
Unlocking the Potential of the Antimicrobial Peptide Gomesin: From Discovery and Structure–Activity Relationships to Therapeutic Applications

Abstract
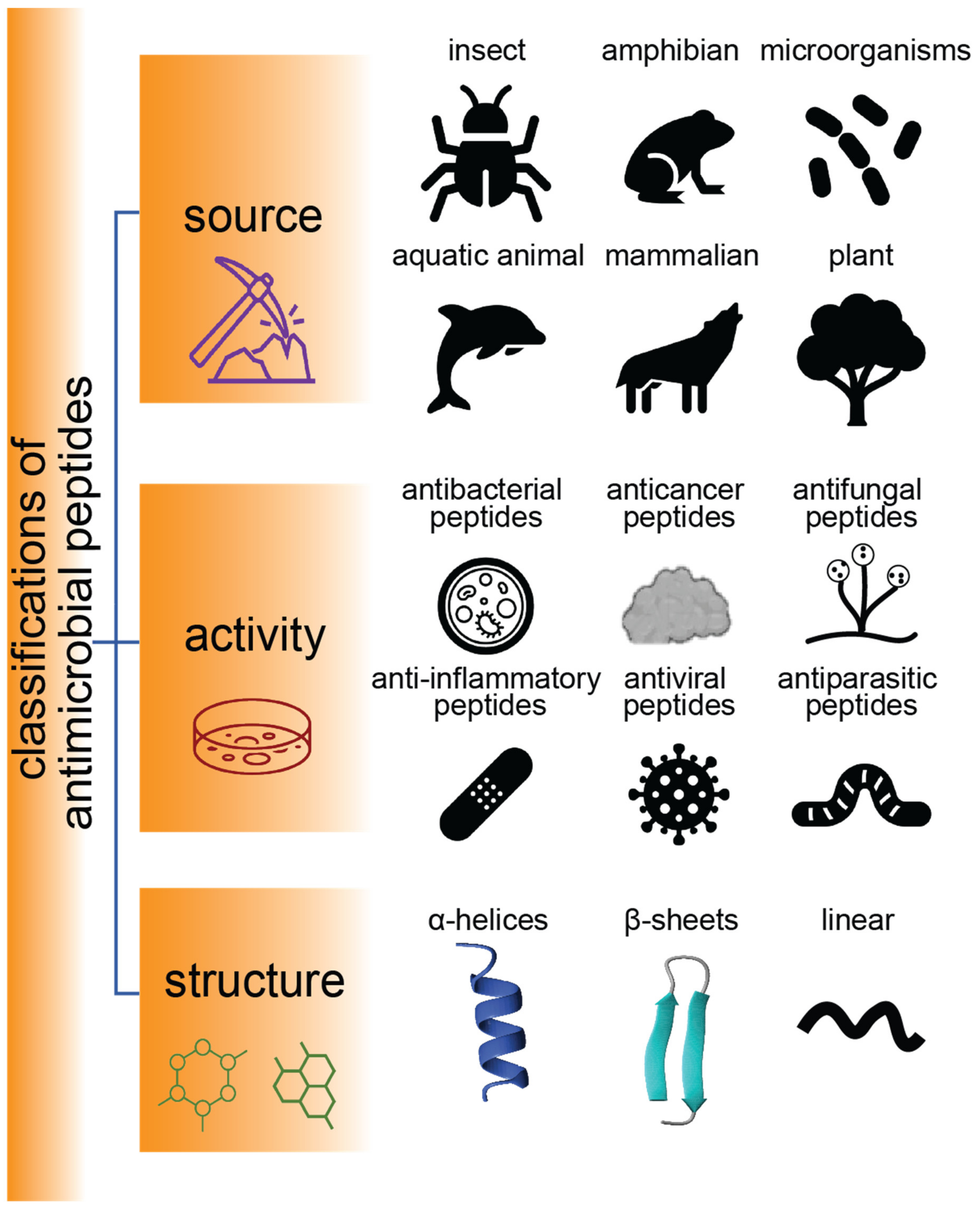
1. Introduction
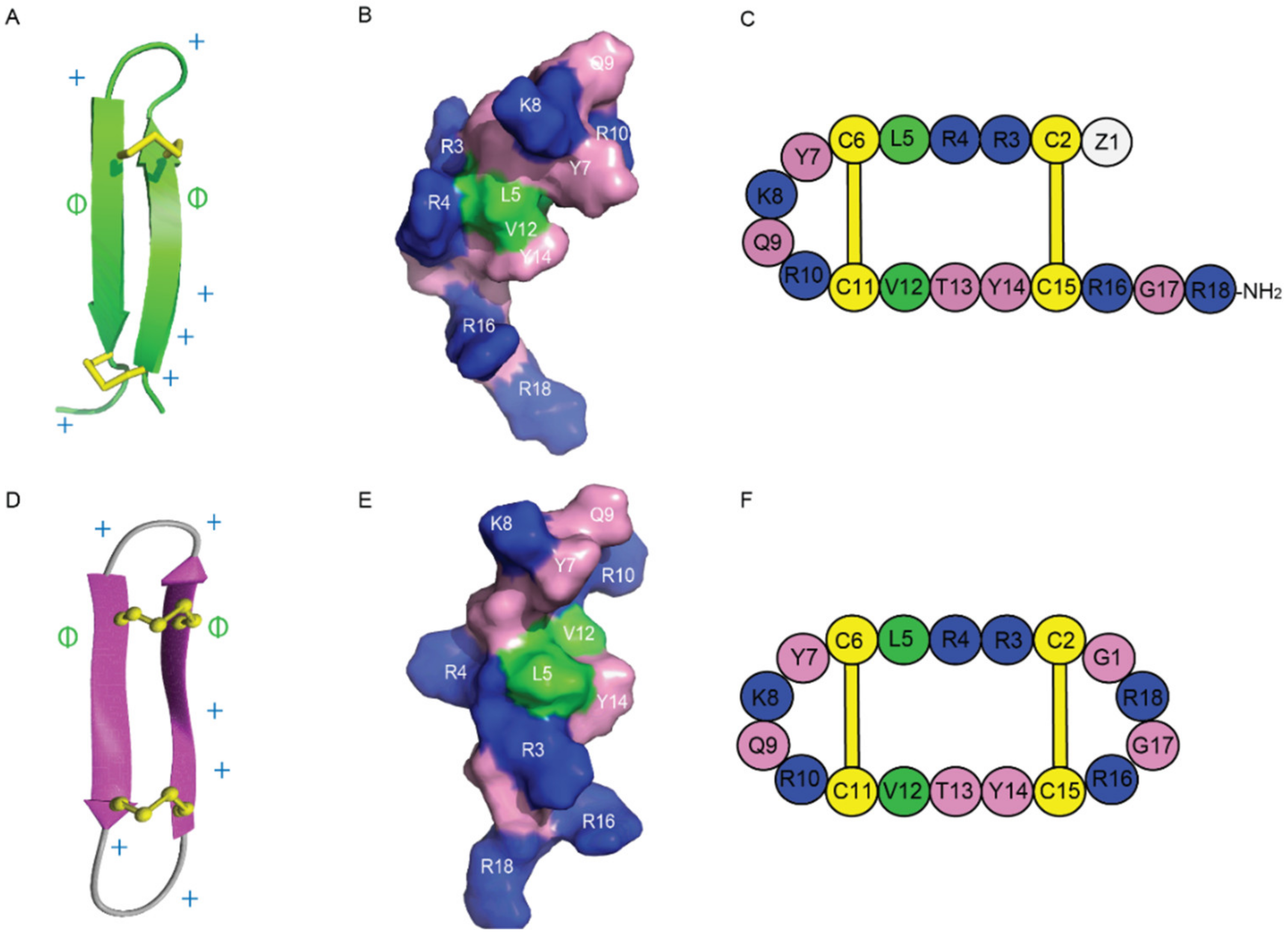
2. Discovery, Synthesis and Structural Characterization of Gomesin
3. Chemical Methods to Derive Structure–Activity Relationships
4. Biophysical Studies to Elucidate the Mechanism of Action Gomesin on Bacterial and Mammalian Cell Membranes
4.1. Membrane Binding and Peptide–Lipid Interactions
4.2. Permeabilization of Cell Membranes and Leakage Activity
5. Modifications of Gm and cGm
6. Biological Activity of Gomesin and Analogues
6.1. Antimicrobial Activity
6.2. Anticancer Activity
6.3. Antiprotozoal Activity
6.4. Therapeutic Efficacy and Safety Profile on Human Red Blood Cells
7. Future Outlook and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Antimicrobial Resistance Collaborators. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- WHO. Antimicrobial Resistance November 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance (accessed on 9 November 2022).
- CDC. About Antibiotic Resistance December 2021. Available online: https://www.cdc.gov/drugresistance/about.html (accessed on 9 November 2022).
