
Berberine Rescues D-Ribose-Induced Alzheimer‘s Pathology via Promoting Mitophagy


Abstract
1. Introduction
2. Results
2.1. Berberine Ameliorates D-Ribose-Induced Mitochondrial Dysfunction via Promoting Mitophagy
2.2. Berberine Revives D-Ribose-Induced Mitophagy Dysfunction through PINK1–Parkin Pathway
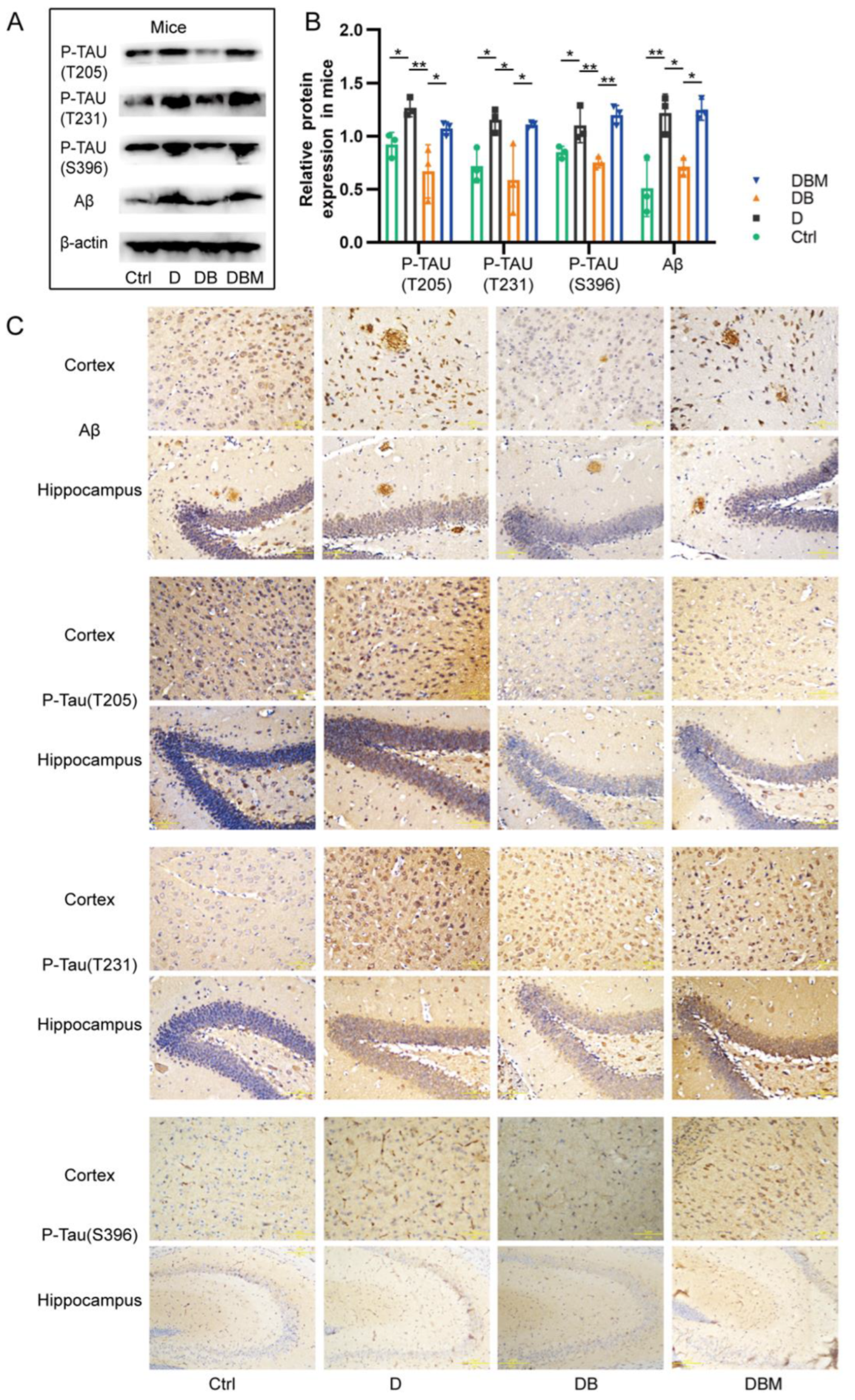
2.3. Berberine Mitigates Alzheimer’s-Like Pathology Induced by D-Ribose via Promoting Mitophagy
2.4. BBR Ameliorates Cognitive Impairment of APP/PS1 Mice Induced by D-Ribose via Mitophagy
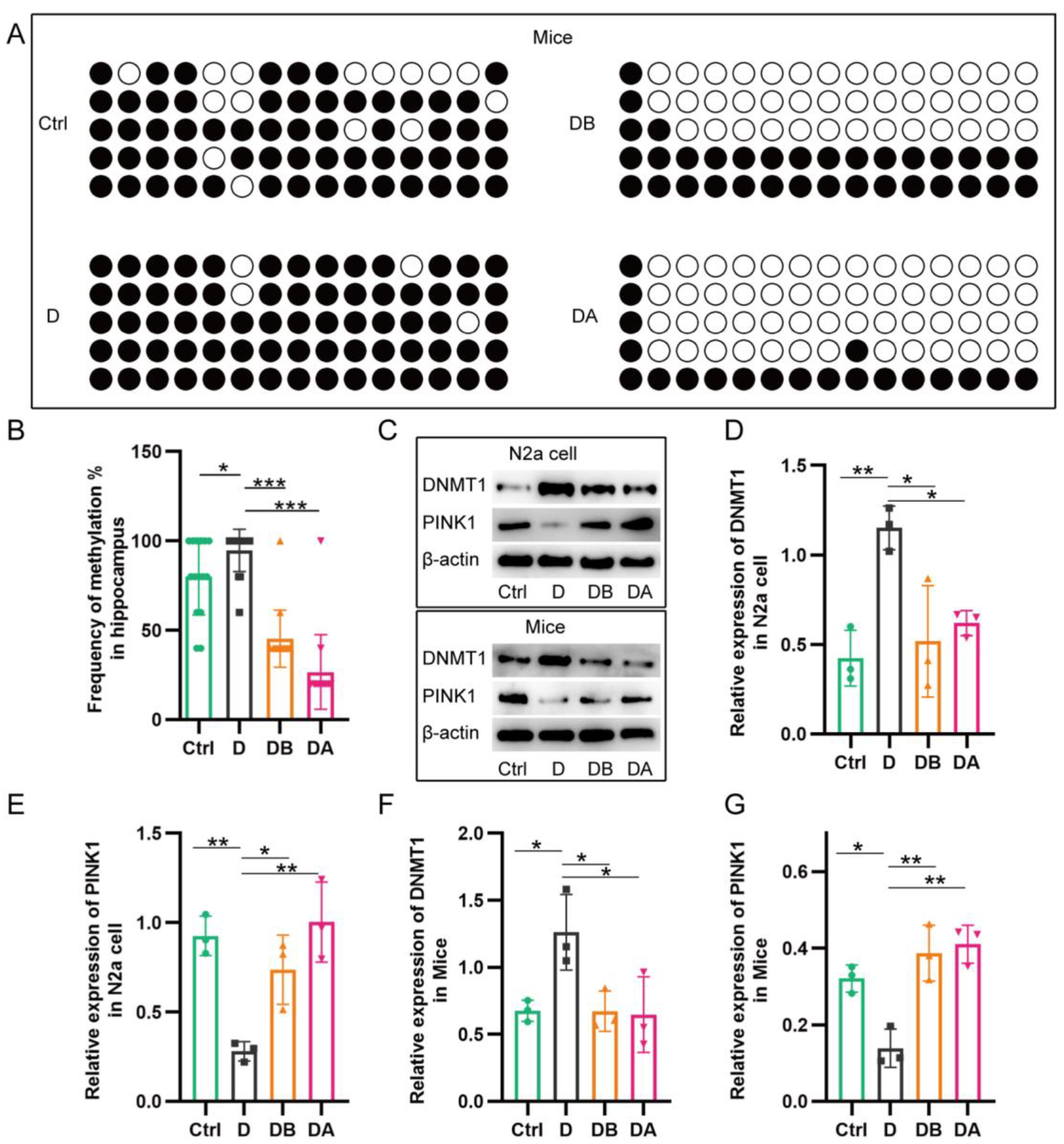
2.5. BBR Inhibits PINK1 Promoter Methylation
3. Discussion
4. Materials and Methods
4.1. Animals, Cell Lines, and Reagents
4.2. Experimental Design and Drug Treatment
4.3. Behavioral Tests
4.4. Mitochondrial Membrane Potential Assay
4.5. Reactive Oxygen Species (ROS) Detection
4.6. Apoptosis Assays
4.7. Mitochondria (Mito) and Lysosome (Lyso) Tracker Staining
4.8. Small Interfering RNA (siRNA) Interference
4.9. Transmission Electron Microscopy (TEM)
4.10. Mitochondrial Isolation and Western Blotting (WB)
4.11. Histological Analysis and Immunohistochemistry
4.12. Immunofluorescent Staining
4.13. Bisulfite Sequencing PCR (BSP)
4.14. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gaugler, J.; James, B.; Johnson, T.; Reimer, J.; Solis, M.; Weuve, J.; Buckley, R.F.; Hohman, T.J. 2022 Alzheimer’s disease facts and figures. Alzheimers Dement. 2022, 18, 700–789. [Google Scholar] [CrossRef]
- Xu, K.; Wang, M.; Zhou, W.; Pu, J.; Wang, H.; Xie, P. Chronic D-ribose and D-mannose overload induce depressive/anxiety-like behavior and spatial memory impairment in mice. Transl. Psychiatry 2021, 11, 90. [Google Scholar] [CrossRef]
- Javed, M.; Ahmad, M.I.; Javed, H.; Naseem, S. D-ribose and pathogenesis of Alzheimer’s disease. Mol. Biol. Rep. 2020, 47, 2289–2299. [Google Scholar] [CrossRef] [PubMed]
- Turck, D.; Bresson, J.L.; Burlingame, B.; Dean, T.; Fairweather-Tait, S.; Heinonen, M.; Hirsch-Ernst, K.I.; Mangelsdorf, I.; McArdle, H.; Naska, A.; et al. Safety of d-ribose as a novel food pursuant to Regulation (EU) 2015/2283. EFSA J. 2018, 16, e05265. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Chen, Y.; Xu, Y.; He, T.; Wei, Y.; He, R. D-ribose is elevated in T1DM patients and can be involved in the onset of encephalopathy. Aging 2019, 11, 4943–4969. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Han, C.; Wang, Y.; Wu, B.; Su, T.; Liu, Y.; He, R. Ribosylation triggering Alzheimer’s disease-like Tau hyperphosphorylation via activation of CaMKII. Aging Cell 2015, 14, 754–763. [Google Scholar] [CrossRef]
