

Investigation of the Presence of DNA in Human Blood Plasma Small Extracellular Vesicles



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
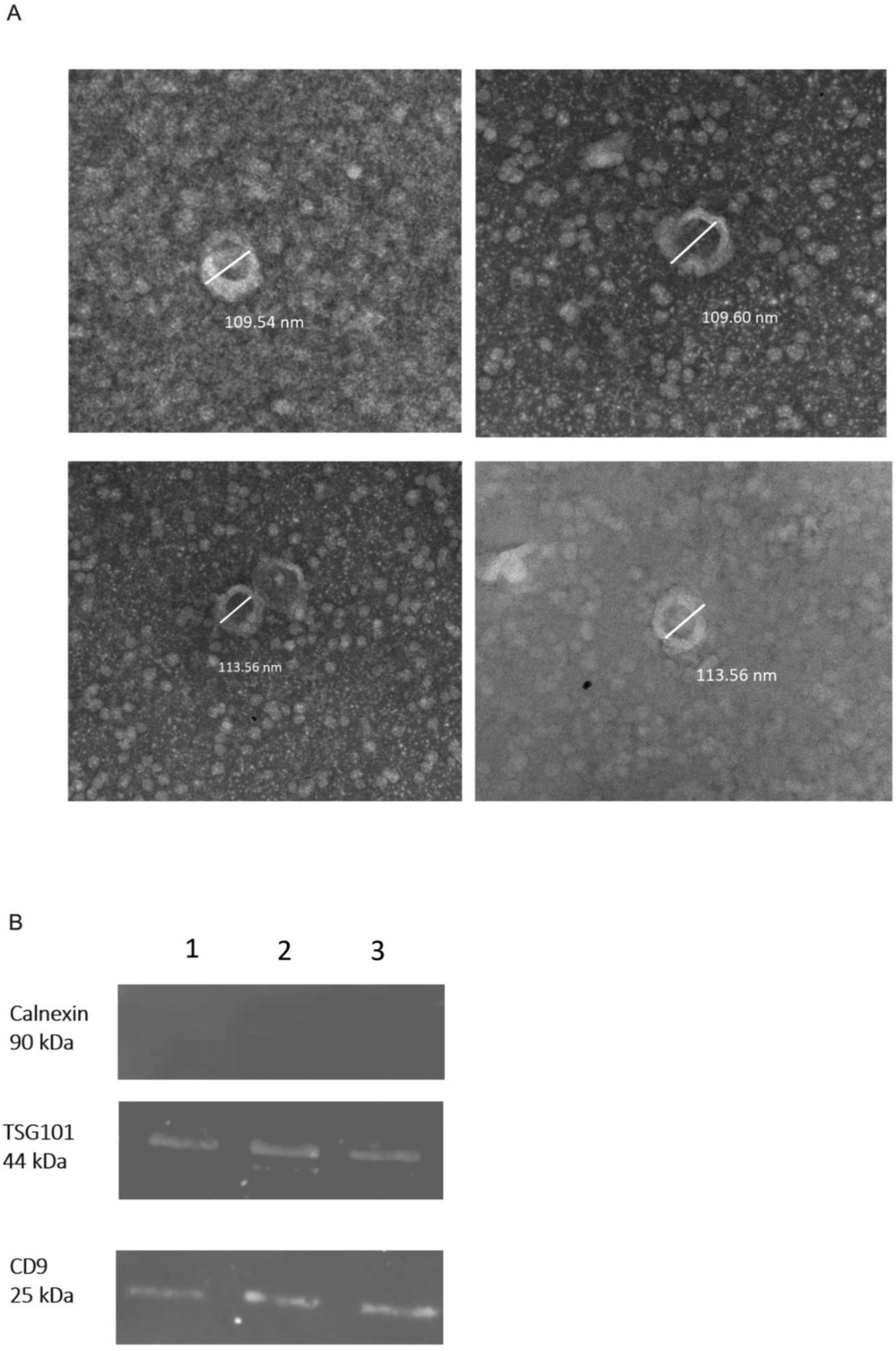
2.1. Confirmation of Isolated sEVs
2.2. Presence of DNA in sEVs
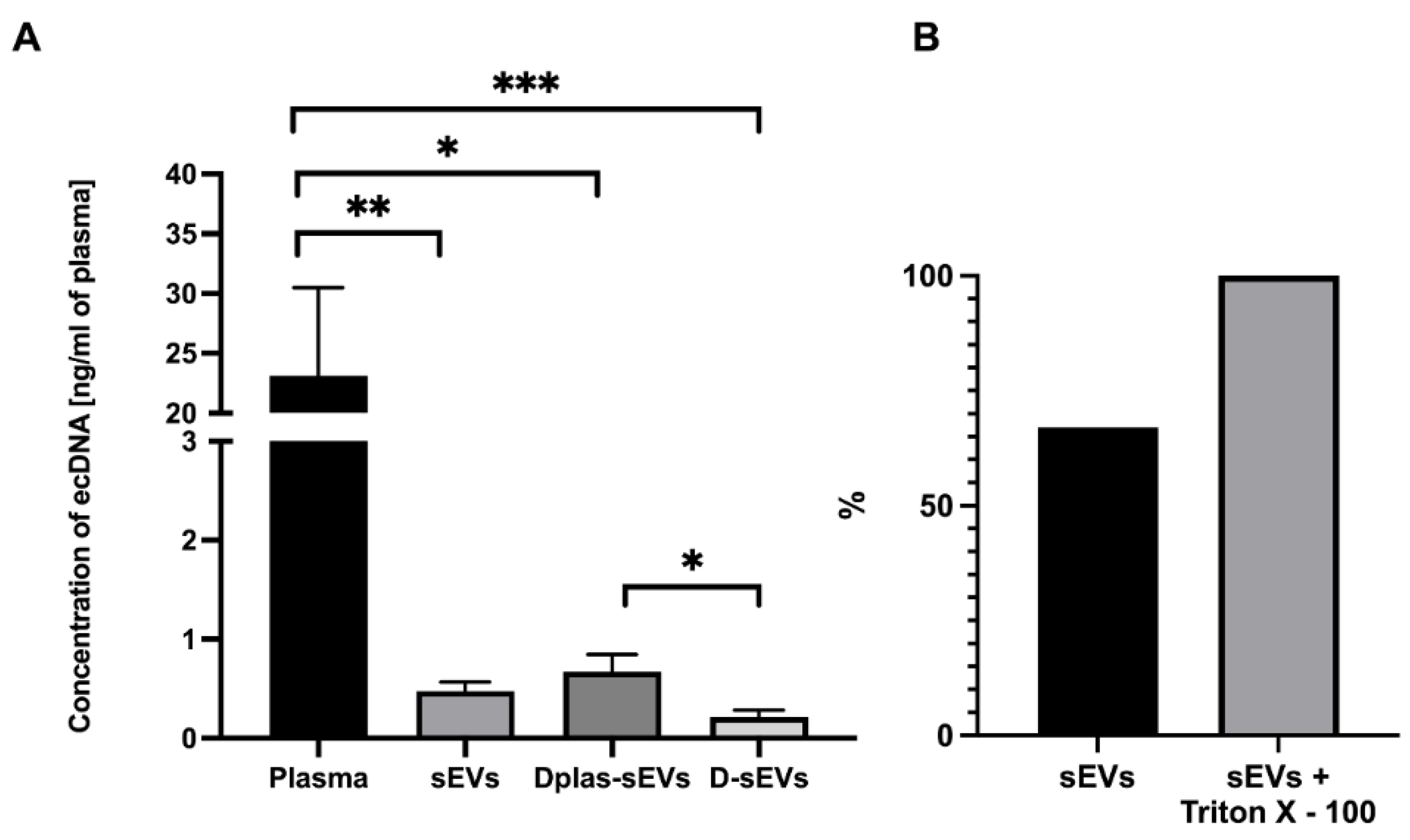
2.3. Concentration of ecDNA in sEVs
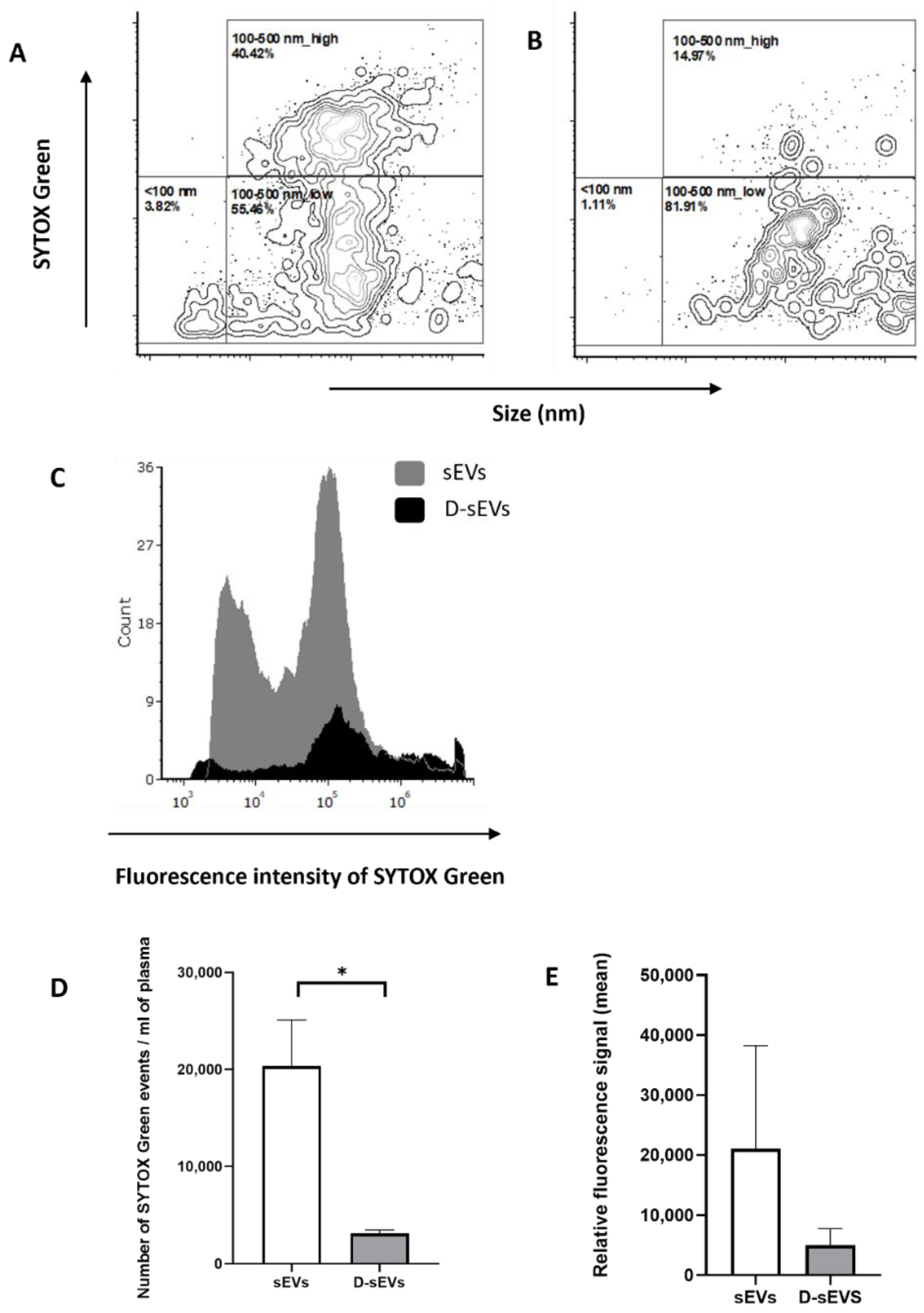
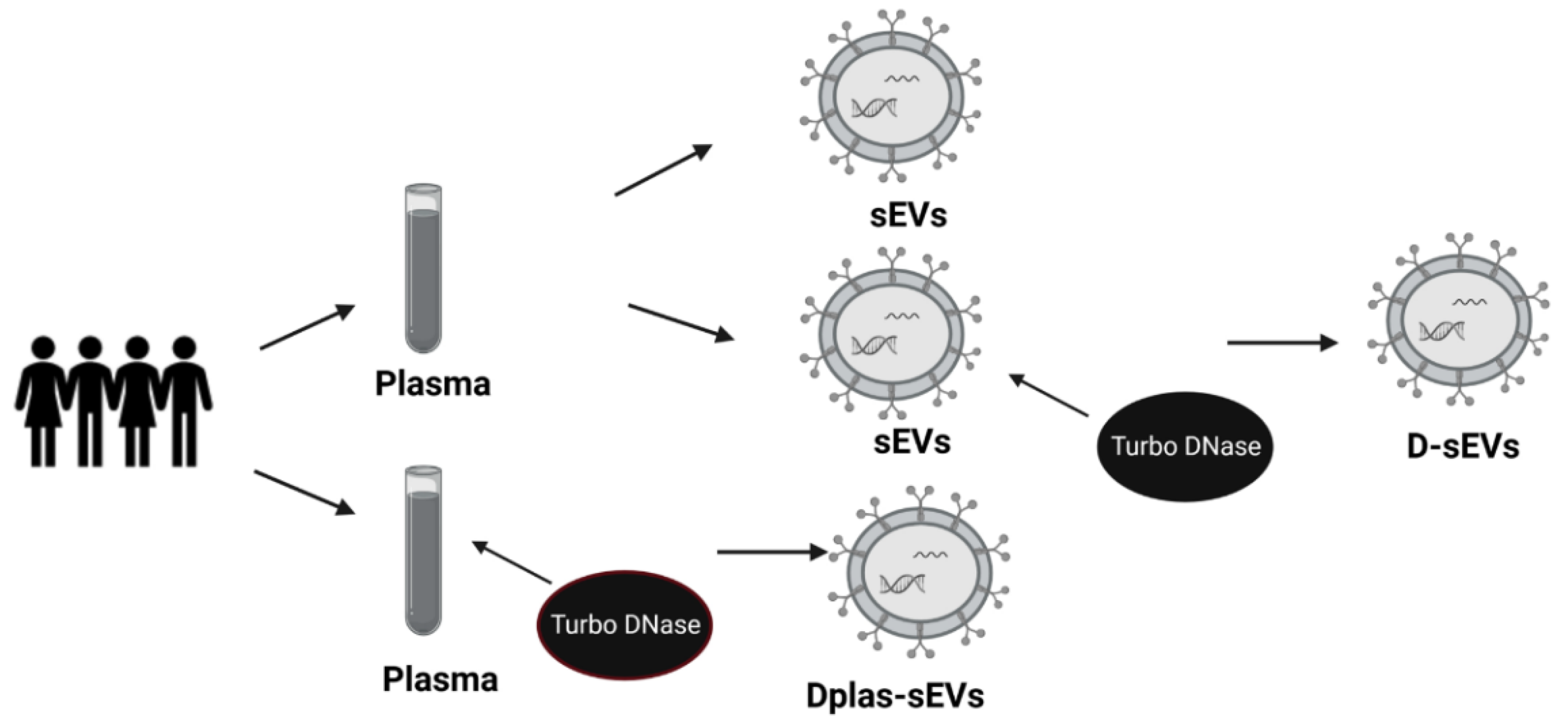
2.4. Protected or Unprotected DNA
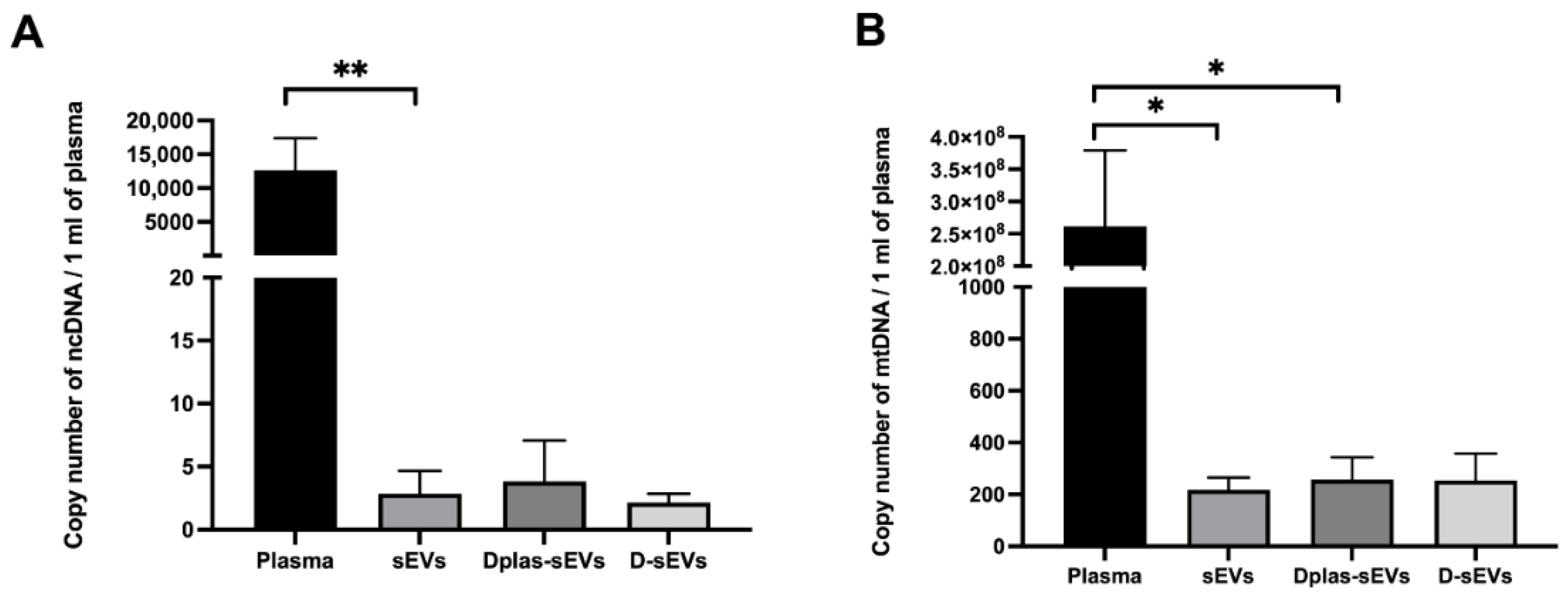
2.5. Subcellular Origin of ecDNA
3. Discussion
4. Materials and Methods
4.1. Plasma Sampling
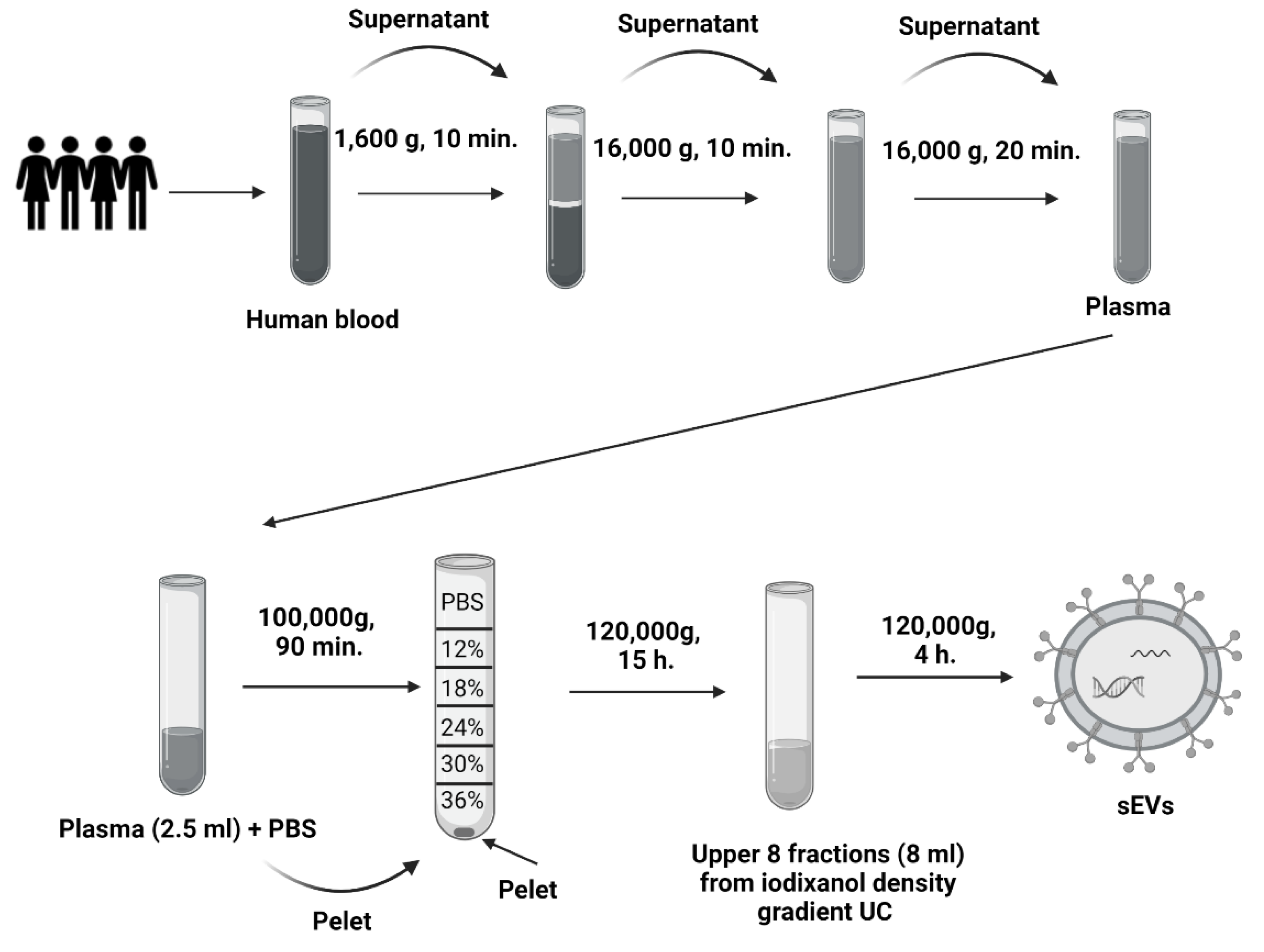
4.2. Isolation of sEVs from Plasma Samples
4.3. Quantification of sEVs Proteins
4.4. Nanoparticle Tracking Analysis
4.5. Western Blot
4.6. Transmission Electron Microscopy
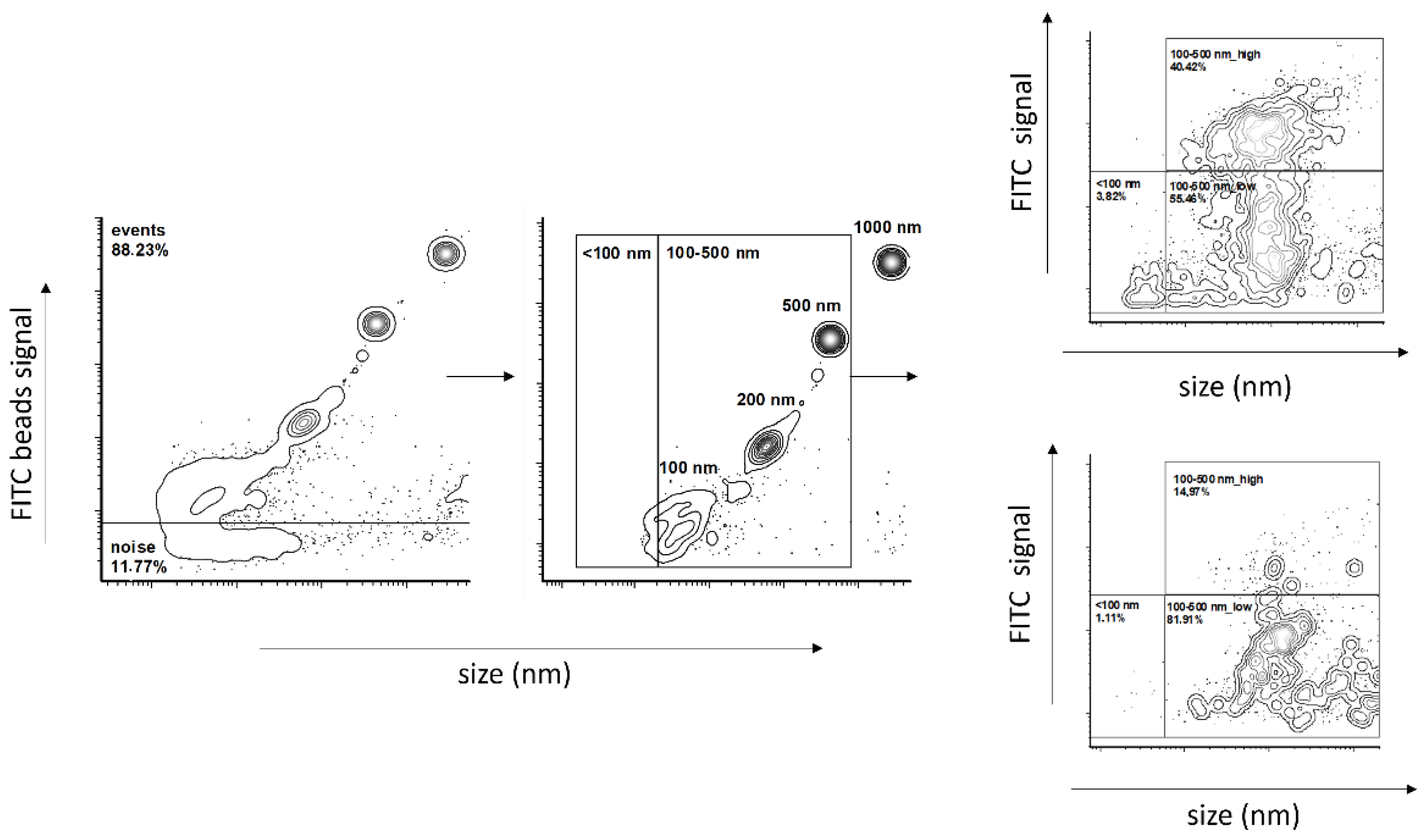
4.7. Flow Cytometry
4.8. Analysis of ecDNA
4.9. Position of ecDNA in sEVs
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mandel, P.; Metais, P. Nuclear Acids In Human Blood Plasma. C. R. Seances Soc. Biol. Fil. 1948, 142, 241–243. [Google Scholar] [PubMed]
- Anker, P.; Stroun, M.; Maurice, P.A. Spontaneous release of DNA by human blood lymphocytes as shown in an in vitro system. Cancer Res. 1975, 35, 2375–2382. [Google Scholar]
- Stroun, M.; Lyautey, J.; Lederrey, C.; Olson-Sand, A.; Anker, P. About the possible origin and mechanism of circulating DNA: Apoptosis and active DNA release. Clin. Chim. Acta 2001, 313, 139–142. [Google Scholar] [CrossRef]