- Khan, S.N.; Khan, A.U. Breaking the Spell: Combating Multidrug Resistant ‘Superbugs’. Front. Microbiol. 2016, 7, 174. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.I.; Prenner, E.J.; Vogel, H.J. Tryptophan- and arginine-rich antimicrobial peptides: Structures and mechanisms of action. Biochim. Biophys. Acta 2006, 1758, 1184–1202. [Google Scholar] [CrossRef] [PubMed]
- Mattiuzzo, M.; Bandiera, A.; Gennaro, R.; Benincasa, M.; Pacor, S.; Antcheva, N.; Scocchi, M. Role of the Escherichia coli SbmA in the antimicrobial activity of proline-rich peptides. Mol. Microbiol. 2007, 66, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Mardirossian, M.; Grzela, R.; Giglione, C.; Meinnel, T.; Gennaro, R.; Mergaert, P.; Scocchi, M. The host antimicrobial peptide Bac71-35 binds to bacterial ribosomal proteins and inhibits protein synthesis. Chem. Biol. 2014, 21, 1639–1647. [Google Scholar] [CrossRef]
- Le, C.F.; Gudimella, R.; Razali, R.; Manikam, R.; Sekaran, S.D. Transcriptome analysis of Streptococcus pneumoniae treated with the designed antimicrobial peptides, DM3. Sci. Rep. 2016, 6, 26828. [Google Scholar] [CrossRef]
- Kragol, G.; Lovas, S.; Varadi, G.; Condie, B.A.; Hoffmann, R.; Otvos, L., Jr. The antibacterial peptide pyrrhocoricin inhibits the ATPase actions of DnaK and prevents chaperone-assisted protein folding. Biochemistry 2001, 40, 3016–3026. [Google Scholar] [CrossRef]
- Le, C.F.; Fang, C.M.; Sekaran, S.D. Intracellular Targeting Mechanisms by Antimicrobial Peptides. Antimicrob. Agents Chemother. 2017, 61, e02340-16. [Google Scholar] [CrossRef]
- Wronska, A.K.; Bogus, M.I. Heat shock proteins (HSP 90, 70, 60, and 27) in Galleria mellonella (Lepidoptera) hemolymph are affected by infection with Conidiobolus coronatus (Entomophthorales). PLoS ONE 2020, 15, e0228556. [Google Scholar] [CrossRef]
- Nikaido, H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656. [Google Scholar] [CrossRef]
- Cabib, E. Two novel techniques for determination of polysaccharide cross-links show that Crh1p and Crh2p attach chitin to both beta(1-6)- and beta(1-3)glucan in the Saccharomyces cerevisiae cell wall. Eukaryot Cell 2009, 8, 1626–1636. [Google Scholar] [CrossRef]
- Spohn, R.; Daruka, L.; Lazar, V.; Martins, A.; Vidovics, F.; Grezal, G.; Mehi, O.; Kintses, B.; Szamel, M.; Jangir, P.K.; et al. Integrated evolutionary analysis reveals antimicrobial peptides with limited resistance. Nat. Commun. 2019, 10, 4538. [Google Scholar] [CrossRef]
- Rautenbach, M.; Troskie, A.M.; Vosloo, J.A. Antifungal peptides: To be or not to be membrane active. Biochimie 2016, 130, 132–145. [Google Scholar] [CrossRef]
- Shu, G.; Chen, Y.; Liu, T.; Ren, S.; Kong, Y. Antimicrobial Peptide Cathelicidin-BF Inhibits Platelet Aggregation by Blocking Protease-Activated Receptor 4. Int. J. Peptide Res. Ther. 2019, 25, 349–358. [Google Scholar] [CrossRef]
- Perlikowska, R.; Silva, J.; Alves, C.; Susano, P.; Pedrosa, R. The Therapeutic Potential of Naturally Occurring Peptides in Counteracting SH-SY5Y Cells Injury. Int. J. Mol. Sci. 2022, 23, 11778. [Google Scholar] [CrossRef]