- Mahoney, D.E.; Hiebert, J.B.; Thimmesch, A.; Pierce, J.T.; Vacek, J.L.; Clancy, R.L.; Sauer, A.J.; Pierce, J.D. Understanding D-Ribose and Mitochondrial Function. Adv. Biosci. Clin. Med. 2018, 6, 1–5. [Google Scholar] [CrossRef]
- Castellazzi, M.; Patergnani, S.; Donadio, M.; Giorgi, C.; Bonora, M.; Bosi, C.; Brombo, G.; Pugliatti, M.; Seripa, D.; Zuliani, G.; et al. Autophagy and mitophagy biomarkers are reduced in sera of patients with Alzheimer’s disease and mild cognitive impairment. Sci. Rep. 2019, 9, 20009. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Goudarzi, S.; Hosseini, A.; Abdollahi, M.; Haghi-Aminjan, H. Insights Into Parkin-Mediated Mitophagy in Alzheimer’s Disease: A Systematic Review. Front. Aging Neurosci. 2021, 13, 674071. [Google Scholar] [CrossRef]
- Han, X.J.; Hu, Y.Y.; Yang, Z.J.; Jiang, L.P.; Shi, S.L.; Li, Y.R.; Guo, M.Y.; Wu, H.L.; Wan, Y.Y. Amyloid beta-42 induces neuronal apoptosis by targeting mitochondria. Mol. Med. Rep. 2017, 16, 4521–4528. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Yan, Q.; Huang, Z.T.; Zou, Q.; Li, J.; Yuan, M.H.; Wu, L.Q.; Cai, Z.Y. Ameliorating Ribosylation-Induced Amyloid-beta Pathology by Berberine via Inhibiting mTOR/p70S6K Signaling. J. Alzheimer Dis. 2021, 79, 833–844. [Google Scholar] [CrossRef]
- Cai, Z.; Wang, C.; Yang, W. Role of berberine in Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2016, 12, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Abudureyimu, M.; Yu, W.; Cao, R.Y.; Zhang, Y.; Liu, H.; Zheng, H. Berberine Promotes Cardiac Function by Upregulating PINK1/Parkin-Mediated Mitophagy in Heart Failure. Front. Physiol. 2020, 11, 565751. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Xue, Q.; Kuang, L.; Xie, L.; Luo, R.; Nie, X. Berberine alleviates cisplatin-induced acute kidney injury by regulating mitophagy via PINK 1/Parkin pathway. Transl. Androl. Urol. 2020, 9, 1712–1724. [Google Scholar] [CrossRef]
- Cui, D.; Xu, X. DNA Methyltransferases, DNA Methylation, and Age-Associated Cognitive Function. Int. J. Mol. Sci. 2018, 19, 1315. [Google Scholar] [CrossRef]
