
The Integrity of the Blood–Brain Barrier as a Critical Factor for Regulating Glutamate Levels in Traumatic Brain Injury

 , ,
, , 
Abstract
1. Introduction
2. Results
2.1. Neurological Performance
2.2. Main and Interaction Effects on Outcomes
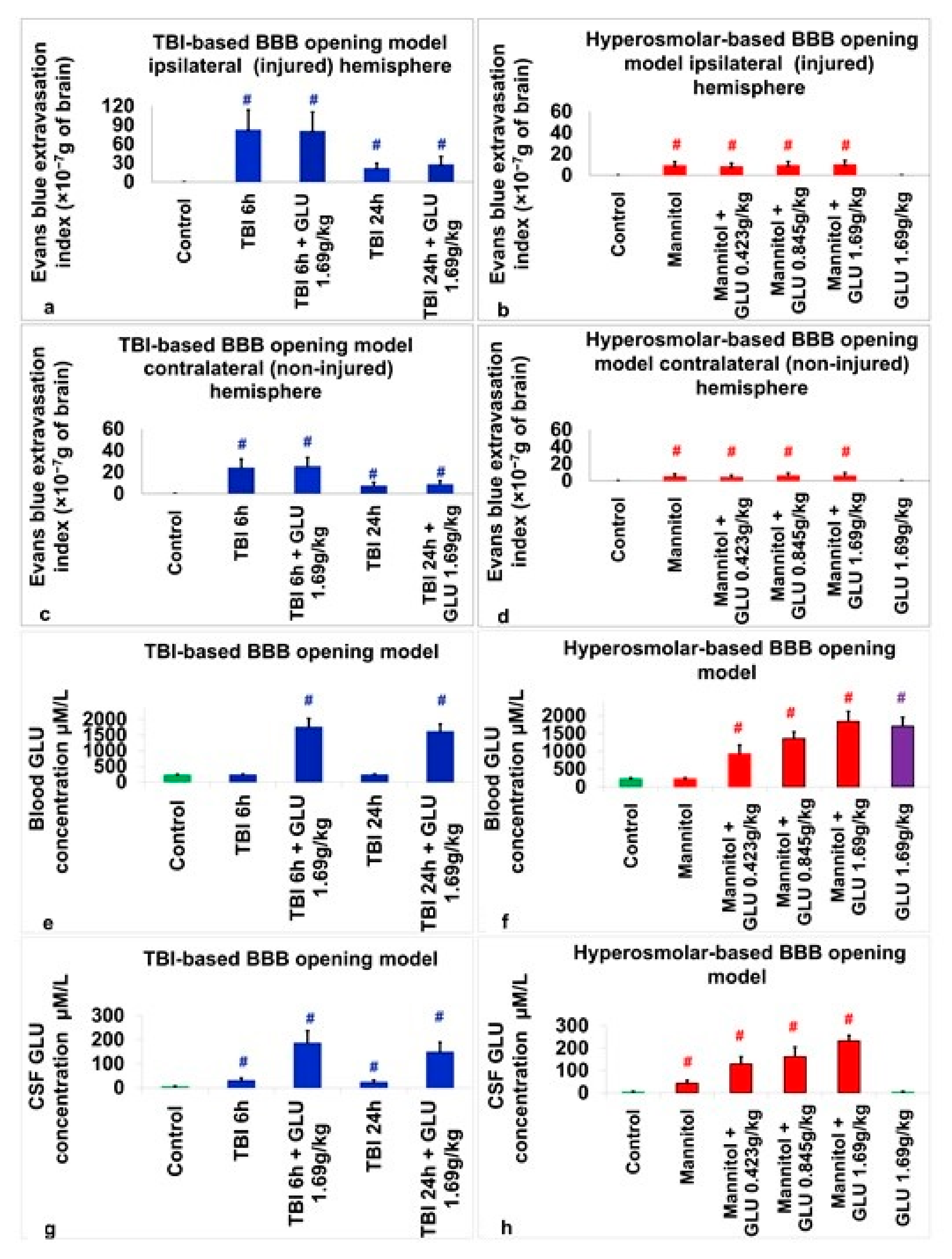
2.2.1. Evans Blue Extravasation Index
2.2.2. Blood Glutamate Level
2.2.3. CSF Glutamate Level
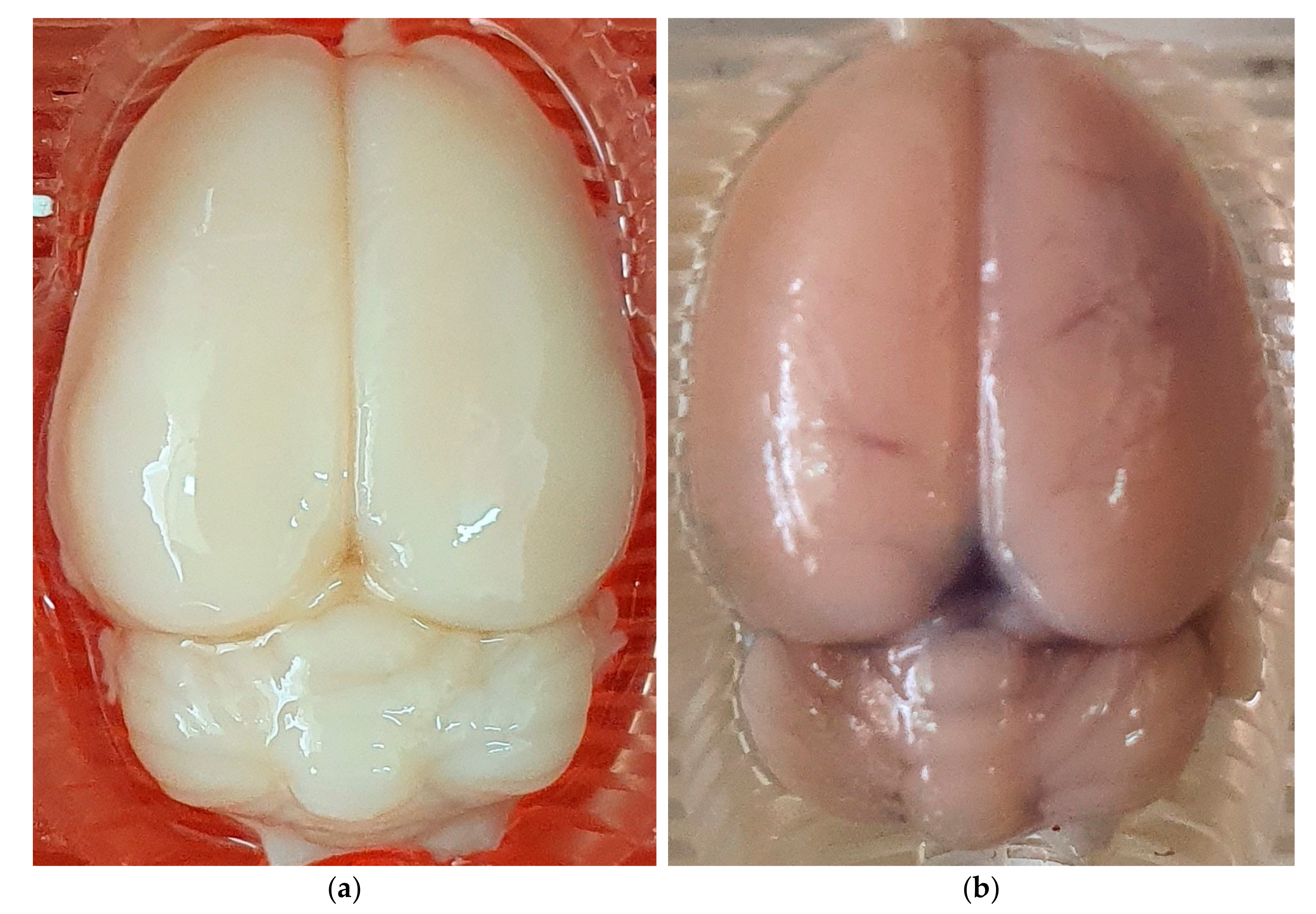
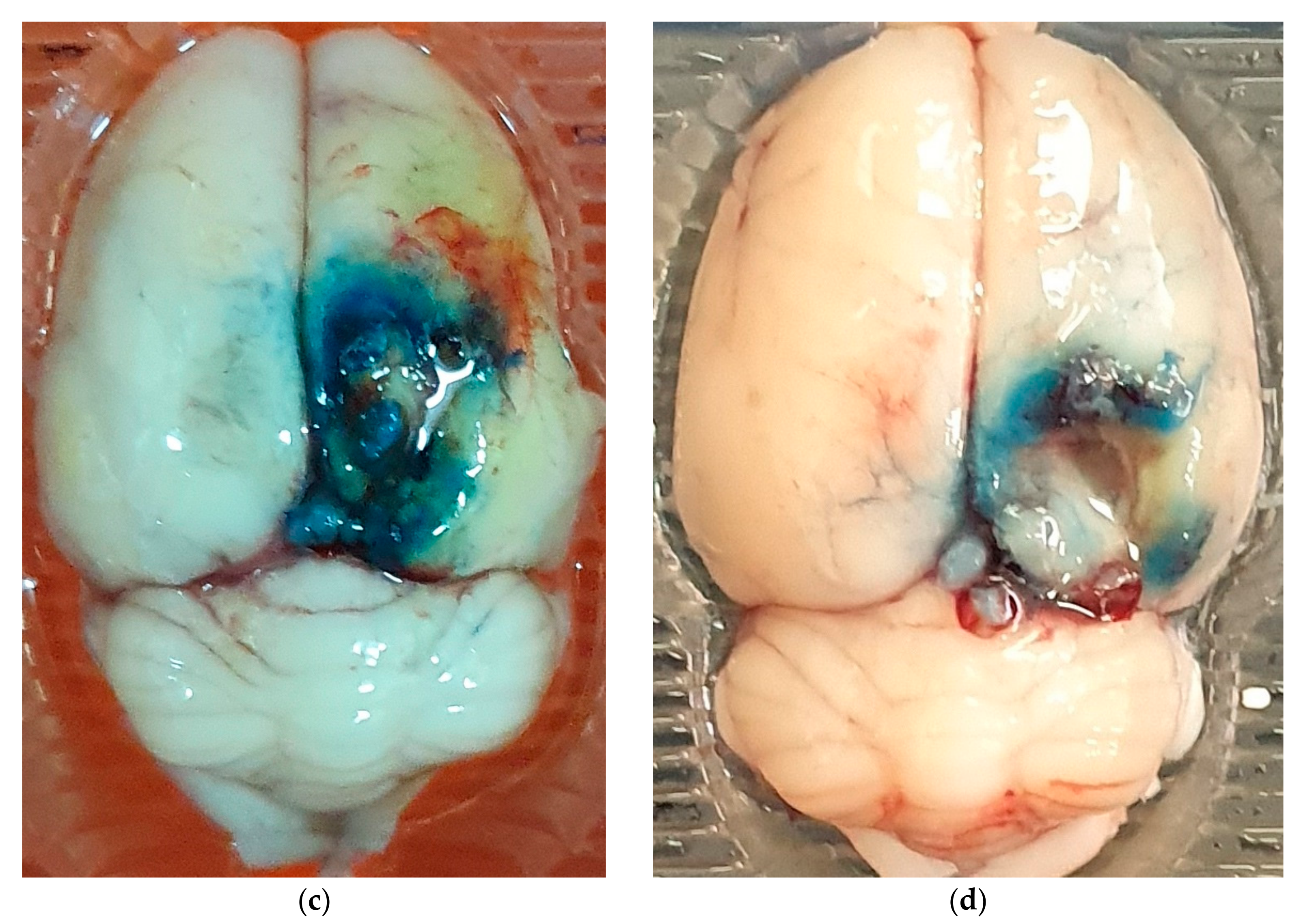
2.3. Assessment of BBB Disruption
2.4. Assessment of Blood Glutamate
2.5. Assessment of CSF Glutamate
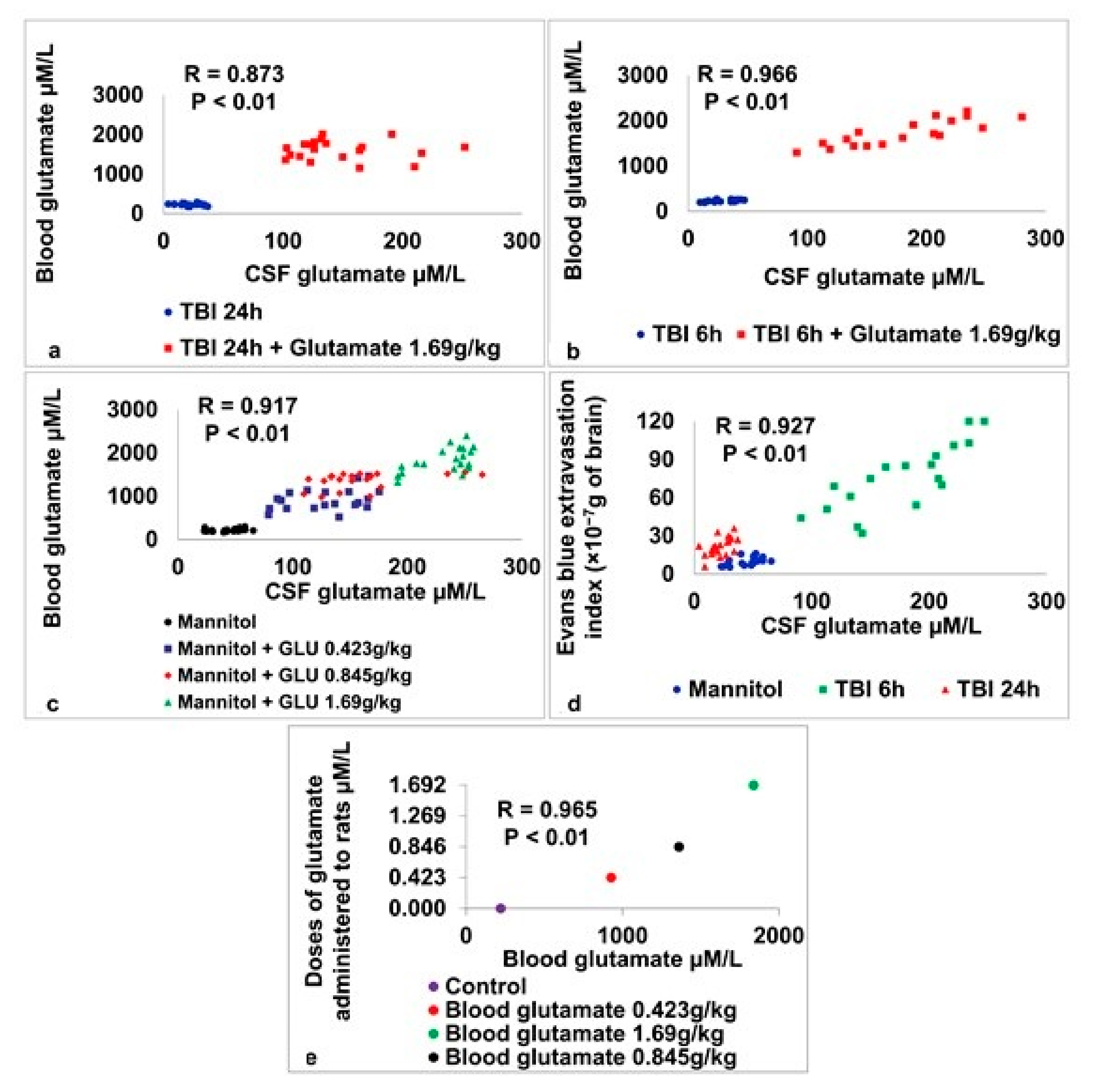
2.6. Correlations between CSF Glutamate, Blood Glutamate, and the Degree of BBB Permeability
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Drugs
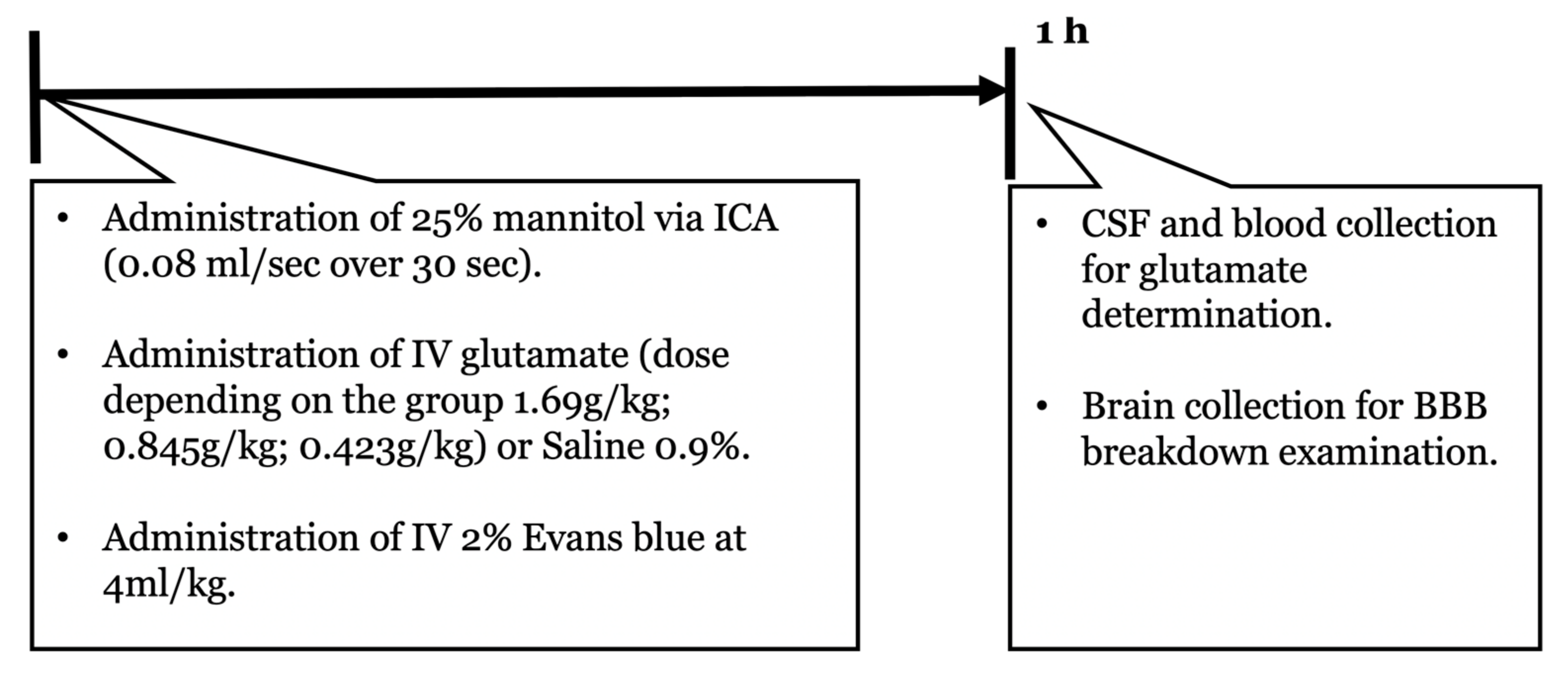
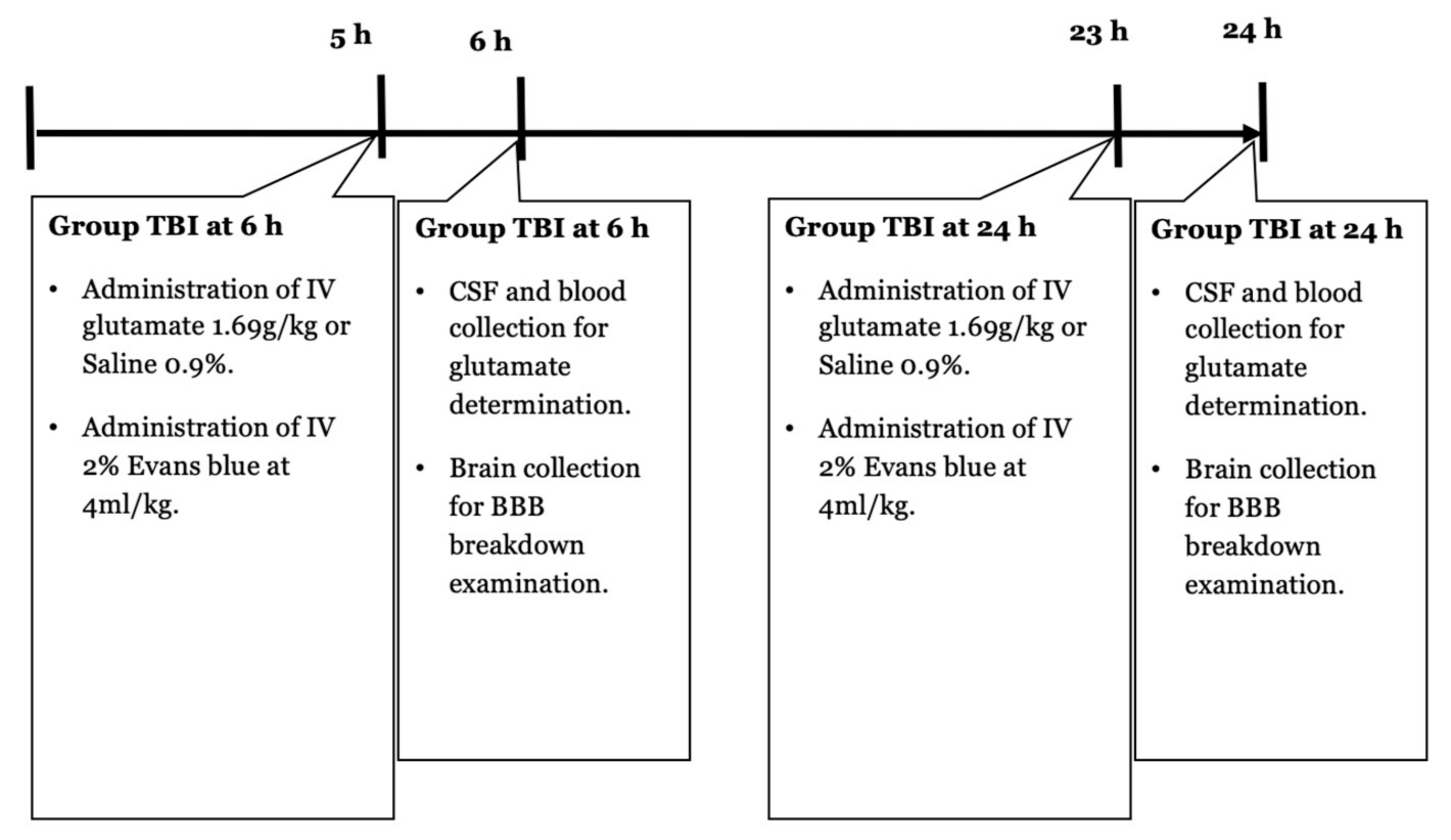
4.3. Experimental Design
4.4. HOBBB
4.5. TBI
4.6. Blood Sample Collection
4.7. Determination of Blood Glutamate
4.8. Cerebrospinal Fluid Collection
4.9. Cerebrospinal Fluid Glutamate
4.10. Assessment of BBB Disruption
4.11. Neurological Performance
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yelamanchi, S.D.; Jayaram, S.; Thomas, J.K.; Gundimeda, S.; Khan, A.A.; Singhal, A.; Keshava Prasad, T.; Pandey, A.; Somani, B.; Gowda, H. A pathway map of glutamate metabolism. J. Cell Commun. Signal. 2016, 10, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Schousboe, A.; Scafidi, S.; Bak, L.K.; Waagepetersen, H.S.; McKenna, M.C. Glutamate metabolism in the brain focusing on astrocytes. In Glutamate and ATP at the Interface of Metabolism and Signaling in the Brain; Springer: Berlin/Heidelberg, Germany, 2014; pp. 13–30. [Google Scholar]
- Beiderbeck, D.I.; Neumann, I.D.; Veenema, A.H. Differences in intermale aggression are accompanied by opposite vasopressin release patterns within the septum in rats bred for low and high anxiety. Eur. J. Neurosci. 2007, 26, 3597–3605. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders: DSM-5; American Psychiatric Association: Washington, DC, USA, 2013; Volume 10. [Google Scholar]
