
Alcohol as a Modifiable Risk Factor for Alzheimer’s Disease—Evidence from Experimental Studies



Abstract
1. Introduction
2. Alcohol Intake and AD: Evidence from Epidemiological Studies
3. Impact of Alcohol Intake on AD: Evidence from Rodent Models
3.1. Alcohol-Feeding Models in AD Mice and Rats
3.1.1. Chronic Alcohol Feeding through Mixing Alcohol with a Liquid Diet (Lieber DeCarli Diet)
3.1.2. Chronic Alcohol Feeding through Mixing Alcohol in Drinking Water
3.1.3. Chronic Alcohol Binges
3.1.4. Intragastric Alcohol Feeding
3.1.5. National Institute on Alcohol Abuse and Alcoholism Chronic and Binge Alcohol Feeding
3.2. Animal Studies Showing an Increase in AD Pathology with Alcohol
3.2.1. Chronic Alcohol Feeding to Mature-Adult 3xTg-AD Mice

{kind=link}
| Strain | Model | Behavioral Testing | Biomarkers Measured | Outcome | Ref. # |
|---|---|---|---|---|---|
| 3xTg and WT mice Age (start): ~3 months Age (end): ~7 months | Oral sweetened alcohol for 4 months (25% w/v alcohol + saccharin 0.1% w/v) | Open field test to assess locomotion. Rotarod to assess motor coordination and balance. Morris water maze test to evaluate spatial memory. Pre-pulse inhibition to assess sensorimotor gating and startle response. Fear conditioning to assess emotional processing and learning and memory. | Aβ: Aβ40, Aβ42, Tau: p-tau Thr181, Ser-199/Ser202, Akt/mTOR phospho-proteins: p70S6K (Thr412), IRS1 (Ser636), IGF1R (Tyr1135 /Tyr1136), PTEN (Ser380), ERK/MAPK ½ (Thr185/Tyr187), RPS6 (Ser235 /Ser236) IR (Tyr1162 /Tyr1163), mTOR (Ser2448), GSK3α (Ser21), GSK3β (Ser9) | Alcohol treatment did not change motor ability, but impaired spatial memory and increased fear response in 3xTg mice. Alcohol also increased the Aβ (42/40) ratio and total tau in LEC and PFC, upregulated pTau (Ser199/Ser202), and dysregulated Akt/mTOR phosphoproteins in the 3xTg mice. Conclusion: Chronic alcohol intake aggravates amyloid and tau pathology in the 3xTg mice. | [29] |
| APP23/PS45 mice Age (start): ~2 months Age (end): ~3 months | Oral sweetened alcohol for 4 weeks (20% v/v alcohol + saccharin 0.07% w/v) | Morris water maze test to assess spatial memory. | Aβ40, Aβ42, APP, APP-CTFs, and BACE-1 | Chronic alcohol intake impaired spatial memory and increased Aβ40, Aβ42, APP, BACE-1, and APP-CTF β-fragments (C-99 and C-89) in APP23/PS45 mice. Conclusion: Alcohol exposure promotes APP processing and aggravates AD pathology. | [30] |
| APPswe/PS1dE9 mice Age (start): ~5.5 months Age (end): ~8 months | 20% alcohol in drinking water for 12 h/day for 4 consecutive days per week for 10 weeks | Open field test and light/dark assay to assess locomotion and anxiety-like behavior. Marble burying to assess repetitive and compulsive behavior. Object location memory task to assess spatial memory. Nest building to assess self-care. | Aβ40 and Aβ42 mRNA levels of NMDA and GABAA receptors | Chronic alcohol treatment increased locomotor activity and modulated mRNA levels of NMDA (increases) and GABAA (decreases) receptors in APP/PS1 mice brains. Alcohol elevated the hippocampal interstitial fluid Aβ40 levels with no changes in the Aβ42 levels. Conclusion: Chronic alcohol consumption aggravates AD-related pathology in the APPswe/PS1dE9 mice. | [31] |
| APPswe/PS1dE9 mice Age (start): ~4 months Age (end): ~6 months | Alcohol dosed orally at 10 mL/kg daily for 2 months | Passive avoidance test to assess learning and memory. Morris water maze test to assess spatial memory. | mRNA of ZO-1, VE-cadherin, Occludin, LRP-1, Mfsd2a, RAGE, AQP4 and Aβ42 | Chronic alcohol intake altered spatial memory and exacerbated cognitive decline in APPswe/PS1dE9 mice. Alcohol decreased mRNA levels of functional and structural proteins in APPswe/PS1dE9 mice brains, with no changes in Aβ42 levels. Conclusion: Long-term intake of alcohol aggravates cognitive decline by causing neural lesions and dysregulating the structure and function of the BBB. | [32] |
| 3xTg mice Age (start): P25–55 Age (end): ~ 6–12 months | 25% alcohol (5 g/kg/d) orally in 2-day on/2-day off AIE regimen from P25 to P55. Unperturbed from P55–P200, followed by behavioral testing in adulthood (P200–270) and sacrifice | Open field test to assess locomotion. Novel object recognition test to assess recognition memory. Three-chamber social choice test. Acoustic startle test (pre-pulse inhibition). Morris water maze test to assess spatial memory. Sleep pattern assessment using Piezo sleep system | Aβ42, p-tau Thr181, IL-1β, MCP-1, IL-6, IFNα, TNFα, TLR4, and microglial DAM genes | AIE altered the mouse behavior and increased Aβ42, p-tau (Thr181), and various cytokines in female 3xTg mice brains. AIE also altered the expression of microglial DAM genes in female 3xTg mice. Conclusion: Alcohol intake in adolescence promotes the early onset of AD pathology by triggering proinflammatory microglial signaling. | [33] |
| APPswe/PS1dE9 mice Age (start): 6/12 months Age (end): 7/13 months | Alcohol (2.5 g/kg, intraperitoneally) intermittently over 6 weeks | Hebb–William’s maze to evaluate spatial memory. | Aβ42 and total hippocampal RNA and protein levels of CB2, DAGLα, and MAGL | Alcohol binge worsened cognitive impairment, accelerated AD pathology, and increased Aβ42 levels. Alcohol lowered CB2 RNA levels and increased the expressions of MAGL, with no change in DAGLα. Conclusion: Binge alcohol drinking during adolescence alters hippocampal ECS activity in adult mice. These altered ECS conditions could contribute to memory impairment. Binge alcohol also accelerates hippocampal Aβ in AD mice. | [34] |
| APP/PS1 mice Age (start): ~2 months Age (end): ~3 months | 0.5, 1, 2, 3, and 4% w/v alcohol-containing liquid diet daily for 5 weeks | Y-maze test to evaluate spatial memory. | Aβ40, Aβ42, β-APP α, γ, and β-secretase activity | High dose (2–4%) of alcohol altered spatial memory, increased Aβ42, Aβ40, and β-APP by altering the γ- and β-secretase activities in the APP/PS1 mice. Low dose (0.5–1%) of alcohol had a protective effect in APP/PS1 mice. Conclusion: Dose-dependent effects of chronic alcohol intake such that higher doses of alcohol (2–4%) aggravate Aβ pathology and lower doses of alcohol (0.5–1%) reduce Aβ pathology. | [35] |