- De Jager, P.L.; Srivastava, G.; Lunnon, K.; Burgess, J.; Schalkwyk, L.C.; Yu, L.; Eaton, M.L.; Keenan, B.T.; Ernst, J.; McCabe, C.; et al. Alzheimer’s disease: Early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat. Neurosci. 2014, 17, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Yan, H.; Fei, J.; Jiang, M.; Zhu, H.; Lu, D.; Gao, X. Berberine reduces methylation of the MTTP promoter and alleviates fatty liver induced by a high-fat diet in rats. J. Lipid Res. 2010, 51, 2504–2515. [Google Scholar] [CrossRef] [PubMed]
- Henrique, A.M.; Gianetti, N.G.; Ferrari, M.F.R. Parkin is downregulated among autophagy-related proteins prior to hyperphosphorylation of Tau in TS65DN mice. Biochem. Biophys. Res. Commun. 2021, 561, 59–64. [Google Scholar] [CrossRef]
- Hou, X.; Watzlawik, J.O.; Cook, C.; Liu, C.C.; Kang, S.S.; Lin, W.L.; DeTure, M.; Heckman, M.G.; Diehl, N.N.; Al-Shaikh, F.S.H.; et al. Mitophagy alterations in Alzheimer’s disease are associated with granulovacuolar degeneration and early tau pathology. Alzheimer Dement. 2020, 17, 417–430. [Google Scholar] [CrossRef]
- Zhu, X.; Wei, Y.; He, Y.; He, R.; Li, J. Urine D-ribose levels correlate with cognitive function in community-dwelling older adults. BMC Geriatr. 2022, 22, 693. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Jiang, H.; Zhang, H.; Li, R.; Zhang, Q.; Luo, D.; Cai, X.; Li, M. Serum oxidized low density lipoprotein serves as a mediator for the inverse relationship between serum d-ribose and cognitive performance in type 2 diabetic patients. Free. Radic. Biol. Med. 2021, 171, 91–98. [Google Scholar] [CrossRef]
- Hakimizadeh, E.; Zamanian, M.; Gimenez-Llort, L.; Sciorati, C.; Nikbakhtzadeh, M.; Kujawska, M.; Kaeidi, A.; Hassanshahi, J.; Fatemi, I. Calcium Dobesilate Reverses Cognitive Deficits and Anxiety-Like Behaviors in the D-Galactose-Induced Aging Mouse Model through Modulation of Oxidative Stress. Antioxidants 2021, 10, 649. [Google Scholar] [CrossRef] [PubMed]
- Boulard, M.; Rucli, S.; Edwards, J.R.; Bestor, T.H. Methylation-directed glycosylation of chromatin factors represses retrotransposon promoters. Proc. Natl. Acad. Sci. USA 2020, 117, 14292–14298. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Yang, Y.; Xiong, R.; Ni, Y.; Ma, X.; Hou, M.; Chen, L.; Xu, Z.; Ji, M. Oral berberine ameliorates high-fat diet-induced obesity by activating TAS2Rs in tuft and endocrine cells in the gut. Life Sci. 2022, 311 Pt A, 121141. [Google Scholar] [CrossRef]