- Marks, D.M.; Park, M.-H.; Ham, B.-J.; Han, C.; Patkar, A.A.; Masand, P.S.; Pae, C.-U. Paroxetine: Safety and tolerability issues. Expert Opin. Drug Saf. 2008, 7, 783–794. [Google Scholar] [CrossRef]
- Jorge, R.E.; Arciniegas, D.B. Neuropsychiatry of traumatic brain injury. Psychiatr. Clin. 2014, 37, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Rauen, K.; Reichelt, L.; Probst, P.; Schäpers, B.; Müller, F.; Jahn, K.; Plesnila, N. Quality of life up to 10 years after traumatic brain injury: A cross-sectional analysis. Health Qual. Life Outcomes 2020, 18, 166. [Google Scholar] [CrossRef]
- Tateno, A.; Jorge, R.E.; Robinson, R.G. Clinical correlates of aggressive behavior after traumatic brain injury. J. Neuropsychiatry Clin. Neurosci. 2003, 15, 155–160. [Google Scholar] [CrossRef]
- Hicks, A.J.; Clay, F.J.; Hopwood, M.; James, A.C.; Jayaram, M.; Perry, L.A.; Batty, R.; Ponsford, J.L. The efficacy and harms of pharmacological interventions for aggression after traumatic brain injury—Systematic review. Front. Neurol. 2019, 10, 1169. [Google Scholar] [CrossRef] [PubMed]
- Hoofien, D.; Gilboa, A.; Vakil, E.; Donovick, P.J. Traumatic brain injury (TBI) 10? 20 years later: A comprehensive outcome study of psychiatric symptomatology, cognitive abilities and psychosocial functioning. Brain Inj. 2001, 15, 189–209. [Google Scholar]
- Koponen, S.; Taiminen, T.; Portin, R.; Himanen, L.; Isoniemi, H.; Heinonen, H.; Hinkka, S.; Tenovuo, O. Axis I and II psychiatric disorders after traumatic brain injury: A 30-year follow-up study. Am. J. Psychiatry 2002, 159, 1315–1321. [Google Scholar] [CrossRef]
- Rivara, F.P.; Koepsell, T.D.; Wang, J.; Temkin, N.; Dorsch, A.; Vavilala, M.S.; Durbin, D.; Jaffe, K.M. Disability 3, 12, and 24 months after traumatic brain injury among children and adolescents. Pediatrics 2011, 128, e1129–e1138. [Google Scholar] [CrossRef]