- Pös, O.; Biró, O.; Szemes, T.; Nagy, B. Circulating cell-free nucleic acids: Characteristics and applications. Eur. J. Hum. Genet. 2018, 26, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Kustanovich, A.; Schwartz, R.; Peretz, T.; Grinshpun, A. Life and death of circulating cell-free DNA. Cancer Biol. Ther. 2019, 20, 1057–1067. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Zhu, L.; Wu, X.; Bao, H.; Wang, X.; Chang, Z.; Shao, Y.W.; Wang, Z. Cell-Free DNA Provides a Good Representation of the Tumor Genome Despite Its Biased Fragmentation Patterns. PLoS ONE 2017, 12, e0169231. [Google Scholar] [CrossRef]
- Pisetsky, D.S.; Perl, A.; Cooke, A.; Lood, C.; Duvvuri, B. Cell-Free DNA as a Biomarker in Autoimmune Rheumatic Diseases. Front. Immunol. 2019, 1, 502. [Google Scholar]
- Kubiritova, Z.; Radvanszky, J.; Gardlik, R. Cell-free nucleic acids and their emerging role in the pathogenesis and clinical management of inflammatory bowel disease. Int. J. Mol. Sci. 2019, 20, 3662. [Google Scholar] [CrossRef]
- Chang, C.P.Y.; Chia, R.H.; Wu, T.L.; Tsao, K.C.; Sun, C.F.; Wu, J.T. Elevated cell-free serum DNA detected in patients with myocardial infarction. Clin. Chim. Acta 2003, 327, 95–101. [Google Scholar] [CrossRef]
- Nie, S.; Lu, J.; Wang, L.; Gao, M. Pro-inflammatory role of cell-free mitochondrial DNA in cardiovascular diseases. IUBMB Life 2020, 72, 1879–1890. [Google Scholar] [CrossRef]