- Xie, W.; Su, F.; Wang, G.; Peng, Z.; Xu, Y.; Zhang, Y.; Xu, N.; Hou, K.; Hu, Z.; Chen, Y.; et al. Glucose-lowering effect of berberine on type 2 diabetes: A systematic review and meta-analysis. Front. Pharmacol. 2022, 13, 1015045. [Google Scholar] [CrossRef]
- Yang, Y.N.; Wang, Q.C.; Xu, W.; Yu, J.; Zhang, H.; Wu, C. The berberine-enriched gut commensal Blautia producta ameliorates high-fat diet (HFD)-induced hyperlipidemia and stimulates liver LDLR expression. Biomed. Pharmacother. 2022, 155, 113749. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Wang, R.; Liu, H.; Wang, L.; Li, J.; Wu, S.; Chen, X.; Yang, X.; Zhao, Y. Berberine regulates macrophage polarization through IL-4-STAT6 signaling pathway in Helicobacter pylori-induced chronic atrophic gastritis. Life Sci. 2021, 266, 118903. [Google Scholar] [CrossRef]
- Yan, S.; Chang, J.; Hao, X.; Liu, J.; Tan, X.; Geng, Z.; Wang, Z. Berberine regulates short-chain fatty acid metabolism and alleviates the colitis-associated colorectal tumorigenesis through remodeling intestinal flora. Phytomedicine 2022, 102, 154217. [Google Scholar] [CrossRef] [PubMed]
- Akbar, M.; Shabbir, A.; Rehman, K.; Akash, M.S.H.; Shah, M.A. Neuroprotective potential of berberine in modulating Alzheimer’s disease via multiple signaling pathways. J. Food Biochem. 2021, 45, e13936. [Google Scholar] [CrossRef] [PubMed]
- Raju, M.; Kunde, S.S.; Auti, S.T.; Kulkarni, Y.A.; Wairkar, S. Berberine loaded nanostructured lipid carrier for Alzheimer’s disease: Design, statistical optimization and enhanced in vivo performance. Life Sci. 2021, 285, 119990. [Google Scholar] [CrossRef]
- He, W.; Wang, C.; Chen, Y.; He, Y.; Cai, Z. Berberine attenuates cognitive impairment and ameliorates tau hyperphosphorylation by limiting the self-perpetuating pathogenic cycle between NF-kappaB signaling, oxidative stress and neuroinflammation. Pharmacol. Rep. 2017, 69, 1341–1348. [Google Scholar] [CrossRef]
- Cai, Z.; Wang, C.; He, W.; Chen, Y. Berberine Alleviates Amyloid-Beta Pathology in the Brain of APP/PS1 Transgenic Mice via Inhibiting beta/gamma-Secretases Activity and Enhancing alpha-Secretases. Curr. Alzheimer Res. 2018, 15, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.Y.; Wang, C.L.; Lu, T.T.; Yang, W.M. Berberine Alleviates Amyloid-beta Pathogenesis Via Activating LKB1/AMPK Signaling in the Brain of APP/PS1 Transgenic Mice. Curr. Mol. Med. 2019, 19, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Yamagishi, S. Involvement of toxic AGEs (TAGE) in the pathogenesis of diabetic vascular complications and Alzheimer’s disease. J. Alzheimer Dis. 2009, 16, 845–858. [Google Scholar] [CrossRef]