- Bodnar, C.N.; Roberts, K.N.; Higgins, E.K.; Bachstetter, A.D. A systematic review of closed head injury models of mild traumatic brain injury in mice and rats. J. Neurotrauma 2019, 36, 1683–1706. [Google Scholar] [CrossRef] [PubMed]
- Nasser, M.; Bejjani, F.; Raad, M.; Abou-El-Hassan, H.; Mantash, S.; Nokkari, A.; Ramadan, N.; Kassem, N.; Mondello, S.; Hamade, E. Traumatic brain injury and blood-brain barrier cross-talk. CNS Neurol. Disord.-Drug Targets Former. Curr. Drug Targets-CNS Neurol. Disord. 2016, 15, 1030–1044. [Google Scholar] [CrossRef]
- Gruenbaum, B.F.; Zlotnik, A.; Fleidervish, I.; Frenkel, A.; Boyko, M. Glutamate Neurotoxicity and Destruction of the Blood–Brain Barrier: Key Pathways for the Development of Neuropsychiatric Consequences of TBI and Their Potential Treatment Strategies. Int. J. Mol. Sci. 2022, 23, 9628. [Google Scholar] [CrossRef] [PubMed]
- Al-Nasser, M.N.; Mellor, I.R.; Carter, W.G. Is L-Glutamate Toxic to Neurons and Thereby Contributes to Neuronal Loss and Neurodegeneration? A Systematic Review. Brain Sci. 2022, 12, 577. [Google Scholar] [CrossRef]
- Gruenbaum, B.F.; Zlotnik, A.; Frenkel, A.; Fleidervish, I.; Boyko, M. Glutamate Efflux across the Blood–Brain Barrier: New Perspectives on the Relationship between Depression and the Glutamatergic System. Metabolites 2022, 12, 459. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, R.A.; Viña, J.R. How glutamate is managed by the blood–brain barrier. Biology 2016, 5, 37. [Google Scholar] [CrossRef]
- Bai, W.; Zhou, Y.-G. Homeostasis of the intraparenchymal-blood glutamate concentration gradient: Maintenance, imbalance, and regulation. Front. Mol. Neurosci. 2017, 10, 400. [Google Scholar] [CrossRef]
- Chodobski, A.; Zink, B.J.; Szmydynger-Chodobska, J. Blood–brain barrier pathophysiology in traumatic brain injury. Transl. Stroke Res. 2011, 2, 492–516. [Google Scholar] [CrossRef]