- Koyama, M.; Kurumizaka, H. Structural diversity of the nucleosome. J. Biochem. 2018, 163, 85–95. [Google Scholar] [CrossRef]
- Gahan, P.B.; Stroun, M. The virtosome-a novel cytosolic informative entity and intercellular messenger. Cell Biochem. Funct. 2010, 28, 529–538. [Google Scholar] [CrossRef]
- Jackson Chornenki, N.L.; Coke, R.; Kwong, A.C.; Dwivedi, D.J.; Xu, M.K.; McDonald, E.; Marshall, J.C.; Fox-Robichaud, A.E.; Charbonney, E.; Liaw, P.C. Comparison of the source and prognostic utility of cfDNA in trauma and sepsis. Intensive Care Med. Exp. 2019, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.M.; Wang, M.Z. Overview of extracellular vesicles, their origin, composition, purpose, and methods for exosome isolation and analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yu, D. Exosomes in cancer development, metastasis, and immunity. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 455–468. [Google Scholar] [CrossRef]
- Ludwig, N.; Whiteside, T.L.; Reichert, T.E. Challenges in exosome isolation and analysis in health and disease. Int. J. Mol. Sci. 2019, 20, 4684. [Google Scholar] [CrossRef]
- Yáñez-Mó, M.; Siljander, P.R.M.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 1–60. [Google Scholar] [CrossRef]
- Van Niel, G.; Porto-Carreiro, I.; Simoes, S.; Raposo, G. Exosomes: A common pathway for a specialized function. J. Biochem. 2006, 140, 13–21. [Google Scholar] [CrossRef]