- Reddy, V.P.; Aryal, P.; Darkwah, E.K. Advanced Glycation End Products in Health and Disease. Microorganisms 2022, 10, 1848. [Google Scholar] [CrossRef]
- Dewanjee, S.; Chakraborty, P.; Bhattacharya, H.; Chacko, L.; Singh, B.; Chaudhary, A.; Javvaji, K.; Pradhan, S.R.; Vallamkondu, J.; Dey, A.; et al. Altered glucose metabolism in Alzheimer’s disease: Role of mitochondrial dysfunction and oxidative stress. Free. Radic. Biol. Med. 2022, 193 Pt 1, 134–157. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, Q.; Li, S.; Li, X.J.; Yang, W.; He, D. Microglial autophagy in Alzheimer’s disease and Parkinson’s disease. Front. Aging Neurosci. 2022, 14, 1065183. [Google Scholar] [CrossRef]
- Nafchi, A.R.; Esmaeili, M.; Myers, O.; Oprea, T.; Bearer, E.L. Autophagy and Herpesvirus: A collaboration Contributing to Alzheimer’s Disease. FASEB J. 2022, 36. [Google Scholar] [CrossRef] [PubMed]
- Mishra, E.; Thakur, M.K. Mitophagy: A promising therapeutic target for neuroprotection during aging and age-related diseases. Br. J. Pharmacol. 2023. published online ahead of print. [Google Scholar] [CrossRef]
- Mary, A.; Eysert, F.; Checler, F.; Chami, M. Mitophagy in Alzheimer’s disease: Molecular defects and therapeutic approaches. Mol. Psychiatry 2023, 28, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Pradeepkiran, J.A.; Baig, J.; Selman, A.; Reddy, P.H. Mitochondria in Aging and Alzheimer’s Disease: Focus on Mitophagy. Neuroscientist 2023. published online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Barazzuol, L.; Giamogante, F.; Brini, M.; Cali, T. PINK1/Parkin Mediated Mitophagy, Ca2+ Signalling, and ER-Mitochondria Contacts in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 1772. [Google Scholar] [CrossRef] [PubMed]
- Vives-Bauza, C.; de Vries, R.L.; Tocilescu, M.; Przedborski, S. PINK1/Parkin direct mitochondria to autophagy. Autophagy 2010, 6, 315–316. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Wang, N.; Kang, J.; Fang, Y. Beta-Asarone improves learning and memory in Aβ1-42 -induced Alzheimer’s disease rats by regulating PINK1-Parkin-mediated mitophagy. Metab. Brain Dis. 2020, 35, 1109–1117. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Shu, J.; Yang, X.; Wei, W.; Yan, A. Exosomes Derived From M2 Microglia Cells Attenuates Neuronal Impairment and Mitochondrial Dysfunction in Alzheimer’s Disease Through the PINK1/Parkin Pathway. Front. Cell. Neurosci. 2022, 16, 874102. [Google Scholar] [CrossRef]