- Yu, M.; Wang, M.; Yang, D.; Wei, X.; Li, W. Dynamics of blood brain barrier permeability and tissue microstructure following controlled cortical impact injury in rat: A dynamic contrast-enhanced magnetic resonance imaging and diffusion kurtosis imaging study. Magn. Reson. Imaging 2019, 62, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, E.A.; Ndode-Ekane, X.E.; Lehto, L.J.; Gorter, J.A.; Andrade, P.; Aronica, E.; Gröhn, O.; Pitkänen, A. Long-lasting blood-brain barrier dysfunction and neuroinflammation after traumatic brain injury. Neurobiol. Dis. 2020, 145, 105080. [Google Scholar] [CrossRef]
- Hawkins, R.A. The blood-brain barrier and glutamate. Am. J. Clin. Nutr. 2009, 90, 867S–874S. [Google Scholar] [CrossRef] [PubMed]
- McGrath, T.; Baskerville, R.; Rogero, M.; Castell, L. Emerging Evidence for the Widespread Role of Glutamatergic Dysfunction in Neuropsychiatric Diseases. Nutrients 2022, 14, 917. [Google Scholar] [CrossRef]
- Lau, A.; Tymianski, M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflügers Arch.-Eur. J. Physiol. 2010, 460, 525–542. [Google Scholar] [CrossRef] [PubMed]
- Avoli, M.; D’Antuono, M.; Louvel, J.; Köhling, R.; Biagini, G.; Pumain, R.; D’Arcangelo, G.; Tancredi, V. Network and pharmacological mechanisms leading to epileptiform synchronization in the limbic system in vitro. Prog. Neurobiol. 2002, 68, 167–207. [Google Scholar] [CrossRef]
- Borbély, É.; Simon, M.; Fuchs, E.; Wiborg, O.; Czéh, B.; Helyes, Z. Novel drug developmental strategies for treatment-resistant depression. Br. J. Pharmacol. 2022, 179, 1146–1186. [Google Scholar] [CrossRef]
- Berman, R.M.; Cappiello, A.; Anand, A.; Oren, D.A.; Heninger, G.R.; Charney, D.S.; Krystal, J.H. Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 2000, 47, 351–354. [Google Scholar] [CrossRef]