| 3xTg mice Age (start): ~4 months Age (end): ~6 months | 6% alcohol in drinking water 3 times per week for 2 months | Barnes maze test to assess spatial memory. | 3xTg mice displayed improved working memory and altered Aβ aggregation with alcohol consumption. Conclusion: Alcohol consumption protects against Aβ-induced toxicity. | [36] | |
| Tg2576 mice Age (start): ~4 months Age (end): ~11 months | Alcohol (6% in drinking water) over 7 months in the form of Cabernet Sauvignon (red wine) or alcohol | Barnes maze test to assess spatial memory. | Aβ40 and Aβ42, and liver panel enzymes (AST, ALT) to assess liver damage | Cabernet Sauvignon improved spatial memory and reduced neocortical Aβ40 and Aβ42 or AD-type plaque burden in the Tg2576 mice. Alcohol treatment had no effect on any of the measured parameters. Cabernet Sauvignon or alcohol did not change the serum levels of bilirubin, AST, and ALT in the Tg2576 mice. Conclusion: Cabernet Sauvignon at moderate doses protects from Aβ-induced neurotoxicity and improves cognitive recognition in the Tg2576 mice, which may be due to the antioxidants and polyphenols such as resveratrol. | [37] |
3.2.2. Chronic Alcohol Feeding to Young APP23/PS45 AD Mice
3.2.3. Chronic Alcohol Feeding to Mature-Adult APP/PS1 AD Mice
3.2.4. Chronic Binge Alcohol Administration to Mature-Adult APP/PS1 AD Mice
3.2.5. Alcohol Feeding to Adolescent 3xTg-AD Mice
3.2.6. Chronic Binge Alcohol Administration to Adolescent APP/PS1 AD Mice
3.3. Studies Showing a Dose-Dependent Effect of Alcohol on AD Pathology
4. Alcohol Intake and AD: Evidence from Cell Culture Studies
| Invitro Model | Assay | Biomarkers Measured | Outcome | Ref. # |
|---|---|---|---|---|
| 2EB2 cells (HEK293 cells overexpressing human Swedish APP and BACE-1), 20E2 cells (expressing human Swedish APP in HEK293), SH105 cells, and SH-SY5Y cells (human neuroblastoma cell line expressing BACE-1) | Cells were treated with 0-, 9-, 17-, 35-, 69-, and 139 mM alcohol for 48 h. | Protein levels of APP, APP-CTFs, ADAM10, BACE-1, and PS1 in 2EB2 cell lysates. Aβ40 and Aβ42 in the media of 20E2 cells. APP and BACE-1 in SH105 and SH-SY5Y cells. | Alcohol treatment increased the expression of: APP, APP-CTFs, and BACE-1 in 2EB2 cells, Aβ40 and Aβ42 levels in 20E2 cell lysates, and APP and BACE-1 in SH105 and SH-SY5Y neuronal cell lines. Conclusion: Alcohol exposure promotes APP processing and aggravates AD-associated phenotypes in a dose-dependent manner. | [30] |
| Primary rat hippocampal 10–12-day cultures | Cells incubated with Aβ (1 and 5 µM) and alcohol (1–100 mM) at 37 °C for 30 min. | Aβ, α-tubulin, synaptophysin, and synaptic vesicle 2 were detected by Western blotting. | Low concentrations (10mM) of alcohol reduced Aβ oligomer-induced neurotoxicity. Alcohol decreased the association of Aβ oligomers with neurons and increased Aβ disaggregation. Conclusion: These results may explain the lower risk of AD with low alcohol consumption. | [36] |
| SK-N-MC cells (human neuroblastoma cells) | Cells incubated with serum-free medium, later exposed to alcohol (0–103 mM) and acetaldehyde (0–215 µM) for 24 h. Parafilm-sealed plates were used to minimize alcohol evaporation. | Aβ and PGE2 levels in the cell culture media after alcohol exposure were measured. BACE-1, C-99, and intracellular ROS levels were also measured. | Alcohol increased BACE-1, C-99 fragment of APP, Aβ secretion, ROS, and PGE2 levels. Acetaldehyde increased BACE-1 expression. Conclusion: Alcohol-induced ER stress affects the PKA/CREB pathway via COX-2-mediated PGE2 formation, which leads to increased BACE-1 expression and Aβ production. | [39] |
| Mouse neuroblastoma N2a cell line expressing human APP (N2a-APP-695) | N2a-APP cells were incubated (16 h) with or without alcohol (130 mM/522 mM) in the presence of vehicle, APOE3 or APOE4 protein. Seal-plate film was used to minimize alcohol evaporation. | Cell viability, apoptotic cells, and cellular oxidative stress. | Synergistic neurotoxic effects of APOE4 and alcohol on N2a-APP cells. APOE4 exacerbated alcohol-induced neuronal apoptosis and cellular oxidative stress in N2a-APP cells. Conclusion: The study revealed the differential roles of APOE isoforms on alcohol-induced neurotoxicity, with APOE4 augmenting cellular oxidative stress and apoptotic cell death. | [40] |
| Primary rat microglial cells (prepared using frontal cortex of postnatal day 2 SD rats of both sexes) | Cells were incubated at 37 °C for 24 h with 75 mM alcohol along with a mixture of proinflammatory cytokines (TNFα, 10 ng/mL, IL-1β, 10 ng/mL, and IFNγ, 10 IU/mL) in a chamber flushed with 5% CO2, 21% O2, and balanced N2. Modular chambers were used to minimize alcohol evaporation. | Aβ phagocytosis was assessed. Nitrite production measured (to assess inflammatory activation of microglia). Microglial mRNA levels were measured. | Exposure of microglial cells to alcohol reduced Aβ42 phagocytosis and increased nitrite levels. Alcohol increased changes in phagocytosis-related mRNAs. Conclusion: Alcohol significantly suppresses Aβ42 phagocytosis in rat microglial cells. | [41] |
| M1C cells (human neuroblastoma BE2-M17D cell line expressing the 4R0N isoform of human tau) | Cells were incubated with different concentrations of alcohol (27, 54, and 109 mM) for up to 4 days. | Tau mRNA and protein levels were measured. Cell viability was assessed. Calpain, cathepsin B, and cathepsin D activity were measured. | Alcohol increased tau protein, decreased cell viability, and reduced calpain activity but had no effect on the activity of cathepsin B and D. Conclusion: Alcohol causes dose-dependent tau accumulation and reduced cell viability in the M1C cells. | [42] |
| HEK293 cells (human embryonic kidney cells), PC12 cells (pheochromocytoma of the rat adrenal medulla cells) | Cells were grown in 48-well plates and exposed for 24 h to Aβ (0.05 to 10 µM) in cell culture medium containing alcohol (0.1 mM, 10 mM, and 50 mM). | Aβ aggregation and cell viability were assessed. | Alcohol reduced Aβ aggregation and the toxic effects of Aβ. Conclusion: Alcohol protects against Aβ toxicity by destabilizing the salt bridge formed between Asp 23 and Lys 28 of Aβ that prevents the formation of complex Aβ fibers. | [43] |