- Sarraf, S.A.; Sideris, D.P.; Giagtzoglou, N.; Ni, L.; Kankel, M.W.; Sen, A.; Bochicchio, L.E.; Huang, C.H.; Nussenzweig, S.C.; Worley, S.H.; et al. PINK1/Parkin Influences Cell Cycle by Sequestering TBK1 at Damaged Mitochondria, Inhibiting Mitosis. Cell Rep. 2019, 29, 225–235.e225. [Google Scholar] [CrossRef]
- Zhao, N.; Zhang, X.; Li, B.; Wang, J.; Zhang, C.; Xu, B. Treadmill Exercise Improves PINK1/Parkin-Mediated Mitophagy Activity Against Alzheimer’s Disease Pathologies by Upregulated SIRT1-FOXO1/3 Axis in APP/PS1 Mice. Mol. Neurobiol. 2023, 60, 277–291. [Google Scholar] [CrossRef]
- Shireby, G.; Dempster, E.L.; Policicchio, S.; Smith, R.G.; Pishva, E.; Chioza, B.; Davies, J.P.; Burrage, J.; Lunnon, K.; Seiler Vellame, D.; et al. DNA methylation signatures of Alzheimer’s disease neuropathology in the cortex are primarily driven by variation in non-neuronal cell-types. Nat. Commun. 2022, 13, 5620. [Google Scholar] [CrossRef]
- Shen, W.B.; Yang, J.J.; Yang, P. RNA Hypomethylation and Unchanged DNA Methylation Levels in the Cortex of ApoE4 Carriers and Alzheimer’s Disease Subjects. Curr. Alzheimer Res. 2022, 19, 530–540. [Google Scholar] [CrossRef]
- Yan, M.H.; Wang, X.; Zhu, X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free. Radic. Biol. Med. 2013, 62, 90–101. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Species Source | Supplier | Identifier | Dilution | ||
|---|---|---|---|---|---|---|
| WB | IHC | IF | ||||
| SQSTM1/p62 | Rabbit | CST, Danvers, MA, USA | 39749 | 1:1000 | ||
| PINK1 | Rabbit | Proteintech, Chicago, IL, USA | 23274-1-AP | 1:1000 | ||
| LC3A/B | Rabbit | CST, Danvers, MA, USA | 12741 | 1:1000 | 1:500 | |
| LC3B | Rabbit | CST, Beverly, MA, USA | 3868 | 1:1000 | ||
| β-actin | Mouse | SAB, College Park, MD, USA | 48139 | 1:2000 | ||
| TOMM20 | Mouse | Abcam, Cambridge, MA, USA | ab283317 | 1:500 | ||
| PARKIN | Rabbit | Proteintech, Hubei, China | 14060-1-AP | 1:1000 | 1:100 | |
| VDAC1 | Rabbit | SAB, College Park, MD, USA | 52646 | 1:1000 | ||
| Phospho-Tau (Thr205) | Rabbit | CST, Danvers, MA, USA | 49561 | 1:1000 | 1:600 | |
| Tau (phospho T231) | Rabbit | Abcam, Cambridge, MA, USA | ab151559 | 1:5000 | 1:500 | |