- Blaylock, R.L.; Faria, M. New concepts in the development of schizophrenia, autism spectrum disorders, and degenerative brain diseases based on chronic inflammation: A working hypothesis from continued advances in neuroscience research. Surg. Neurol. Int. 2021, 12, 556. [Google Scholar] [PubMed]
- Belov Kirdajova, D.; Kriska, J.; Tureckova, J.; Anderova, M. Ischemia-triggered glutamate excitotoxicity from the perspective of glial cells. Front. Cell. Neurosci. 2020, 14, 51. [Google Scholar] [CrossRef]
- Lewerenz, J.; Maher, P. Chronic glutamate toxicity in neurodegenerative diseases—What is the evidence? Front. Neurosci. 2015, 9, 469. [Google Scholar] [CrossRef]
- Meldrum, B.S. The role of glutamate in epilepsy and other CNS disorders. Neurology 1994, 44 (Suppl. S8), S14–S23. [Google Scholar] [PubMed]
- Cash, A.; Theus, M.H. Mechanisms of blood–brain barrier dysfunction in traumatic brain injury. Int. J. Mol. Sci. 2020, 21, 3344. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.; Gruenbaum, B.F.; Shelef, I.; Zvenigorodsky, V.; Benjamin, Y.; Shapoval, O.; Gal, R.; Zlotnik, A.; Melamed, I.; Boyko, M. A novel histological technique to assess severity of traumatic brain injury in rodents: Comparisons to neuroimaging and neurological outcomes. Front. Neurosci. 2021, 15, 733115. [Google Scholar] [CrossRef] [PubMed]
- Kozler, P.; Pokorny, J. Altered blood-brain barrier permeability and its effect on the distribution of Evans blue and sodium fluorescein in the rat brain applied by intracarotid injection. Physiol. Res. 2003, 52, 607–614. [Google Scholar]
- Boyko, M.; Zlotnik, A.; Gruenbaum, B.F.; Gruenbaum, S.E.; Ohayon, S.; Kuts, R.; Melamed, I.; Regev, A.; Shapira, Y.; Teichberg, V.I. Pyruvate’s blood glutamate scavenging activity contributes to the spectrum of its neuroprotective mechanisms in a rat model of stroke. Eur. J. Neurosci. 2011, 34, 1432–1441. [Google Scholar] [CrossRef]