- Fernando, M.R.; Jiang, C.; Krzyzanowski, G.D.; Ryan, W.L. New evidence that a large proportion of human blood plasma cell-free DNA is localized in exosomes. PLoS ONE 2017, 12, e0183915. [Google Scholar] [CrossRef] [PubMed]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of Exosome Composition. Cell 2019, 177, 428–445.e18. [Google Scholar] [CrossRef]
- Yokoi, A.; Villar-Prados, A.; OliphInt, P.A.; Zhang, J.; Song, X.; DeHoff, P.; Morey, R.; Liu, J.; Roszik, J.; Clise-Dwyer, K.; et al. Mechanisms of nuclear content loading to exosomes. Sci. Adv. 2019, 5, eaax8849. [Google Scholar] [CrossRef]
- Repiská, G.; Konečná, B.; Shelke, G.V.; Lässer, C.; Vlková, B.I.; Minárik, G. Is the DNA of placental origin packaged in exosomes isolated from plasma and serum of pregnant women? Clin. Chem. Lab. Med. 2018, 56, e150–e153. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Fang, L.; Bai, J.; Wang, Z. Characteristics and Changes of DNA in Extracellular Vesicles. DNA Cell Biol. 2020, 39, 1486–1493. [Google Scholar] [CrossRef] [PubMed]
- Lazo, S.; Noren Hooten, N.; Green, J.; Eitan, E.; Mode, N.A.; Liu, Q.R.; Zonderman, A.B.; Ezike, N.; Mattson, M.P.; Ghosh, P.; et al. Mitochondrial DNA in extracellular vesicles declines with age. Aging Cell 2021, 20, e13283. [Google Scholar] [CrossRef]
- Elzanowska, J.; Semira, C.; Costa-Silva, B. DNA in extracellular vesicles: Biological and clinical aspects. Mol. Oncol. 2021, 15, 1701–1714. [Google Scholar] [CrossRef]