| Rat hippocampal-entorhinal brain slice cultures | 3-week hippocampal-entorhinal slices cultured in 6-well plates were exposed to 30 mM alcohol, incubated for 6 days in a desiccator with 90 mM alcohol to minimize alcohol loss. Later the cultures were treated with pre-aggregated Aβ42 (20 µM) for 24 h. | Cell lysis and cell death were assessed by determining lactate dehydrogenase (LDH) activity. Neuronal cell death in the brain slices was visualized using propidium iodide (PI) along with Aβ42 over 24 h incubation period. Apoptotic DNA fragmentation was detected with bisbenzimide (DNA-binding dye Hoechst 33,342) for 2 min. Inducible heat shock protein70 (hsp70) in the brain slice mixture was determined by ELISA. | Moderate alcohol exposure in the hippocampal brain slice cultures protected the neuronal cells against Aβ42 neurotoxicity by lowering LDH activity and by increasing hsp70 levels. Conclusion: Moderate alcohol exposure protects the hippocampal-entorhinal cortical neurons against Aβ toxicity. | [44] |
| Primary neuronal cultures (cortical and hippocampal neurons obtained from mouse brains) | Neuronal cultures pre-treated with different concentrations of alcohol (4–20 mM) for 1 h were exposed to synthetic peptide biotin-Aβ42 for 1 to 24 h. | Biotin-Aβ42, synaptophysin, and activated cytoplasmic phospholipase-A2 (cPLA2) levels were measured. Free calcium levels in the synaptosomes were also measured. | Alcohol (4–20 mM) protected the neurons against Aβ-induced synapse damage, reduced activated cPLA2, and increased the Aβ-induced increase in calcium levels in the synaptosomes. Conclusion: Low alcohol concentrations (9 mM) protect the cortical and hippocampal neurons against Aβ42 toxicity by inhibiting cPLA2 activation at the synapse. | [45] |
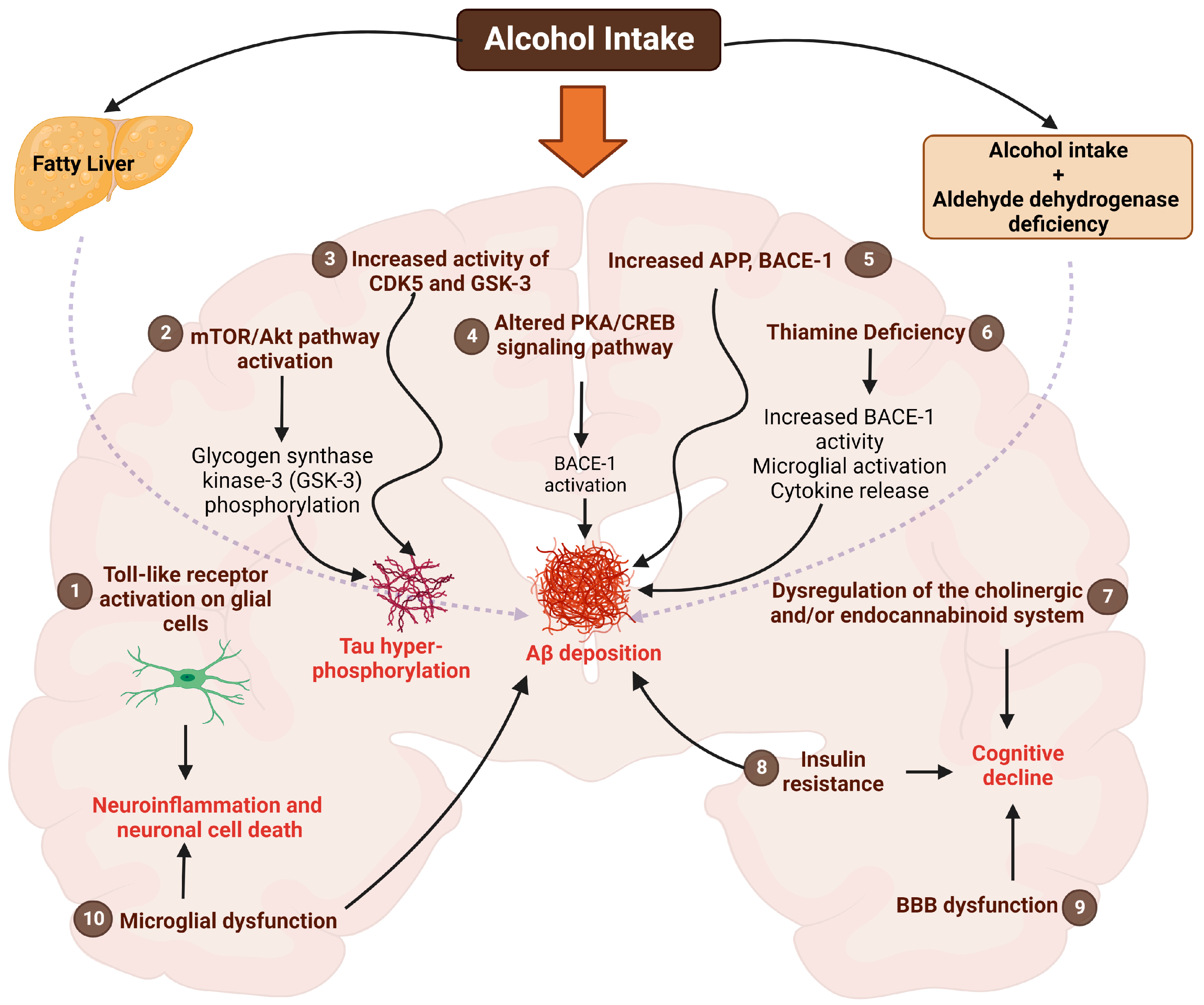
5. Alcohol Use and AD: Mechanistic Insights into the Brain
5.1. Toll-like Receptors (TLRs)
5.2. Akt/mTOR Pathway
5.3. Cyclic-AMP Response Element Binding Protein (CREB) Pathway
5.4. CDK5 and GSK Pathway
5.5. Insulin Resistance
5.6. Thiamine Deficiency
5.7. Endocannabinoid System
5.8. Cholinergic System
5.9. Blood–Brain Barrier (BBB) Alterations
5.10. Microglia-Mediated Effects
5.11. Aldehyde Dehydrogenase (ALDH)
5.12. Aβ Production and Other Mechanisms
6. Alcohol Use and AD: Peripheral Effects of Alcohol
6.1. Impact of Peripheral Aβ on AD
6.2. Impact of the Liver on Peripheral Aβ Levels
6.3. Alcohol Promotion of Peripheral Inflammation
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alzheimer’s Association. 2022 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2022, 18, 700–789. [Google Scholar] [CrossRef]
- Edwards, G.A., III; Gamez, N.; Escobedo, G., Jr.; Calderon, O.; Moreno-Gonzalez, I. Modifiable Risk Factors for Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 146. [Google Scholar] [CrossRef]
- Loeffler, D.A. Modifiable, Non-Modifiable, and Clinical Factors Associated with Progression of Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 80, 1–27. [Google Scholar] [CrossRef]
- Ritchie, H.; Roser, M. Alcohol Consumption. Our World in Data 2018. Available online: https://ourworldindata.org/alcohol-consumption (accessed on 15 February 2023).
- Wiegmann, C.; Mick, I.; Brandl, E.J.; Heinz, A.; Gutwinski, S. Alcohol and Dementia—What is the Link? A Systematic Review. Neuropsychiatr. Dis. Treat. 2020, 16, 87–99. [Google Scholar] [CrossRef]
- Heymann, D.; Stern, Y.; Cosentino, S.; Tatarina-Nulman, O.; Dorrejo, J.N.; Gu, Y. The Association between Alcohol Use and the Progression of Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 1356–1362. [Google Scholar] [CrossRef]
- Beydoun, M.A.; Beydoun, H.A.; Gamaldo, A.A.; Teel, A.; Zonderman, A.B.; Wang, Y. Epidemiologic studies of modifiable factors associated with cognition and dementia: Systematic review and meta-analysis. BMC Public Health 2014, 14, 643. [Google Scholar] [CrossRef]
- Chan, K.K.; Chiu, K.C.; Chu, L.W. Association between alcohol consumption and cognitive impairment in Southern Chinese older adults. Int. J. Geriatr. Psychiatry 2010, 25, 1272–1279. [Google Scholar] [CrossRef]
- Jeon, K.H.; Han, K.; Jeong, S.M.; Park, J.; Yoo, J.E.; Yoo, J.; Lee, J.; Kim, S.; Shin, D.W. Changes in Alcohol Consumption and Risk of Dementia in a Nationwide Cohort in South Korea. JAMA Netw. Open 2023, 6, e2254771. [Google Scholar] [CrossRef]
- Koch, M.; Fitzpatrick, A.L.; Rapp, S.R.; Nahin, R.L.; Williamson, J.D.; Lopez, O.L.; DeKosky, S.T.; Kuller, L.H.; Mackey, R.H.; Mukamal, K.J.; et al. Alcohol Consumption and Risk of Dementia and Cognitive Decline Among Older Adults with or without Mild Cognitive Impairment. JAMA Netw. Open 2019, 2, e1910319. [Google Scholar] [CrossRef]
- Letenneur, L.; Larrieu, S.; Barberger-Gateau, P. Alcohol and tobacco consumption as risk factors of dementia: A review of epidemiological studies. Biomed. Pharmacother. 2004, 58, 95–99. [Google Scholar] [CrossRef]
- Rehm, J.; Hasan, O.S.M.; Black, S.E.; Shield, K.D.; Schwarzinger, M. Alcohol use and dementia: A systematic scoping review. Alzheimer’s Res. Ther. 2019, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Schwarzinger, M.; Pollock, B.G.; Hasan, O.S.M.; Dufouil, C.; Rehm, J. Contribution of alcohol use disorders to the burden of dementia in France 2008-13: A nationwide retrospective cohort study. Lancet Public Health 2018, 3, e124–e132. [Google Scholar] [CrossRef]
- Rosen, J.; Colantonio, A.; Becker, J.T.; Lopez, O.L.; DeKosky, S.T.; Moss, H.B. Effects of a history of heavy alcohol consumption on Alzheimer’s disease. Br. J. Psychiatry 1993, 163, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Avitan, I.; Halperin, Y.; Saha, T.; Bloch, N.; Atrahimovich, D.; Polis, B.; Samson, A.O.; Braitbard, O. Towards a Consensus on Alzheimer’s Disease Comorbidity? J. Clin. Med. 2021, 10, 4360. [Google Scholar] [CrossRef] [PubMed]
- Piazza-Gardner, A.K.; Gaffud, T.J.B.; Barry, A.E. The impact of alcohol on Alzheimer’s disease: A systematic review. Aging Ment. Health 2013, 17, 133–146. [Google Scholar] [CrossRef]
- Ilomaki, J.; Jokanovic, N.; Tan, E.C.; Lonnroos, E. Alcohol Consumption, Dementia and Cognitive Decline: An Overview of Systematic Reviews. Curr. Clin. Pharmacol. 2015, 10, 204–212. [Google Scholar] [CrossRef]