- Miethke, M.; Pieroni, M.; Weber, T.; Bronstrup, M.; Hammann, P.; Halby, L.; Arimondo, P.B.; Glaser, P.; Aigle, B.; Bode, H.B.; et al. Towards the sustainable discovery and development of new antibiotics. Nat. Rev. Chem. 2021, 5, 726–749. [Google Scholar] [CrossRef]
- Huan, Y.; Kong, Q.; Mou, H.; Yi, H. Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front. Microbiol. 2020, 11, 582779. [Google Scholar] [CrossRef]
- Duwadi, D.; Shrestha, A.; Yilma, B.; Kozlovski, I.; Sa-Eed, M.; Dahal, N.; Jukosky, J. Identification and screening of potent antimicrobial peptides in arthropod genomes. Peptides 2018, 103, 26–30. [Google Scholar] [CrossRef]
- Lofgren, S.E.; Miletti, L.C.; Steindel, M.; Bachere, E.; Barracco, M.A. Trypanocidal and leishmanicidal activities of different antimicrobial peptides (AMPs) isolated from aquatic animals. Exp. Parasitol. 2008, 118, 197–202. [Google Scholar] [CrossRef]
- Wu, Q.; Patocka, J.; Kuca, K. Insect Antimicrobial Peptides, a Mini Review. Toxins 2018, 10, 461. [Google Scholar] [CrossRef]
- Silva, P.I., Jr.; Daffre, S.; Bulet, P. Isolation and characterization of gomesin, an 18-residue cysteine-rich defense peptide from the spider Acanthoscurria gomesiana hemocytes with sequence similarities to horseshoe crab antimicrobial peptides of the tachyplesin family. J. Biol. Chem. 2000, 275, 33464–33470. [Google Scholar] [CrossRef]
- Sun, J.; Wei, Q.; Zhou, Y.; Wang, J.; Liu, Q.; Xu, H. A systematic analysis of FDA-approved anticancer drugs. BMC Syst. Biol. 2017, 11, 87. [Google Scholar] [CrossRef] [PubMed]
- De la torre, B.; Albericio, F. Peptide Therapeutics 2.0. Molecules 2020, 25, 2293. [Google Scholar] [CrossRef]
- Mandard, N.; Bulet, P.; Caille, A.; Daffre, S.; Vovelle, F. The solution structure of gomesin, an antimicrobial cysteine-rich peptide from the spider. Eur. J. Biochem. 2002, 269, 1190–1198. [Google Scholar] [CrossRef] [PubMed]
- Fazio, M.A.; Oliveira, V.X., Jr.; Bulet, P.; Miranda, M.T.; Daffre, S.; Miranda, A. Structure-activity relationship studies of gomesin: Importance of the disulfide bridges for conformation, bioactivities, and serum stability. Biopolymers 2006, 84, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Chalker, J.M.; Davis, B.G. Chemical mutagenesis: Selective post-expression interconversion of protein amino acid residues. Curr. Opin. Chem. Biol. 2010, 14, 781–789. [Google Scholar] [CrossRef]
- Chalker, J.M.; Bernardes, G.J.; Lin, Y.A.; Davis, B.G. Chemical modification of proteins at cysteine: Opportunities in chemistry and biology. Chem. Asian J. 2009, 4, 630–640. [Google Scholar] [CrossRef]
- Malins, L.R. Peptide modification and cyclization via transition-metal catalysis. Curr. Opin. Chem. Biol. 2018, 46, 25–32. [Google Scholar] [CrossRef]
- Chan, L.Y.; Zhang, V.M.; Huang, Y.H.; Waters, N.C.; Bansal, P.S.; Craik, D.J.; Daly, N.L. Cyclization of the antimicrobial peptide gomesin with native chemical ligation: Influences on stability and bioactivity. ChemBioChem 2013, 14, 617–624. [Google Scholar] [CrossRef]