- Fortin, D.; Adams, R.; Gallez, A. A blood-brain barrier disruption model eliminating the hemodynamic effect of ketamine. Can. J. Neurol. Sci. 2004, 31, 248–253. [Google Scholar] [CrossRef]
- Boyko, M.; Gruenbaum, B.F.; Shelef, I.; Zvenigorodsky, V.; Severynovska, O.; Binyamin, Y.; Knyazer, B.; Frenkel, A.; Frank, D.; Zlotnik, A. Traumatic brain injury-induced submissive behavior in rats: Link to depression and anxiety. Transl. Psychiatry 2022, 12, 239. [Google Scholar] [CrossRef] [PubMed]
- Boyko, M.; Kuts, R.; Gruenbaum, B.F.; Melamed, I.; Gruenbaum, S.E.; Klein, M.; Shapira, Y.; Zlotnik, A. The role of hypothermia in the regulation of blood glutamate levels in naive rats. J. Neurosurg. Anesthesiol. 2013, 25, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Boyko, M.; Stepensky, D.; Gruenbaum, B.F.; Gruenbaum, S.E.; Melamed, I.; Ohayon, S.; Glazer, M.; Shapira, Y.; Zlotnik, A. Pharmacokinetics of glutamate–oxaloacetate transaminase and glutamate–pyruvate transaminase and their blood glutamate-lowering activity in naïve rats. Neurochem. Res. 2012, 37, 2198–2205. [Google Scholar] [CrossRef]
- Boyko, M.; Melamed, I.; Gruenbaum, B.F.; Gruenbaum, S.E.; Ohayon, S.; Leibowitz, A.; Brotfain, E.; Shapira, Y.; Zlotnik, A. The effect of blood glutamate scavengers oxaloacetate and pyruvate on neurological outcome in a rat model of subarachnoid hemorrhage. Neurotherapeutics 2012, 9, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.; Kuts, R.; Tsenter, P.; Gruenbaum, B.F.; Grinshpun, Y.; Zvenigorodsky, V.; Shelef, I.; Natanel, D.; Brotfain, E.; Zlotnik, A. The effect of pyruvate on the development and progression of post-stroke depression: A new therapeutic approach. Neuropharmacology 2019, 155, 173–184. [Google Scholar] [CrossRef]
- Frank, D.; Gruenbaum, B.F.; Shelef, I.; Severynovska, O.; Gal, R.; Dubilet, M.; Zlotnik, A.; Kofman, O.; Boyko, M. Blood glutamate scavenging with pyruvate as a novel preventative and therapeutic approach for depressive-like behavior following traumatic brain injury in a rat model. Front. Neurosci. 2022, 16, 21. [Google Scholar] [CrossRef] [PubMed]