| Tau (phospho S396) | Rabbit | Abcam, Cambridge, MA, USA | ab109390 | 1:10,000 | 1:1000 | |
| β-Amyloid (1-42) | Rabbit | CST, Danvers, MA, USA | 14974 | 1:1000 | ||
| beta Amyloid | Rabbit | Abcam, Cambridge, MA, USA | ab201060 | 1:1000 | ||
| PINK1 | Rabbit | SAB, College Park, MD, USA | 29297 | 1:1000 | ||
| PARKIN | Mouse | CST, Danvers, MA, USA | 4211 | 1:1000 | ||
| DNMT1 | Rabbit | Beyotime, Beijing, China | AF5150 | 1:500 | ||
| Cytochrome | Mouse | Beyotime, Beijing, China | AC909 | 1:200 | ||
| PINK1 | Mouse | Bioss, Beijing, China | Bsm-51265M | 1:100 | ||
| Groups | APP/PS1 Mice | N2a Cell |
|---|---|---|
| Control (Ctrl) | PBS, intraperitoneally | No treatment |
| D-ribose (D) | D-ribose, 2 g/kg/d, 30 days, intraperitoneally | D-ribose, 10 mM, 24 h |
| D-ribose + BBR (DB) | D-ribose (same as above); BBR, 100 mg/kg/d, 30 days, oral gavage | D-ribose (same as above); BBR, 1 μM, 24 h |
| D-ribose + 5-azacytidine (DA) | D-ribose (same as above); 5-azacytidine, 10 mg/kg/d, 3 times/week, final 3 weeks, intraperitoneally | D-ribose (same as above); 5-azacytidine, 10 μM, 24 h |
| D-ribose + BBR + Mdivi-1 (DBM) | D-ribose (same as above); BBR (same as above); Mdivi-1, 1 mg/kg, every other day, 30 days, intraperitoneally | D-ribose (same as above); BBR (same as above); Mdivi-1, 25 μM, 24 h |
| D-ribose + BBR + si-PINK1 (DBS) | - | D-ribose (same as above); BBR (same as above); si-PINK1, 50 nM, 48 h |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.; Zou, Q.; Pu, Y.; Cai, Z.; Tang, Y. Berberine Rescues D-Ribose-Induced Alzheimer‘s Pathology via Promoting Mitophagy. Int. J. Mol. Sci. 2023, 24, 5896. https://doi.org/10.3390/ijms24065896
Wang C, Zou Q, Pu Y, Cai Z, Tang Y. Berberine Rescues D-Ribose-Induced Alzheimer‘s Pathology via Promoting Mitophagy. International Journal of Molecular Sciences. 2023; 24(6):5896. https://doi.org/10.3390/ijms24065896
Chicago/Turabian StyleWang, Chuanling, Qian Zou, Yinshuang Pu, Zhiyou Cai, and Yong Tang. 2023. "Berberine Rescues D-Ribose-Induced Alzheimer‘s Pathology via Promoting Mitophagy" International Journal of Molecular Sciences 24, no. 6: 5896. https://doi.org/10.3390/ijms24065896
APA StyleWang, C., Zou, Q., Pu, Y., Cai, Z., & Tang, Y. (2023). Berberine Rescues D-Ribose-Induced Alzheimer‘s Pathology via Promoting Mitophagy. International Journal of Molecular Sciences, 24(6), 5896. https://doi.org/10.3390/ijms24065896