res. 2009, 37, D885–D890. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.X.; Son, E.W.; Yao, R. iDEP: An integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinform. 2018, 19, 534. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Horvath, S.; Dong, J. Geometric interpretation of gene coexpression network analysis. PLoS Comput. Biol. 2008, 4, e1000117. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Hagberg, A.; Swart, P.; S Chult, D. Exploring network structure, dynamics, and function using NetworkX. In Proceedings of the SCIPY 08, Pasadena, CA, USA, 21 August 2008. [Google Scholar]
- Palaniswamy, S.K.; James, S.; Sun, H.; Lamb, R.S.; Davuluri, R.V.; Grotewold, E. AGRIS and AtRegNet. a platform to link cis-regulatory elements and transcription factors into regulatory networks. Plant Physiol. 2006, 140, 818–829. [Google Scholar] [CrossRef]
- O’Malley, R.C.; Huang, S.C.; Song, L.; Lewsey, M.G.; Bartlett, A.; Nery, J.R.; Galli, M.; Gallavotti, A.; Ecker, J.R. Cistrome and Epicistrome Features Shape the Regulatory DNA Landscape. Cell 2016, 166, 1598. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.P.; Lin, J.J.; Li, W.H. Positional distribution of transcription factor binding sites in Arabidopsis thaliana. Sci. Rep. 2016, 6, 25164. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.R.; Vaneechoutte, D.; Van de Velde, J.; Vandepoele, K. TF2Network: Predicting transcription factor regulators and gene regulatory networks in Arabidopsis using publicly available binding site information. Nucleic Acids Res. 2018, 46, e31. [Google Scholar] [CrossRef]
- Jin, J.; He, K.; Tang, X.; Li, Z.; Lv, L.; Zhao, Y.; Luo, J.; Gao, G. An Arabidopsis Transcriptional Regulatory Map Reveals Distinct Functional and Evolutionary Features of Novel Transcription Factors. Mol. Biol. Evol. 2015, 32, 1767–1773. [Google Scholar] [CrossRef]
- Rhee, S.Y.; Beavis, W.; Berardini, T.Z.; Chen, G.; Dixon, D.; Doyle, A.; Garcia-Hernandez, M.; Huala, E.; Lander, G.; Montoya, M.; et al. The Arabidopsis Information Resource (TAIR): A model organism database providing a centralized, curated gateway to Arabidopsis biology, research materials and community. Nucleic Acids Res. 2003, 31, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Cote, R.; Reisinger, F.; Martens, L.; Barsnes, H.; Vizcaino, J.A.; Hermjakob, H. The Ontology Lookup Service: Bigger and better. Nucleic Acids Res. 2010, 38, W155–W160. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, N.; Mishra, B.K.; Liu, J.; Mohan, B.; Thingujam, D.; Pajerowska-Mukhtar, K.M.; Mukhtar, M.S. Network Biology Analyses and Dynamic Modeling of Gene Regulatory Networks under Drought Stress Reveal Major Transcriptional Regulators in Arabidopsis. Int. J. Mol. Sci. 2023, 24, 7349. https://doi.org/10.3390/ijms24087349
Kumar N, Mishra BK, Liu J, Mohan B, Thingujam D, Pajerowska-Mukhtar KM, Mukhtar MS. Network Biology Analyses and Dynamic Modeling of Gene Regulatory Networks under Drought Stress Reveal Major Transcriptional Regulators in Arabidopsis. International Journal of Molecular Sciences. 2023; 24(8):7349. https://doi.org/10.3390/ijms24087349
Chicago/Turabian StyleKumar, Nilesh, Bharat K. Mishra, Jinbao Liu, Binoop Mohan, Doni Thingujam, Karolina M. Pajerowska-Mukhtar, and M. Shahid Mukhtar. 2023. "Network Biology Analyses and Dynamic Modeling of Gene Regulatory Networks under Drought Stress Reveal Major Transcriptional Regulators in Arabidopsis" International Journal of Molecular Sciences 24, no. 8: 7349. https://doi.org/10.3390/ijms24087349
APA StyleKumar, N., Mishra, B. K., Liu, J., Mohan, B., Thingujam, D., Pajerowska-Mukhtar, K. M., & Mukhtar, M. S. (2023). Network Biology Analyses and Dynamic Modeling of Gene Regulatory Networks under Drought Stress Reveal Major Transcriptional Regulators in Arabidopsis. International Journal of Molecular Sciences, 24(8), 7349. https://doi.org/10.3390/ijms24087349