- Tsukamoto, H.; French, S.W.; Benson, N.; Delgado, G.; Rao, G.A.; Larkin, E.C.; Largman, C. Severe and progressive steatosis and focal necrosis in rat liver induced by continuous intragastric infusion of ethanol and low fat diet. Hepatology 1985, 5, 224–232. [Google Scholar] [CrossRef]
- Lieber, C.S.; Decarli, L.M. Animal models of ethanol dependence and liver injury in rats and baboons. Fed. Proc. 1976, 35, 1232–1236. [Google Scholar]
- Bertola, A.; Mathews, S.; Ki, S.H.; Wang, H.; Gao, B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat. Protoc. 2013, 8, 627–637. [Google Scholar] [CrossRef]
- Shinohara, M.; Ji, C.; Kaplowitz, N. Differences in betaine-homocysteine methyltransferase expression, endoplasmic reticulum stress response, and liver injury between alcohol-fed mice and rats. Hepatology 2010, 51, 796–805. [Google Scholar] [CrossRef]
- Thiele, T.E.; Navarro, M. “Drinking in the dark” (DID) procedures: A model of binge-like ethanol drinking in non-dependent mice. Alcohol 2014, 48, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Brandon-Warner, E.; Schrum, L.W.; Schmidt, C.M.; McKillop, I.H. Rodent models of alcoholic liver disease: Of mice and men. Alcohol 2012, 46, 715–725. [Google Scholar] [CrossRef]
- Brandon-Warner, E.; Walling, T.L.; Schrum, L.W.; McKillop, I.H. Chronic ethanol feeding accelerates hepatocellular carcinoma progression in a sex-dependent manner in a mouse model of hepatocarcinogenesis. Alcohol. Clin. Exp. Res. 2012, 36, 641–653. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, H.; French, S.W.; Reidelberger, R.D.; Largman, C. Cyclical pattern of blood alcohol levels during continuous intragastric ethanol infusion in rats. Alcohol. Clin. Exp. Res. 1985, 9, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Johnson, H.S.; Rao, M.P.; Martin, G.; Sancheti, H.; Silkwood, K.H.; Decker, C.W.; Nguyen, K.T.; Casian, J.G.; Cadenas, E.; et al. Mitochondrial remodeling in the liver following chronic alcohol feeding to rats. Free Radic. Biol. Med. 2017, 102, 100–110. [Google Scholar] [CrossRef]
- Han, D.; Ybanez, M.D.; Johnson, H.S.; McDonald, J.N.; Mesropyan, L.; Sancheti, H.; Martin, G.; Martin, A.; Lim, A.M.; Dara, L.; et al. Dynamic adaptation of liver mitochondria to chronic alcohol feeding in mice: Biogenesis, remodeling, and functional alterations. J. Biol. Chem. 2012, 287, 42165–42179. [Google Scholar] [CrossRef]
- Garcia, J.; Chang, R.; Steinberg, R.A.; Arce, A.; Yang, J.; Van Der Eb, P.; Abdullah, T.; Chandrashekar, D.V.; Eck, S.M.; Meza, P.; et al. Modulation of hepatic amyloid precursor protein and lipoprotein receptor-related protein 1 by chronic alcohol intake: Potential link between liver steatosis and amyloid-β. Front. Physiol. 2022, 13, 930402. [Google Scholar] [CrossRef]
- Hoffman, J.L.; Faccidomo, S.; Kim, M.; Taylor, S.M.; Agoglia, A.E.; May, A.M.; Smith, E.N.; Wong, L.C.; Hodge, C.W. Alcohol drinking exacerbates neural and behavioral pathology in the 3xTg-AD mouse model of Alzheimer’s disease. Int. Rev. Neurobiol. 2019, 148, 169–230. [Google Scholar] [CrossRef]
- Huang, D.; Yu, M.; Yang, S.; Lou, D.; Zhou, W.; Zheng, L.; Wang, Z.; Cai, F.; Zhou, W.; Li, T.; et al. Ethanol Alters APP Processing and Aggravates Alzheimer-Associated Phenotypes. Mol. Neurobiol. 2018, 55, 5006–5018. [Google Scholar] [CrossRef]
- Day, S.M.; Gironda, S.C.; Clarke, C.W.; Snipes, J.A.; Nicol, N.I.; Kamran, H.; Vaughan, W.; Weiner, J.L.; Macauley, S.L. Ethanol exposure alters Alzheimer’s-related pathology, behavior, and metabolism in APP/PS1 mice. Neurobiol. Dis. 2022, 177, 105967. [Google Scholar] [CrossRef]
- Wei, J.; Qin, L.; Fu, Y.; Dai, Y.; Wen, Y.; Xu, S. Long-term consumption of alcohol exacerbates neural lesions by destroying the functional integrity of the blood-brain barrier. Drug Chem. Toxicol. 2022, 45, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Barnett, A.; David, E.; Rohlman, A.; Nikolova, V.D.; Moy, S.S.; Vetreno, R.P.; Coleman, L.G., Jr. Adolescent Binge Alcohol Enhances Early Alzheimer’s Disease Pathology in Adulthood Through Proinflammatory Neuroimmune Activation. Front. Pharmacol. 2022, 13, 884170. [Google Scholar] [CrossRef] [PubMed]
- Ledesma, J.C.; Rodríguez-Arias, M.; Gavito, A.L.; Sánchez-Pérez, A.M.; Viña, J.; Medina Vera, D.; Rodríguez de Fonseca, F.; Miñarro, J. Adolescent binge-ethanol accelerates cognitive impairment and β-amyloid production and dysregulates endocannabinoid signaling in the hippocampus of APP/PSE mice. Addict. Biol. 2021, 26, e12883. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.-S.; Hou, F.-L.; Guo, J.; Lin, L.; Zhu, F.-Y. Effects of alcohol intake on cognitive function and β-amyloid protein in APP/PS1 transgenic mice. Food Chem. Toxicol. 2021, 151, 112105. [Google Scholar] [CrossRef]
- Muñoz, G.; Urrutia, J.C.; Burgos, C.F.; Silva, V.; Aguilar, F.; Sama, M.; Yeh, H.H.; Opazo, C.; Aguayo, L.G. Low concentrations of ethanol protect against synaptotoxicity induced by Aβ in hippocampal neurons. Neurobiol. Aging 2015, 36, 845–856. [Google Scholar] [CrossRef]
- Wang, J.; Ho, L.; Zhao, Z.; Seror, I.; Humala, N.; Dickstein, D.L.; Thiyagarajan, M.; Percival, S.S.; Talcott, S.T.; Pasinetti, G.M. Moderate consumption of Cabernet Sauvignon attenuates Abeta neuropathology in a mouse model of Alzheimer’s disease. FASEB J. 2006, 20, 2313–2320. [Google Scholar] [CrossRef]
- Yoneyama, N.; Crabbe, J.C.; Ford, M.M.; Murillo, A.; Finn, D.A. Voluntary ethanol consumption in 22 inbred mouse strains. Alcohol 2008, 42, 149–160. [Google Scholar] [CrossRef]
- Gabr, A.A.; Lee, H.J.; Onphachanh, X.; Jung, Y.H.; Kim, J.S.; Chae, C.W.; Han, H.J. Ethanol-induced PGE(2) up-regulates Aβ production through PKA/CREB signaling pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2942–2953. [Google Scholar] [CrossRef]
- Li, J.; Cheng, J. Apolipoprotein E4 exacerbates ethanol-induced neurotoxicity through augmentation of oxidative stress and apoptosis in N2a-APP cells. Neurosci. Lett. 2018, 665, 1–6. [Google Scholar] [CrossRef]
- Kalinin, S.; González-Prieto, M.; Scheiblich, H.; Lisi, L.; Kusumo, H.; Heneka, M.T.; Madrigal, J.L.M.; Pandey, S.C.; Feinstein, D.L. Transcriptome analysis of alcohol-treated microglia reveals downregulation of beta amyloid phagocytosis. J. Neuroinflamm. 2018, 15, 141. [Google Scholar] [CrossRef]