- Miranda, A.; Jouvensal, L.; Vovelle, F.; Bulet, P.; Daffre, S. A powerful antimicrobial peptide isolated from the Brazilian tarantula spider Acanthoscurria gomesiana. In Animal Toxins: State of the Art. Perspectives in Halth and Biotechnology; de Lima, M.E., de Castro Pimenta, A.M., Martin-Eauclaire, M.F., Zingali, R.B., Rochat, H., Eds.; UFMG: Belo Horizonte, Brazil, 2009; pp. 227–248. [Google Scholar]
- Chan, W.; White, P. Fmoc Solid Phase Peptide Synthesis: A Practical Approach; Oxford University Press: Oxford, UK, 1999. [Google Scholar] [CrossRef]
- Machado, A.; Fazio, M.A.; Miranda, A.; Daffre, S.; Machini, M.T. Synthesis and properties of cyclic gomesin and analogues. J. Pept. Sci. 2012, 18, 588–598. [Google Scholar] [CrossRef]
- Kawano, K.; Yoneya, T.; Miyata, T.; Yoshikawa, K.; Tokunaga, F.; Terada, Y.; Iwanaga, S. Antimicrobial peptide, tachyplesin I, isolated from hemocytes of the horseshoe crab (Tachypleus tridentatus). NMR determination of the beta-sheet structure. J. Biol. Chem. 1990, 265, 15365–15367. [Google Scholar] [CrossRef]
- Nakamura, T.; Furunaka, H.; Miyata, T.; Tokunaga, F.; Muta, T.; Iwanaga, S.; Niwa, M.; Takao, T.; Shimonishi, Y. Tachyplesin, a class of antimicrobial peptide from the hemocytes of the horseshoe crab (Tachypleus tridentatus). Isolation and chemical structure. J. Biol. Chem. 1988, 263, 16709–16713. [Google Scholar] [CrossRef]
- Miyata, T.; Tokunaga, F.; Yoneya, T.; Yoshikawa, K.; Iwanaga, S.; Niwa, M.; Takao, T.; Shimonishi, Y. Antimicrobial peptides, isolated from horseshoe crab hemocytes, tachyplesin II, and polyphemusins I and II: Chemical structures and biological activity. J. Biochem. 1989, 106, 663–668. [Google Scholar] [CrossRef]
- Mandard, N.; Sy, D.; Maufrais, C.; Bonmatin, J.M.; Bulet, P.; Hetru, C.; Vovelle, F. Androctonin, a novel antimicrobial peptide from scorpion Androctonus australis: Solution structure and molecular dynamics simulations in the presence of a lipid monolayer. J. Biomol. Struct. Dyn. 1999, 17, 367–380. [Google Scholar] [CrossRef]
- Kokryakov, V.N.; Harwig, S.S.; Panyutich, E.A.; Shevchenko, A.A.; Aleshina, G.M.; Shamova, O.V.; Korneva, H.A.; Lehrer, R.I. Protegrins: Leukocyte antimicrobial peptides that combine features of corticostatic defensins and tachyplesins. FEBS Lett. 1993, 327, 231–236. [Google Scholar] [CrossRef]
- Deplazes, E.; Chin, Y.K.; King, G.F.; Mancera, R.L. The unusual conformation of cross-strand disulfide bonds is critical to the stability of beta-hairpin peptides. Proteins 2020, 88, 485–502. [Google Scholar] [CrossRef]
- Tanner, J.D.; Deplazes, E.; Mancera, R.L. The Biological and Biophysical Properties of the Spider Peptide Gomesin. Molecules 2018, 23, 1733. [Google Scholar] [CrossRef]
- Troeira Henriques, S.; Lawrence, N.; Chaousis, S.; Ravipati, A.S.; Cheneval, O.; Benfield, A.H.; Elliott, A.G.; Kavanagh, A.M.; Cooper, M.A.; Chan, L.Y.; et al. Redesigned Spider Peptide with Improved Antimicrobial and Anticancer Properties. ACS Chem. Biol. 2017, 12, 2324–2334. [Google Scholar] [CrossRef]
- Benfield, A.H.; Defaus, S.; Lawrence, N.; Chaousis, S.; Condon, N.; Cheneval, O.; Huang, Y.H.; Chan, L.Y.; Andreu, D.; Craik, D.J.; et al. Cyclic gomesin, a stable redesigned spider peptide able to enter cancer cells. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183480. [Google Scholar] [CrossRef]