- Vagner, T.; Spinelli, C.; Minciacchi, V.R.; Bala, J.L.; Zandian, M.; Conley, A.; Zijlstra, A.; Freeman, M.R.; Demichelis, F.; De, S.; et al. Large extracellular vesicles carry most of the tumour DNA circulating in prostate cancer patient plasma. J. Extracell. Vesicles 2018, 7, 1505403. [Google Scholar] [CrossRef]
- Toda, Y.; Takata, K.; Nakagawa, Y.; Kawakami, H.; Fujioka, S.; Kobayashi, K.; Hattori, Y.; Kitamura, Y.; Akaji, K.; Ashihara, E. Effective internalization of U251-MG-secreted exosomes into cancer cells and characterization of their lipid components. Biochem. Biophys. Res. Commun. 2015, 456, 768–773. [Google Scholar] [CrossRef]
- Laulagnier, K.; Motta, C.; Hamdi, S.; Roy, S.; Fauvelle, F.; Pageaux, J.F.; Kobayashi, T.; Salles, J.-P.; Perret, B.; Bonnerot, C.; et al. Mast cell- and dendritic cell-derived display a specific lipid composition and an unusual membrane organization. Biochem. J. 2004, 380, 161–171. [Google Scholar] [CrossRef]
- Parolini, I.; Federici, C.; Raggi, C.; Lugini, L.; Palleschi, S.; De Milito, A.; Coscia, C.; Iessi, E.; Logozzi, M.; Molinari, A.; et al. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J. Biol. Chem. 2009, 284, 34211–34222. [Google Scholar] [CrossRef] [PubMed]
- Bui, V.C.; Nguyen, T.H. DNA aggregation induced by Mg2+ ions under different conditions. J. Mol. Recognit. 2018, 31, e2721. [Google Scholar] [CrossRef] [PubMed]