- Gendron, T.F.; McCartney, S.; Causevic, E.; Ko, L.W.; Yen, S.H. Ethanol enhances tau accumulation in neuroblastoma cells that inducibly express tau. Neurosci. Lett. 2008, 443, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Ormeño, D.; Romero, F.; López-Fenner, J.; Avila, Á.; Martínez-Torres, A.; Parodi, J. Ethanol Reduces Amyloid Aggregation In Vitro and Prevents Toxicity in Cell Lines. Arch. Med. Res. 2013, 44, 1–7. [Google Scholar] [CrossRef]
- Belmadani, A.; Kumar, S.; Schipma, M.; Collins, M.A.; Neafsey, E.J. Inhibition of amyloid-beta-induced neurotoxicity and apoptosis by moderate ethanol preconditioning. Neuroreport 2004, 15, 2093–2096. [Google Scholar] [CrossRef] [PubMed]
- Bate, C.; Williams, A. Ethanol protects cultured neurons against amyloid-β and α-synuclein-induced synapse damage. Neuropharmacology 2011, 61, 1406–1412. [Google Scholar] [CrossRef]
- Brust, J.C. Ethanol and cognition: Indirect effects, neurotoxicity and neuroprotection: A review. Int. J. Environ. Res. Public Health 2010, 7, 1540–1557. [Google Scholar] [CrossRef]
- de la Monte, S.M.; Vonsattel, J.P.; Richardson, E.P., Jr. Morphometric demonstration of atrophic changes in the cerebral cortex, white matter, and neostriatum in Huntington’s disease. J. Neuropathol. Exp. Neurol. 1988, 47, 516–525. [Google Scholar] [CrossRef]
- Sullivan, E.V.; Marsh, L.; Mathalon, D.H.; Lim, K.O.; Pfefferbaum, A. Anterior hippocampal volume deficits in nonamnesic, aging chronic alcoholics. Alcohol. Clin. Exp. Res. 1995, 19, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Mann, K.; Agartz, I.; Harper, C.; Shoaf, S.; Rawlings, R.R.; Momenan, R.; Hommer, D.W.; Pfefferbaum, A.; Sullivan, E.V.; Anton, R.F.; et al. Neuroimaging in alcoholism: Ethanol and brain damage. Alcohol. Clin. Exp. Res. 2001, 25, 104S–109S. [Google Scholar] [CrossRef]
- Peng, B.; Yang, Q.; Joshi, R.B.; Liu, Y.; Akbar, M.; Song, B.J.; Zhou, S.; Wang, X. Role of Alcohol Drinking in Alzheimer’s Disease, Parkinson’s Disease, and Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2020, 21, 2316. [Google Scholar] [CrossRef]
- Venkataraman, A.; Kalk, N.; Sewell, G.; Ritchie, C.W.; Lingford-Hughes, A. Alcohol and Alzheimer’s Disease-Does Alcohol Dependence Contribute to Beta-Amyloid Deposition, Neuroinflammation and Neurodegeneration in Alzheimer’s Disease? Alcohol Alcohol. 2017, 52, 151–158. [Google Scholar] [CrossRef]
- Yi, K.; Shi, Y.; Zhao, Y. Effects of ethanol on the pathogenesis of Alzheimer’s disease. IOP Conf. Ser. Mater. Sci. Eng. 2018, 394, 022024. [Google Scholar] [CrossRef]
- Kamal, H.; Tan, G.C.; Ibrahim, S.F.; Shaikh, M.F.; Mohamed, I.N.; Mohamed, R.M.P.; Hamid, A.A.; Ugusman, A.; Kumar, J. Alcohol Use Disorder, Neurodegeneration, Alzheimer’s and Parkinson’s Disease: Interplay between Oxidative Stress, Neuroimmune Response and Excitotoxicity. Front. Cell. Neurosci. 2020, 14, 282. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Lizarbe, S.; Pascual, M.; Guerri, C. Critical role of TLR4 response in the activation of microglia induced by ethanol. J. Immunol. 2009, 183, 4733–4744. [Google Scholar] [CrossRef] [PubMed]
- Blanco, A.M.; Vallés, S.L.; Pascual, M.; Guerri, C. Involvement of TLR4/type I IL-1 receptor signaling in the induction of inflammatory mediators and cell death induced by ethanol in cultured astrocytes. J. Immunol. 2005, 175, 6893–6899. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Minoretti, P.; Gazzaruso, C.; Vito, C.D.; Emanuele, E.; Bianchi, M.; Coen, E.; Reino, M.; Geroldi, D. Effect of the functional toll-like receptor 4 Asp299Gly polymorphism on susceptibility to late-onset Alzheimer’s disease. Neurosci. Lett. 2006, 391, 147–149. [Google Scholar] [CrossRef]
- Coleman, L.G.; Zou, J.; Crews, F.T. Microglial-derived miRNA let-7 and HMGB1 contribute to ethanol-induced neurotoxicity via TLR7. J. Neuroinflamm. 2017, 14, 22. [Google Scholar] [CrossRef]
- Laguesse, S.; Morisot, N.; Phamluong, K.; Ron, D. Region specific activation of the AKT and mTORC1 pathway in response to excessive alcohol intake in rodents. Addict. Biol. 2017, 22, 1856–1869. [Google Scholar] [CrossRef]
- Sen, N. ER Stress, CREB, and Memory: A Tangled Emerging Link in Disease. Neuroscientist 2018, 25, 420–433. [Google Scholar] [CrossRef]
- Sharma, V.K.; Singh, T.G. CREB: A Multifaceted Target for Alzheimer’s Disease. Curr. Alzheimer Res. 2020, 17, 1280–1293. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, Y.; Gao, R.; Li, H.; Dunn, T.; Wu, P.; Smith, R.G.; Sarkar, P.S.; Fang, X. Ethanol suppresses PGC-1α expression by interfering with the cAMP-CREB pathway in neuronal cells. PLoS ONE 2014, 9, e104247. [Google Scholar] [CrossRef] [PubMed]
- Giese, K.P.; Ris, L.; Plattner, F. Is there a role of the cyclin-dependent kinase 5 activator p25 in Alzheimer’s disease? Neuroreport 2005, 16, 1725–1730. [Google Scholar] [CrossRef]
- Kimura, T.; Ishiguro, K.; Hisanaga, S. Physiological and pathological phosphorylation of tau by Cdk5. Front. Mol. Neurosci. 2014, 7, 65. [Google Scholar] [CrossRef]
- Rajgopal, Y.; Vemuri, M.C. Ethanol induced changes in cyclin-dependent kinase-5 activity and its activators, P35, P67 (Munc-18) in rat brain. Neurosci. Lett. 2001, 308, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Patrick, G.N.; Zukerberg, L.; Nikolic, M.; de la Monte, S.; Dikkes, P.; Tsai, L.H. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 1999, 402, 615–622. [Google Scholar] [CrossRef]
- Joshi, V.; Subbanna, S.; Shivakumar, M.; Basavarajappa, B.S. CB1R regulates CDK5 signaling and epigenetically controls Rac1 expression contributing to neurobehavioral abnormalities in mice postnatally exposed to ethanol. Neuropsychopharmacology 2019, 44, 514–525. [Google Scholar] [CrossRef]
- Fang, H. [P1–084]: Long-term drinking induces tau hyperphosphorylation and cognitive deficit. Alzheimer’s Dement. 2017, 13, P270. [Google Scholar] [CrossRef]
- Ji, Z.; Yuan, L.; Lu, X.; Ding, H.; Luo, J.; Ke, Z.-J. Binge Alcohol Exposure Causes Neurobehavioral Deficits and GSK3β Activation in the Hippocampus of Adolescent Rats. Sci. Rep. 2018, 8, 3088. [Google Scholar] [CrossRef]