- Castro, J.R.; Fuzo, C.A.; Degreve, L.; Caliri, A. The role of disulfide bridges in the 3-D structures of the antimicrobial peptides gomesin and protegrin-1: A molecular dynamics study. Genet. Mol. Res. 2008, 7, 1070–1088. [Google Scholar] [CrossRef]
- Rodrigues, E.G.; Dobroff, A.S.; Cavarsan, C.F.; Paschoalin, T.; Nimrichter, L.; Mortara, R.A.; Santos, E.L.; Fazio, M.A.; Miranda, A.; Daffre, S.; et al. Effective topical treatment of subcutaneous murine B16F10-Nex2 melanoma by the antimicrobial peptide gomesin. Neoplasia 2008, 10, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Rojo, M.A.; Deplazes, E.; Pineda, S.S.; Brust, A.; Marth, T.; Wilhelm, P.; Martel, N.; Ramm, G.A.; Mancera, R.L.; Alewood, P.F.; et al. Gomesin peptides prevent proliferation and lead to the cell death of devil facial tumour disease cells. Cell Death Discov. 2018, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Moraes, L.G.; Fazio, M.A.; Vieira, R.F.; Nakaie, C.R.; Miranda, M.T.; Schreier, S.; Daffre, S.; Miranda, A. Conformational and functional studies of gomesin analogues by CD, EPR and fluorescence spectroscopies. Biochim. Biophys. Acta 2007, 1768, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Moreira, C.K.; Rodrigues, F.G.; Ghosh, A.; Varotti Fde, P.; Miranda, A.; Daffre, S.; Jacobs-Lorena, M.; Moreira, L.A. Effect of the antimicrobial peptide gomesin against different life stages of Plasmodium spp. Exp. Parasitol. 2007, 116, 346–353. [Google Scholar] [CrossRef]
- Nadal-Bufi, F.; Mason, J.M.; Chan, L.Y.; Craik, D.J.; Kaas, Q.; Troeira Henriques, S. Designed beta-Hairpins Inhibit LDH5 Oligomerization and Enzymatic Activity. J. Med. Chem. 2021, 64, 3767–3779. [Google Scholar] [CrossRef]
- Dias, S.A.; Pinto, S.N.; Silva-Herdade, A.S.; Cheneval, O.; Craik, D.J.; Coutinho, A.; Castanho, M.; Henriques, S.T.; Veiga, A.S. A designed cyclic analogue of gomesin has potent activity against Staphylococcus aureus biofilms. J. Antimicrob. Chemother. 2022, 77, 3256–3264. [Google Scholar] [CrossRef]
- Domingues, T.M.; Riske, K.A.; Miranda, A. Revealing the lytic mechanism of the antimicrobial peptide gomesin by observing giant unilamellar vesicles. Langmuir 2010, 26, 11077–11084. [Google Scholar] [CrossRef]
- Domingues, T.M.; Perez, K.R.; Miranda, A.; Riske, K.A. Comparative study of the mechanism of action of the antimicrobial peptide gomesin and its linear analogue: The role of the beta-hairpin structure. Biochim. Biophys. Acta 2015, 1848, 2414–2421. [Google Scholar] [CrossRef]
- Freire, J.M.; Gaspar, D.; Veiga, A.S.; Castanho, M.A. Shifting gear in antimicrobial and anticancer peptides biophysical studies: From vesicles to cells. J. Pept. Sci. 2015, 21, 178–185. [Google Scholar] [CrossRef]
- Soletti, R.C.; del Barrio, L.; Daffre, S.; Miranda, A.; Borges, H.L.; Moura-Neto, V.; Lopez, M.G.; Gabilan, N.H. Peptide gomesin triggers cell death through L-type channel calcium influx, MAPK/ERK, PKC and PI3K signaling and generation of reactive oxygen species. Chem. Biol. Interact. 2010, 186, 135–143. [Google Scholar] [CrossRef]