- Németh, A.; Orgovan, N.; Sódar, B.W.; Osteikoetxea, X.; Pálóczi, K.; Szabó-Taylor, K.; Vukman, K.V.; Kittel, A.; Turiák, L.; Wiener, Z.; et al. Antibiotic-induced release of small extracellular vesicles (exosomes) with surface-associated DNA. Sci. Rep. 2017, 7, 8202. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Tian, Y.; Xue, C.; Niu, Q.; Chen, C.; Yan, X. Analysis of extracellular vesicle DNA at the single-vesicle level by nano-flow cytometry. J. Extracell. Vesicles 2022, 11, e12206. [Google Scholar] [CrossRef] [PubMed]
- Guan, S.; Yu, H.; Yan, G.; Gao, M.; Sun, W.; Zhang, X. Characterization of Urinary Exosomes Purified with Size Exclusion Chromatography and Ultracentrifugation. J. Proteome Res. 2020, 19, 2217–2225. [Google Scholar] [CrossRef]
- Riley, J.S.; Tait, S.W. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 2020, 21, e49799. [Google Scholar] [CrossRef]
- Malkin, E.Z.; Bratman, S.V. Bioactive DNA from extracellular vesicles and particles. Cell Death Dis. 2020, 11, 584. [Google Scholar] [CrossRef]
- Hong, E.E.; Okitsu, C.Y.; Smith, A.D.; Hsieh, C.L. Regionally Specific and Genome-Wide Analyses Conclusively Demonstrate the Absence of CpG Methylation in Human Mitochondrial DNA. Mol. Cell Biol. 2013, 33, 2683–2690. [Google Scholar] [CrossRef] [PubMed]
- Collins, L.V.; Hajizadeh, S.; Holme, E.; Jonsson, I.M.; Tarkowski, A. Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J. Leukoc. Biol. 2004, 75, 995–1000. [Google Scholar] [CrossRef]