- Kuts, R.; Frank, D.; Gruenbaum, B.F.; Grinshpun, J.; Melamed, I.; Knyazer, B.; Tarabrin, O.; Zvenigorodsky, V.; Shelef, I.; Zlotnik, A. A novel method for assessing cerebral edema, infarcted zone and blood-brain barrier breakdown in a single post-stroke rodent brain. Front. Neurosci. 2019, 13, 1105. [Google Scholar] [CrossRef] [PubMed]
- Boyko, M.; Ohayon, S.; Goldsmith, T.; Novack, L.; Novack, V.; Perry, Z.H.; Gruenbaum, B.F.; Gruenbaum, S.E.; Steiner, O.; Shapira, Y. Morphological and neuro-behavioral parallels in the rat model of stroke. Behav. Brain Res. 2011, 223, 17–23. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Effect | TBI 6 h vs. Control | TBI 24 h vs. Control | HOBBB vs. Control |
|---|---|---|---|
| Models of BBB disruption | p < 0.01 | p < 0.01 | p < 0.01 |
| Treatment | n-s | n-s | n-s |
| Sex | n-s | n-s | n-s |
| Models of BBB disruption × treatment | n-s | n-s | n-s |
| Models of BBB disruption × sex | n-s | n-s | n-s |
| Models of BBB disruption × sex × treatment | n-s | n-s | n-s |
| Treatment × sex | n-s | n-s | n-s |
| Effect | Glutamate 0.423 g/kg vs. Placebo | Glutamate 0.845 g/kg vs. Placebo | Glutamate 1.69 g/kg vs. Placebo |
|---|---|---|---|
| Models of BBB disruption | n-s | n-s | n-s |
| Treatment | p < 0.01 | p < 0.01 | p < 0.01 |
| Sex | n-s | n-s | n-s |
| Models of BBB disruption × treatment | n-s | n-s | n-s |
| Models of BBB disruption × sex | n-s | n-s | n-s |
| Models of BBB disruption × sex × treatment | n-s | n-s | n-s |
| Treatment × sex | n-s | n-s | n-s |
| Effect | Glutamate 0.423 g/kg vs. Placebo | Glutamate 0.845 g/kg vs. Placebo | Glutamate 1.69 g/kg vs. Placebo | TBI 6 h vs. Control | TBI 24 h vs. Control | HOBBB vs. Control |
|---|---|---|---|---|---|---|
| Models of BBB disruption | p < 0.01 | p < 0.01 | p < 0.01 | p < 0.01 | p < 0.01 | p < 0.01 |