- Fazio, M.A.; Jouvensal, L.; Vovelle, F.; Bulet, P.; Miranda, M.T.; Daffre, S.; Miranda, A. Biological and structural characterization of new linear gomesin analogues with improved therapeutic indices. Biopolymers 2007, 88, 386–400. [Google Scholar] [CrossRef]
- Imai, K.; Takaoka, A. Comparing antibody and small-molecule therapies for cancer. Nat. Rev. Cancer 2006, 6, 714–727. [Google Scholar] [CrossRef]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Signal Transduct. Target Ther. 2022, 7, 48. [Google Scholar] [CrossRef]
- Smith, A.J. New horizons in therapeutic antibody discovery: Opportunities and challenges versus small-molecule therapeutics. J. Biomol. Screen 2015, 20, 437–453. [Google Scholar] [CrossRef]
- Mahlapuu, M.; Björn, C.; Ekblom, J. Antimicrobial peptides as therapeutic agents: Opportunities and challenges. Crit. Rev. Biotechnol. 2020, 40, 978–992. [Google Scholar] [CrossRef]
- Elad, S.; Epstein, J.B.; Raber-Durlacher, J.; Donnelly, P.; Strahilevitz, J. The antimicrobial effect of Iseganan HCl oral solution in patients receiving stomatotoxic chemotherapy: Analysis from a multicenter, double-blind, placebo-controlled, randomized, phase III clinical trial. J. Oral Pathol. Med. 2012, 41, 229–234. [Google Scholar] [CrossRef]
- Rader, A.F.B.; Reichart, F.; Weinmuller, M.; Kessler, H. Improving oral bioavailability of cyclic peptides by N-methylation. Bioorg. Med. Chem. 2018, 26, 2766–2773. [Google Scholar] [CrossRef]
- Bech, E.M.; Pedersen, S.L.; Jensen, K.J. Chemical Strategies for Half-Life Extension of Biopharmaceuticals: Lipidation and Its Alternatives. ACS Med. Chem. Lett. 2018, 9, 577–580. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
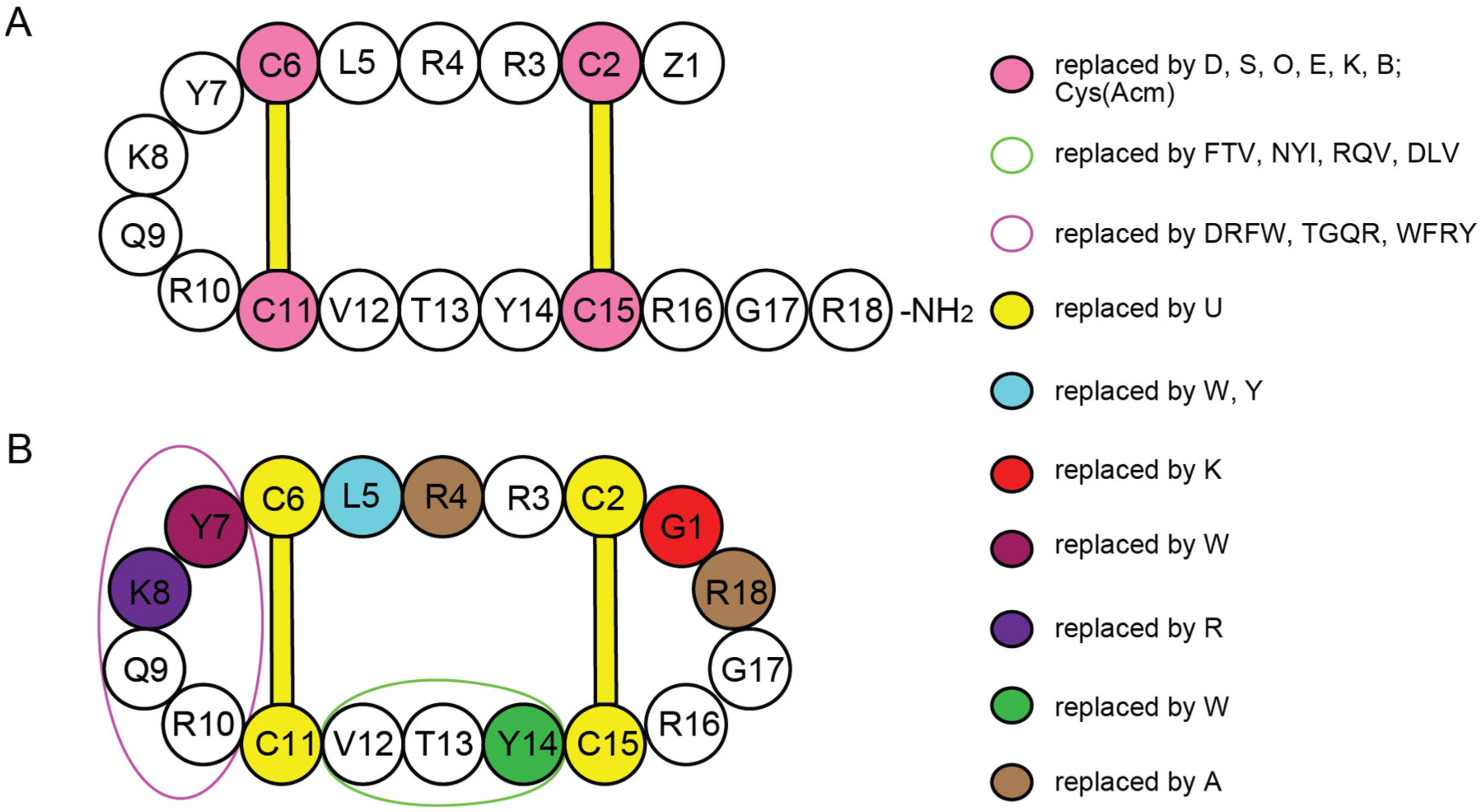{kind=link}
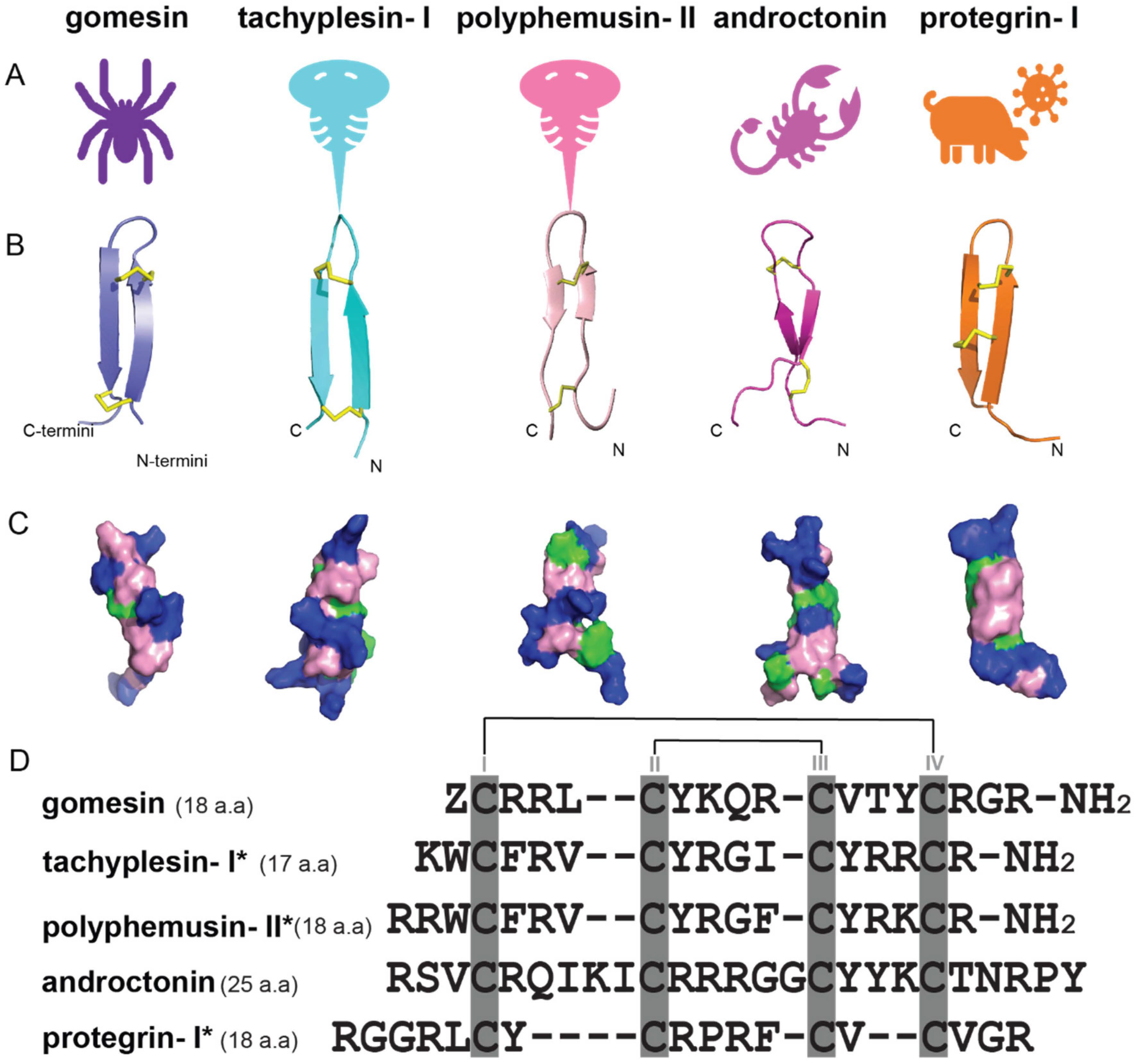
| Antimicrobial Peptides | Function/Activity | Disulfide Connectivity | Cysα-Cysα Distances (nm) | Types of β-Sheets | Total Net Charge |
|---|---|---|---|---|---|
| gomesin | Antimicrobial, anticancer | Cys2-Cys15, Cys6-Cys11 | 0.37 ± 0.10, 0.38 ± 0.10 (<0.45) [41] | right-handed rotamer, antiparallel β-sheets | +6 |
| tachyplesin-I | Antimicrobial, antifungal, and anticancer | Cys3-Cys16, Cys7-Cys12 | 0.43 ± 0.20, 0.39 ± 0.20 [40] | right-handed rotamer, antiparallel β-sheets | +6 |
| polyphemusin-II | Antimicrobial and antifungal | Cys4-Cys17, Cys8-Cys13 | 0.43 ± 0.50, 0.37 ± 0.10 [40] | right-handed rotamer, antiparallel β-sheets | +7 |
| androctonin | Inhibits the growth of Gram-positive bacteria | Cys4-Cys20, Cys10-Cys13 | n.a. | right-handed rotamer, antiparallel β-sheets | +8 |
| protegrins-I | Antimicrobial and antifungal | Cys6-Cys15, Cys8-Cys13 | 0.38 ± 0.30, 0.35 ± 0.10 [40] | right-handed rotamer, antiparallel β-sheets | +6 |
| Peptide | Applications | CC50/IC50/MIC * | References |
|---|---|---|---|
gomesin | anticancer, antimicrobial, antifungal | K-562; 3.8 ± 0.3 μM E. coli ATCC 25922; 4 μM C. albicans ATCC 90028; 8–16 μM | [42] |
cyclic gomesin | anticancer, antimicrobial, antifungal | K-562; 2.7 ± 0.1 μM E. coli ATCC 25922; 4 μM C. albicans ATCC 90028; 4–8 μM | [42,49] |