- Rahman, M.S.; Hossain, K.S.; Das, S.; Kundu, S.; Adegoke, E.O.; Rahman, M.A.; Hannan, M.A.; Uddin, M.J.; Pang, M.-G. Role of Insulin in Health and Disease: An Update. Int. J. Mol. Sci. 2021, 22, 6403. [Google Scholar] [CrossRef]
- Ivan, D.C.; Berve, K.C.; Walthert, S.; Monaco, G.; Borst, K.; Bouillet, E.; Ferreira, F.; Lee, H.; Steudler, J.; Buch, T.; et al. Insulin-like growth factor-1 receptor controls the function of CNS-resident macrophages and their contribution to neuroinflammation. Acta Neuropathol. Commun. 2023, 11, 35. [Google Scholar] [CrossRef]
- de la Monte, S.M.; Tong, M.; Cohen, A.C.; Sheedy, D.; Harper, C.; Wands, J.R. Insulin and insulin-like growth factor resistance in alcoholic neurodegeneration. Alcohol. Clin. Exp. Res. 2008, 32, 1630–1644. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.C.; Tong, M.; Wands, J.R.; de la Monte, S.M. Insulin and insulin-like growth factor resistance with neurodegeneration in an adult chronic ethanol exposure model. Alcohol. Clin. Exp. Res. 2007, 31, 1558–1573. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M.; Neely, T.R.; Cannon, J.; Wands, J.R. Ethanol impairs insulin-stimulated mitochondrial function in cerebellar granule neurons. Cell. Mol. Life Sci. 2001, 58, 1950–1960. [Google Scholar] [CrossRef]
- Soscia, S.J.; Tong, M.; Xu, X.J.; Cohen, A.C.; Chu, J.; Wands, J.R.; de la Monte, S.M. Chronic gestational exposure to ethanol causes insulin and IGF resistance and impairs acetylcholine homeostasis in the brain. Cell. Mol. Life Sci. 2006, 63, 2039–2056. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Cholerton, B.; Baker, L.D. Insulin and Alzheimer’s disease: Untangling the web. J. Alzheimer’s Dis. 2013, 33 (Suppl. S1), S263–S275. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, R.; Haeri, A.; Dargahi, L.; Mohamed, Z.; Ahmadiani, A. Insulin in the brain: Sources, localization and functions. Mol. Neurobiol. 2013, 47, 145–171. [Google Scholar] [CrossRef]
- Rivera, E.J.; Goldin, A.; Fulmer, N.; Tavares, R.; Wands, J.R.; de la Monte, S.M. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: Link to brain reductions in acetylcholine. J. Alzheimer’s Dis. 2005, 8, 247–268. [Google Scholar] [CrossRef]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease--is this type 3 diabetes? J. Alzheimer’s Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef]
- Liu, D.; Ke, Z.; Luo, J. Thiamine Deficiency and Neurodegeneration: The Interplay Among Oxidative Stress, Endoplasmic Reticulum Stress, and Autophagy. Mol. Neurobiol. 2017, 54, 5440–5448. [Google Scholar] [CrossRef]
- Langlais, P.J. Alcohol-Related Thiamine Deficiency: Impact on Cognitive and Memory Functioning. Alcohol Health Res. World 1995, 19, 113–121. [Google Scholar]
- Karuppagounder, S.S.; Xu, H.; Shi, Q.; Chen, L.H.; Pedrini, S.; Pechman, D.; Baker, H.; Beal, M.F.; Gandy, S.E.; Gibson, G.E. Thiamine deficiency induces oxidative stress and exacerbates the plaque pathology in Alzheimer’s mouse model. Neurobiol. Aging 2009, 30, 1587–1600. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yang, G.; Li, W.; Fan, Z.; Sun, A.; Luo, J.; Ke, Z.J. Thiamine deficiency increases β-secretase activity and accumulation of β-amyloid peptides. Neurobiol. Aging 2011, 32, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Henderson-Redmond, A.N.; Guindon, J.; Morgan, D.J. Roles for the endocannabinoid system in ethanol-motivated behavior. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2016, 65, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Marin, L.; Pavon, F.J.; Decara, J.; Suarez, J.; Gavito, A.; Castilla-Ortega, E.; Rodriguez de Fonseca, F.; Serrano, A. Effects of Intermittent Alcohol Exposure on Emotion and Cognition: A Potential Role for the Endogenous Cannabinoid System and Neuroinflammation. Front. Behav. Neurosci. 2017, 11, 15. [Google Scholar] [CrossRef]
- Alvarez-Jaimes, L.; Stouffer, D.G.; Parsons, L.H. Chronic ethanol treatment potentiates ethanol-induced increases in interstitial nucleus accumbens endocannabinoid levels in rats. J. Neurochem. 2009, 111, 37–48. [Google Scholar] [CrossRef] [PubMed]
- González, S.; Grazia Cascio, M.; Fernández-Ruiz, J.; Fezza, F.; Di Marzo, V.; Ramos, J.A. Changes in endocannabinoid contents in the brain of rats chronically exposed to nicotine, ethanol or cocaine. Brain Res. 2002, 954, 73–81. [Google Scholar] [CrossRef]
- Altamura, C.; Ventriglia, M.; Martini, M.G.; Montesano, D.; Errante, Y.; Piscitelli, F.; Scrascia, F.; Quattrocchi, C.; Palazzo, P.; Seccia, S.; et al. Elevation of Plasma 2-Arachidonoylglycerol Levels in Alzheimer’s Disease Patients as a Potential Protective Mechanism against Neurodegenerative Decline. J. Alzheimer’s Dis. 2015, 46, 497–506. [Google Scholar] [CrossRef]
- Chen, R.; Zhang, J.; Wu, Y.; Wang, D.; Feng, G.; Tang, Y.P.; Teng, Z.; Chen, C. Monoacylglycerol lipase is a therapeutic target for Alzheimer’s disease. Cell Rep. 2012, 2, 1329–1339. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, M.; Liu, W.; Ma, Z.; Wu, J. Activation of cannabinoid receptor 2 protects rat hippocampal neurons against Aβ-induced neuronal toxicity. Neurosci. Lett. 2020, 735, 135207. [Google Scholar] [CrossRef]
- Solas, M.; Francis, P.T.; Franco, R.; Ramirez, M.J. CB2 receptor and amyloid pathology in frontal cortex of Alzheimer’s disease patients. Neurobiol. Aging 2013, 34, 805–808. [Google Scholar] [CrossRef]
- Tolón, R.M.; Núñez, E.; Pazos, M.R.; Benito, C.; Castillo, A.I.; Martínez-Orgado, J.A.; Romero, J. The activation of cannabinoid CB2 receptors stimulates in situ and in vitro beta-amyloid removal by human macrophages. Brain Res. 2009, 1283, 148–154. [Google Scholar] [CrossRef] [PubMed]
- van der Stelt, M.; Mazzola, C.; Esposito, G.; Matias, I.; Petrosino, S.; De Filippis, D.; Micale, V.; Steardo, L.; Drago, F.; Iuvone, T.; et al. Endocannabinoids and beta-amyloid-induced neurotoxicity in vivo: Effect of pharmacological elevation of endocannabinoid levels. Cell. Mol. Life Sci. 2006, 63, 1410–1424. [Google Scholar] [CrossRef]
- Aso, E.; Juvés, S.; Maldonado, R.; Ferrer, I. CB2 cannabinoid receptor agonist ameliorates Alzheimer-like phenotype in AβPP/PS1 mice. J. Alzheimer’s Dis. 2013, 35, 847–858. [Google Scholar] [CrossRef]