- Neuberger, E.W.I.; Hillen, B.; Mayr, K.; Simon, P.; Krämer-Albers, E.M.; Brahmer, A. Kinetics and Topology of DNA Associated with Circulating Extracellular Vesicles Released during Exercise. Genes 2021, 12, 522. [Google Scholar] [CrossRef]
- Takahashi, A.; Okada, R.; Nagao, K.; Kawamata, Y.; Hanyu, A.; Yoshimoto, S.; Takasugi, M.; Watanabe, S.; Kanemaki, M.T.; Obuse, C.; et al. Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat. Commun. 2017, 8, 15287. [Google Scholar] [CrossRef]
- Chen, E.; Cario, C.L.; Leong, L.; Lopez, K.; Márquez, C.P.; Chu, C.; Li, P.S.; Oropeza, E.; Tenggara, I.; Cowan, J.; et al. Cell-free DNA concentration and fragment size as a biomarker for prostate cancer. Sci. Rep. 2021, 11, 5040. [Google Scholar] [CrossRef]
- Hur, J.Y.; Lee, K.Y. Characteristics and clinical application of extracellular vesicle-derived dna. Cancers 2021, 13, 3827. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Cornils, K.; Speiseder, T.; Badbaran, A.; Reimer, R.; Indenbirken, D.; Grundhoff, A.; Brunswig-Spickenheier, B.; Alawi, M.; Lange, C. Indication of horizontal DNA gene transfer by extracellular vesicles. PLoS ONE 2016, 11, e0163665. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Wu, G.; Jose, P.A.; Zeng, C. Functional transferred DNA within extracellular vesicles. Exp. Cell Res. 2016, 349, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Chen, K.; Wang, Z.; Wang, Y.; Liu, J.; Lin, L.; Shao, Y.; Gao, L.; Yin, H.; Cui, C.; et al. DNA in serum extracellular vesicles is stable under different storage conditions. BMC Cancer 2016, 16, 753. [Google Scholar] [CrossRef]