| Treatment | p < 0.01 | p < 0.01 | p < 0.01 | p < 0.01 | p < 0.01 | p < 0.01 |
| Sex | n-s | n-s | n-s | n-s | n-s | n-s |
| Models of BBB disruption × treatment | p < 0.01 | p < 0.01 | p < 0.01 | p < 0.01 | p < 0.01 | p < 0.01 |
| Models of BBB disruption × sex | n-s | n-s | n-s | n-s | n-s | n-s |
| Models of BBB disruption × sex × treatment | n-s | n-s | n-s | n-s | n-s | n-s |
| Treatment × sex | n-s | n-s | n-s | n-s | n-s | n-s |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boyko, M.; Gruenbaum, B.F.; Frank, D.; Natanel, D.; Negev, S.; Azab, A.N.; Barsky, G.; Knyazer, B.; Kofman, O.; Zlotnik, A. The Integrity of the Blood–Brain Barrier as a Critical Factor for Regulating Glutamate Levels in Traumatic Brain Injury. Int. J. Mol. Sci. 2023, 24, 5897. https://doi.org/10.3390/ijms24065897
Boyko M, Gruenbaum BF, Frank D, Natanel D, Negev S, Azab AN, Barsky G, Knyazer B, Kofman O, Zlotnik A. The Integrity of the Blood–Brain Barrier as a Critical Factor for Regulating Glutamate Levels in Traumatic Brain Injury. International Journal of Molecular Sciences. 2023; 24(6):5897. https://doi.org/10.3390/ijms24065897
Chicago/Turabian StyleBoyko, Matthew, Benjamin F. Gruenbaum, Dmitry Frank, Dmitry Natanel, Shahar Negev, Abed N. Azab, Guy Barsky, Boris Knyazer, Ora Kofman, and Alexander Zlotnik. 2023. "The Integrity of the Blood–Brain Barrier as a Critical Factor for Regulating Glutamate Levels in Traumatic Brain Injury" International Journal of Molecular Sciences 24, no. 6: 5897. https://doi.org/10.3390/ijms24065897
APA StyleBoyko, M., Gruenbaum, B. F., Frank, D., Natanel, D., Negev, S., Azab, A. N., Barsky, G., Knyazer, B., Kofman, O., & Zlotnik, A. (2023). The Integrity of the Blood–Brain Barrier as a Critical Factor for Regulating Glutamate Levels in Traumatic Brain Injury. International Journal of Molecular Sciences, 24(6), 5897. https://doi.org/10.3390/ijms24065897