- Wu, J.; Hocevar, M.; Foss, J.F.; Bie, B.; Naguib, M. Activation of CB(2) receptor system restores cognitive capacity and hippocampal Sox2 expression in a transgenic mouse model of Alzheimer’s disease. Eur. J. Pharmacol. 2017, 811, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Casamenti, F.; Scali, C.; Vannucchi, M.G.; Bartolini, L.; Pepeu, G. Long-term ethanol consumption by rats: Effect on acetylcholine release in vivo, choline acetyltransferase activity, and behavior. Neuroscience 1993, 56, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Henn, C.; Löffelholz, K.; Klein, J. Stimulatory and inhibitory effects of ethanol on hippocampal acetylcholine release. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1998, 357, 640–647. [Google Scholar] [CrossRef]
- Cadete-Leite, A.; Andrade, J.P.; Sousa, N.; Ma, W.; Ribeiro-Da-Silva, A. Effects of chronic alcohol consumption on the cholinergic innervation of the rat hippocampal formation as revealed by choline acetyltransferase immunocytochemistry. Neuroscience 1995, 64, 357–374. [Google Scholar] [CrossRef]
- Floyd, E.A.; Young-Seigler, A.C.; Ford, B.D.; Reasor, J.D.; Moore, E.L.; Townsel, J.G.; Rucker, H.K. Chronic ethanol ingestion produces cholinergic hypofunction in rat brain. Alcohol 1997, 14, 93–98. [Google Scholar] [CrossRef]
- Nordberg, A.; Larsson, C.; Perdahl, E.; Winblad, B. Changes in cholinergic activity in human hippocampus following chronic alcohol abuse. Pharmacol. Biochem. Behav. 1983, 18 (Suppl. S1), 397–400. [Google Scholar] [CrossRef]
- Archie, S.R.; Al Shoyaib, A.; Cucullo, L. Blood-Brain Barrier Dysfunction in CNS Disorders and Putative Therapeutic Targets: An Overview. Pharmaceutics 2021, 13, 1779. [Google Scholar] [CrossRef]
- Montagne, A.; Zhao, Z.; Zlokovic, B.V. Alzheimer’s disease: A matter of blood-brain barrier dysfunction? J. Exp. Med. 2017, 214, 3151–3169. [Google Scholar] [CrossRef] [PubMed]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Hussain, B.; Fang, C.; Chang, J. Blood-Brain Barrier Breakdown: An Emerging Biomarker of Cognitive Impairment in Normal Aging and Dementia. Front. Neurosci. 2021, 15, 688090. [Google Scholar] [CrossRef] [PubMed]
- Haorah, J.; Ramirez, S.H.; Schall, K.; Smith, D.; Pandya, R.; Persidsky, Y. Oxidative stress activates protein tyrosine kinase and matrix metalloproteinases leading to blood-brain barrier dysfunction. J. Neurochem. 2007, 101, 566–576. [Google Scholar] [CrossRef]
- Abdul Muneer, P.M.; Alikunju, S.; Szlachetka, A.M.; Haorah, J. The mechanisms of cerebral vascular dysfunction and neuroinflammation by MMP-mediated degradation of VEGFR-2 in alcohol ingestion. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Araiz, A.; Porcu, F.; Pérez-Hernández, M.; García-Gutiérrez, M.S.; Aracil-Fernández, M.A.; Gutierrez-López, M.D.; Guerri, C.; Manzanares, J.; O’Shea, E.; Colado, M.I. Disruption of blood-brain barrier integrity in postmortem alcoholic brain: Preclinical evidence of TLR4 involvement from a binge-like drinking model. Addict. Biol. 2017, 22, 1103–1116. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Spangenberg, E.; Severson, P.L.; Hohsfield, L.A.; Crapser, J.; Zhang, J.; Burton, E.A.; Zhang, Y.; Spevak, W.; Lin, J.; Phan, N.Y.; et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat. Commun. 2019, 10, 3758. [Google Scholar] [CrossRef]
- McClain, J.A.; Morris, S.A.; Deeny, M.A.; Marshall, S.A.; Hayes, D.M.; Kiser, Z.M.; Nixon, K. Adolescent binge alcohol exposure induces long-lasting partial activation of microglia. Brain Behav. Immun. 2011, 25 (Suppl. S1), S120–S128. [Google Scholar] [CrossRef]
- Marshall, S.A.; McClain, J.A.; Wooden, J.I.; Nixon, K. Microglia Dystrophy Following Binge-Like Alcohol Exposure in Adolescent and Adult Male Rats. Front. Neuroanat. 2020, 14, 52. [Google Scholar] [CrossRef]
- Lowe, P.P.; Morel, C.; Ambade, A.; Iracheta-Vellve, A.; Kwiatkowski, E.; Satishchandran, A.; Furi, I.; Cho, Y.; Gyongyosi, B.; Catalano, D.; et al. Chronic alcohol-induced neuroinflammation involves CCR2/5-dependent peripheral macrophage infiltration and microglia alterations. J. Neuroinflamm. 2020, 17, 296. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.; Meireles, M.; Silva, S.M. Chronic ethanol intake induces partial microglial activation that is not reversed by long-term ethanol withdrawal in the rat hippocampal formation. Neurotoxicology 2017, 60, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Huang, W.; Cheng, C.-H.; Zhou, L.; Jiang, G.-B.; Hu, Y.-Y. Association between Aldehyde dehydrogenase-2 Polymorphisms and Risk of Alzheimer’s Disease and Parkinson’s Disease: A Meta-Analysis Based on 5315 Individuals. Front. Neurol. 2019, 10, 290. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Chen, J.; Chen, L.; Histen, G.; Lin, Z.; Gross, S.; Hixon, J.; Chen, Y.; Kung, C.; Chen, Y.; et al. ALDH2(E487K) mutation increases protein turnover and promotes murine hepatocarcinogenesis. Proc. Natl. Acad. Sci. USA 2015, 112, 9088–9093. [Google Scholar] [CrossRef]
- Joshi, A.U.; Van Wassenhove, L.D.; Logas, K.R.; Minhas, P.S.; Andreasson, K.I.; Weinberg, K.I.; Chen, C.-H.; Mochly-Rosen, D. Aldehyde dehydrogenase 2 activity and aldehydic load contribute to neuroinflammation and Alzheimer’s disease related pathology. Acta Neuropathol. Commun. 2019, 7, 190. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.W.; Thompson, R.; Zhang, H.; Xu, H. APP processing in Alzheimer’s disease. Mol. Brain 2011, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Jeong, H.Y.; Yang, S.; Choi, S.P.; Seo, M.Y.; Yun, Y.K.; Choi, Y.; Baik, S.H.; Park, J.S.; Gwon, A.R.; et al. Effects of chronic alcohol consumption on expression levels of APP and Aβ-producing enzymes. BMB Rep. 2011, 44, 135–139. [Google Scholar] [CrossRef]
- Brar, G.; Tsukamoto, H. Alcoholic and non-alcoholic steatohepatitis: Global perspective and emerging science. J. Gastroenterol. 2019, 54, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Bataller, R.; Cortez-Pinto, H.; Gao, B.; Gual, A.; Lackner, C.; Mathurin, P.; Mueller, S.; Szabo, G.; Tsukamoto, H. Alcoholic liver disease. Nat. Rev. Dis. Prim. 2018, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.V.; Matyas, C.; Paloczi, J.; Pacher, P. Alcohol Misuse and Kidney Injury: Epidemiological Evidence and Potential Mechanisms. Alcohol Res. 2017, 38, 283–288. [Google Scholar]