- Jiang, P.; Chan, C.W.M.; Chan, K.C.A.; Cheng, S.H.; Wong, J.; Wong, V.W.S.; Wong, G.L.H.; Chan, S.L.; Mok, T.S.K.; Chan, H.L.Y.; et al. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc. Natl. Acad. Sci. USA 2015, 112, E1317–E1325. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lichá, K.; Pastorek, M.; Repiská, G.; Celec, P.; Konečná, B. Investigation of the Presence of DNA in Human Blood Plasma Small Extracellular Vesicles. Int. J. Mol. Sci. 2023, 24, 5915. https://doi.org/10.3390/ijms24065915
Lichá K, Pastorek M, Repiská G, Celec P, Konečná B. Investigation of the Presence of DNA in Human Blood Plasma Small Extracellular Vesicles. International Journal of Molecular Sciences. 2023; 24(6):5915. https://doi.org/10.3390/ijms24065915
Chicago/Turabian StyleLichá, Kristína, Michal Pastorek, Gabriela Repiská, Peter Celec, and Barbora Konečná. 2023. "Investigation of the Presence of DNA in Human Blood Plasma Small Extracellular Vesicles" International Journal of Molecular Sciences 24, no. 6: 5915. https://doi.org/10.3390/ijms24065915
APA StyleLichá, K., Pastorek, M., Repiská, G., Celec, P., & Konečná, B. (2023). Investigation of the Presence of DNA in Human Blood Plasma Small Extracellular Vesicles. International Journal of Molecular Sciences, 24(6), 5915. https://doi.org/10.3390/ijms24065915