[Y7W]cGm | anticancer antimicrobial | K-562; 3.9 ± 0.2 μM E. coli ATCC 25922; 1–2 μM | [42,49] |
[Y14W]cGm | anticancer, antimicrobial | K-562; 2.7 ± 0.1 μM E. coli ATCC 25922; 1–2 μM | [42] |
[K8R]cGm | anticancer, antimicrobial | K-562; 3.1 ± 0.1 μM E. coli ATCC 25922; 1–2 μM | [42] |
[Y7W,K8R,Y14W]cGm | anticancer, antimicrobial | K-562; 3.9 ± 0.1 μM E. coli ATCC 25922; 1 μM | [42] |
[R4A,R18A]cGm | anticancer, antimicrobial, antifungal | K-562; 11.5 ± 0.6 μM E. coli ATCC 25922; 8 μM C. albicans ATCC 90028; 32 μM | [42] |
[G1K,K8R]cGm | anticancer, antimicrobial, antifungal | K-562; 2.1 ± 0.2 μM E. coli ATCC 25922; 0.5–1 μM C. albicans ATCC 90028; 2 μM | [42] |
[C/U]cGm | anticancer, antimicrobial, antifungal | K-562; 1.4 ± 0.2 μM E. coli ATCC 25922; 4–8 μM C. albicans ATCC 90028; 4–8 μM | [42] |
[L5W]cGm | anticancer, antimicrobial | K-562; 1.0 ± 0.1 μM E. coli ATCC 25922; 2–4 μM | [42] |
[DPLP]cGm | anticancer, antimicrobial, antifungal | K-562; 3.9 ± 0.2 μM E. coli ATCC 25922; 4 μM C. albicans ATCC 90028; 8 μM | [42] |
[G1K,L5Y,K8R]cGm | anticancer, antimicrobial, antifungal | K-562; 1.3 ± 0.1 μM E. coli ATCC 25922; 0.5–1 μM C. albicans ATCC 90028; 4 μM | [42] |
[C/U,G1K,L5Y,K8R]cGm | anticancer, antimicrobial, antifungal | K-562; 6.4 ± 0.6 μM E. coli ATCC 25922; 1–2 μM C. albicans ATCC 90028; 4 μM | [42] |
cGmC4 | anticancer | LDH5 inhibitor; 27.3 μM | [49] |
cGmC5 | anticancer | LDH5 inhibitor; 24.0 μM | [49] |
cGmN5 | anticancer | LDH5 inhibitor; >40 μM | [49] |
cGmN6 | anticancer | LDH5 inhibitor; 41.4 μM | [49] |
cGmC6 | anticancer | LDH5 inhibitor; 10.4 μM | [49] |
cGmC7 | anticancer | LDH5 inhibitor; >40 μM | [49] |
cGmC8 | anticancer | LDH5 inhibitor; 24.8 μM | [49] |
cGmC9 | anticancer | LDH5 inhibitor; 2.5 μM | [49] |
cGmC10 | anticancer | LDH5 inhibitor; >40 μM | [49] |
cGmC11 | anticancer | LDH5 inhibitor; 4.5 μM | [49] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Henriques, S.T.; Craik, D.J.; Chan, L.Y. Unlocking the Potential of the Antimicrobial Peptide Gomesin: From Discovery and Structure–Activity Relationships to Therapeutic Applications. Int. J. Mol. Sci. 2023, 24, 5893. https://doi.org/10.3390/ijms24065893
Liu X, Henriques ST, Craik DJ, Chan LY. Unlocking the Potential of the Antimicrobial Peptide Gomesin: From Discovery and Structure–Activity Relationships to Therapeutic Applications. International Journal of Molecular Sciences. 2023; 24(6):5893. https://doi.org/10.3390/ijms24065893
Chicago/Turabian StyleLiu, Xiaorong, Sónia T. Henriques, David J. Craik, and Lai Yue Chan. 2023. "Unlocking the Potential of the Antimicrobial Peptide Gomesin: From Discovery and Structure–Activity Relationships to Therapeutic Applications" International Journal of Molecular Sciences 24, no. 6: 5893. https://doi.org/10.3390/ijms24065893
APA StyleLiu, X., Henriques, S. T., Craik, D. J., & Chan, L. Y. (2023). Unlocking the Potential of the Antimicrobial Peptide Gomesin: From Discovery and Structure–Activity Relationships to Therapeutic Applications. International Journal of Molecular Sciences, 24(6), 5893. https://doi.org/10.3390/ijms24065893