- Zlokovic, B.V.; Ghiso, J.; Mackic, J.B.; McComb, J.G.; Weiss, M.H.; Frangione, B. Blood-brain barrier transport of circulating Alzheimer’s amyloid beta. Biochem. Biophys. Res. Commun. 1993, 197, 1034–1040. [Google Scholar] [CrossRef]
- Henderson, S.J.; Andersson, C.; Narwal, R.; Janson, J.; Goldschmidt, T.J.; Appelkvist, P.; Bogstedt, A.; Steffen, A.C.; Haupts, U.; Tebbe, J.; et al. Sustained peripheral depletion of amyloid-beta with a novel form of neprilysin does not affect central levels of amyloid-beta. Brain 2014, 137, 553–564. [Google Scholar] [CrossRef]
- Georgievska, B.; Gustavsson, S.; Lundkvist, J.; Neelissen, J.; Eketjall, S.; Ramberg, V.; Bueters, T.; Agerman, K.; Jureus, A.; Svensson, S.; et al. Revisiting the peripheral sink hypothesis: Inhibiting BACE1 activity in the periphery does not alter beta-amyloid levels in the CNS. J. Neurochem. 2015, 132, 477–486. [Google Scholar] [CrossRef]
- DeMattos, R.B.; Bales, K.R.; Cummins, D.J.; Dodart, J.C.; Paul, S.M.; Holtzman, D.M. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 8850–8855. [Google Scholar] [CrossRef] [PubMed]
- Shigemori, K.; Nomura, S.; Umeda, T.; Takeda, S.; Tomiyama, T. Peripheral Aβ acts as a negative modulator of insulin secretion. Proc. Natl. Acad. Sci. USA 2022, 119, e2117723119. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.S.; Shen, L.L.; Bu, X.L.; Zhang, W.W.; Chen, S.H.; Huang, Z.L.; Xiong, J.X.; Gao, C.Y.; Dong, Z.; He, Y.N.; et al. Peritoneal dialysis reduces amyloid-beta plasma levels in humans and attenuates Alzheimer-associated phenotypes in an APP/PS1 mouse model. Acta Neuropathol. 2017, 134, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Urayama, A.; Moreno-Gonzalez, I.; Morales-Scheihing, D.; Kharat, V.; Pritzkow, S.; Soto, C. Preventive and therapeutic reduction of amyloid deposition and behavioral impairments in a model of Alzheimer’s disease by whole blood exchange. Mol. Psychiatry 2022, 27, 4285–4296. [Google Scholar] [CrossRef]
- Xiang, Y.; Bu, X.L.; Liu, Y.H.; Zhu, C.; Shen, L.L.; Jiao, S.S.; Zhu, X.Y.; Giunta, B.; Tan, J.; Song, W.H.; et al. Physiological amyloid-beta clearance in the periphery and its therapeutic potential for Alzheimer’s disease. Acta Neuropathol. 2015, 130, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Matsui, T. Role of receptor for advanced glycation end products (RAGE) in liver disease. Eur. J. Med. Res. 2015, 20, 15. [Google Scholar] [CrossRef]
- Mohamed, L.A.; Kaddoumi, A. In Vitro Investigation of Amyloid-β Hepatobiliary Disposition in Sandwich-Cultured Primary Rat Hepatocytes. Drug Metab. Dispos. 2013, 41, 1787–1796. [Google Scholar] [CrossRef]
- Lam, V.; Takechi, R.; Hackett, M.J.; Francis, R.; Bynevelt, M.; Celliers, L.M.; Nesbit, M.; Mamsa, S.; Arfuso, F.; Das, S.; et al. Synthesis of human amyloid restricted to liver results in an Alzheimer disease-like neurodegenerative phenotype. PLoS Biol. 2021, 19, e3001358. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, A.; Cacabelos, R.; Sanpedro, C.; García-Fantini, M.; Aleixandre, M. Serum TNF-alpha levels are increased and correlate negatively with free IGF-I in Alzheimer disease. Neurobiol. Aging 2007, 28, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Paouri, E.; Tzara, O.; Kartalou, G.I.; Zenelak, S.; Georgopoulos, S. Peripheral Tumor Necrosis Factor-Alpha (TNF-alpha) Modulates Amyloid Pathology by Regulating Blood-Derived Immune Cells and Glial Response in the Brain of AD/TNF Transgenic Mice. J. Neurosci. 2017, 37, 5155–5171. [Google Scholar] [CrossRef] [PubMed]
- Zeng, T.; Zhang, C.L.; Xiao, M.; Yang, R.; Xie, K.Q. Critical Roles of Kupffer Cells in the Pathogenesis of Alcoholic Liver Disease: From Basic Science to Clinical Trials. Front. Immunol. 2016, 7, 538. [Google Scholar] [CrossRef]
- Lin, H.Z.; Yang, S.Q.; Zeldin, G.; Diehl, A.M. Chronic ethanol consumption induces the production of tumor necrosis factor-alpha and related cytokines in liver and adipose tissue. Alcohol. Clin. Exp. Res. 1998, 22, 231S–237S. [Google Scholar] [CrossRef] [PubMed]
- Tarkowski, E.; Andreasen, N.; Tarkowski, A.; Blennow, K. Intrathecal inflammation precedes development of Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2003, 74, 1200–1205. [Google Scholar] [CrossRef]
- Tarkowski, E.; Blennow, K.; Wallin, A.; Tarkowski, A. Intracerebral production of tumor necrosis factor-alpha, a local neuroprotective agent, in Alzheimer disease and vascular dementia. J. Clin. Immunol. 1999, 19, 223–230. [Google Scholar] [CrossRef]
- Chang, R.; Knox, J.; Chang, J.; Derbedrossian, A.; Vasilevko, V.; Cribbs, D.; Boado, R.J.; Pardridge, W.M.; Sumbria, R.K. Blood-Brain Barrier Penetrating Biologic TNF-alpha Inhibitor for Alzheimer’s Disease. Mol. Pharm. 2017, 14, 2340–2349. [Google Scholar] [CrossRef]
- Gutierrez, E.G.; Banks, W.A.; Kastin, A.J. Murine tumor necrosis factor alpha is transported from blood to brain in the mouse. J. Neuroimmunol. 1993, 47, 169–176. [Google Scholar] [CrossRef]
- Banks, W.A.; Moinuddin, A.; Morley, J.E. Regional transport of TNF-alpha across the blood-brain barrier in young ICR and young and aged SAMP8 mice. Neurobiol. Aging 2001, 22, 671–676. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chandrashekar, D.V.; Steinberg, R.A.; Han, D.; Sumbria, R.K. Alcohol as a Modifiable Risk Factor for Alzheimer’s Disease—Evidence from Experimental Studies. Int. J. Mol. Sci. 2023, 24, 9492. https://doi.org/10.3390/ijms24119492
Chandrashekar DV, Steinberg RA, Han D, Sumbria RK. Alcohol as a Modifiable Risk Factor for Alzheimer’s Disease—Evidence from Experimental Studies. International Journal of Molecular Sciences. 2023; 24(11):9492. https://doi.org/10.3390/ijms24119492
Chicago/Turabian StyleChandrashekar, Devaraj V., Ross A. Steinberg, Derick Han, and Rachita K. Sumbria. 2023. "Alcohol as a Modifiable Risk Factor for Alzheimer’s Disease—Evidence from Experimental Studies" International Journal of Molecular Sciences 24, no. 11: 9492. https://doi.org/10.3390/ijms24119492
APA StyleChandrashekar, D. V., Steinberg, R. A., Han, D., & Sumbria, R. K. (2023). Alcohol as a Modifiable Risk Factor for Alzheimer’s Disease—Evidence from Experimental Studies. International Journal of Molecular Sciences, 24(11), 9492. https://doi.org/10.3390/ijms24119492