


Kinetic Characterization and Catalytic Mechanism of N-Acetylornithine Aminotransferase Encoded by slr1022 Gene from Synechocystis sp. PCC6803



Abstract
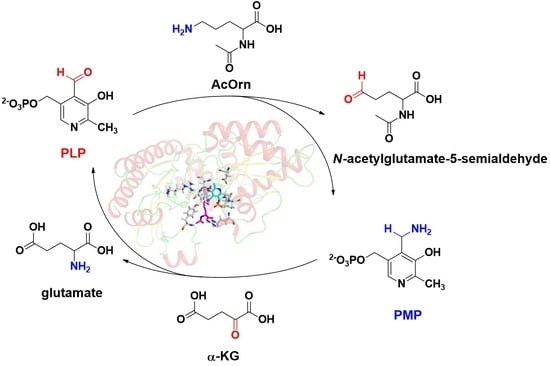
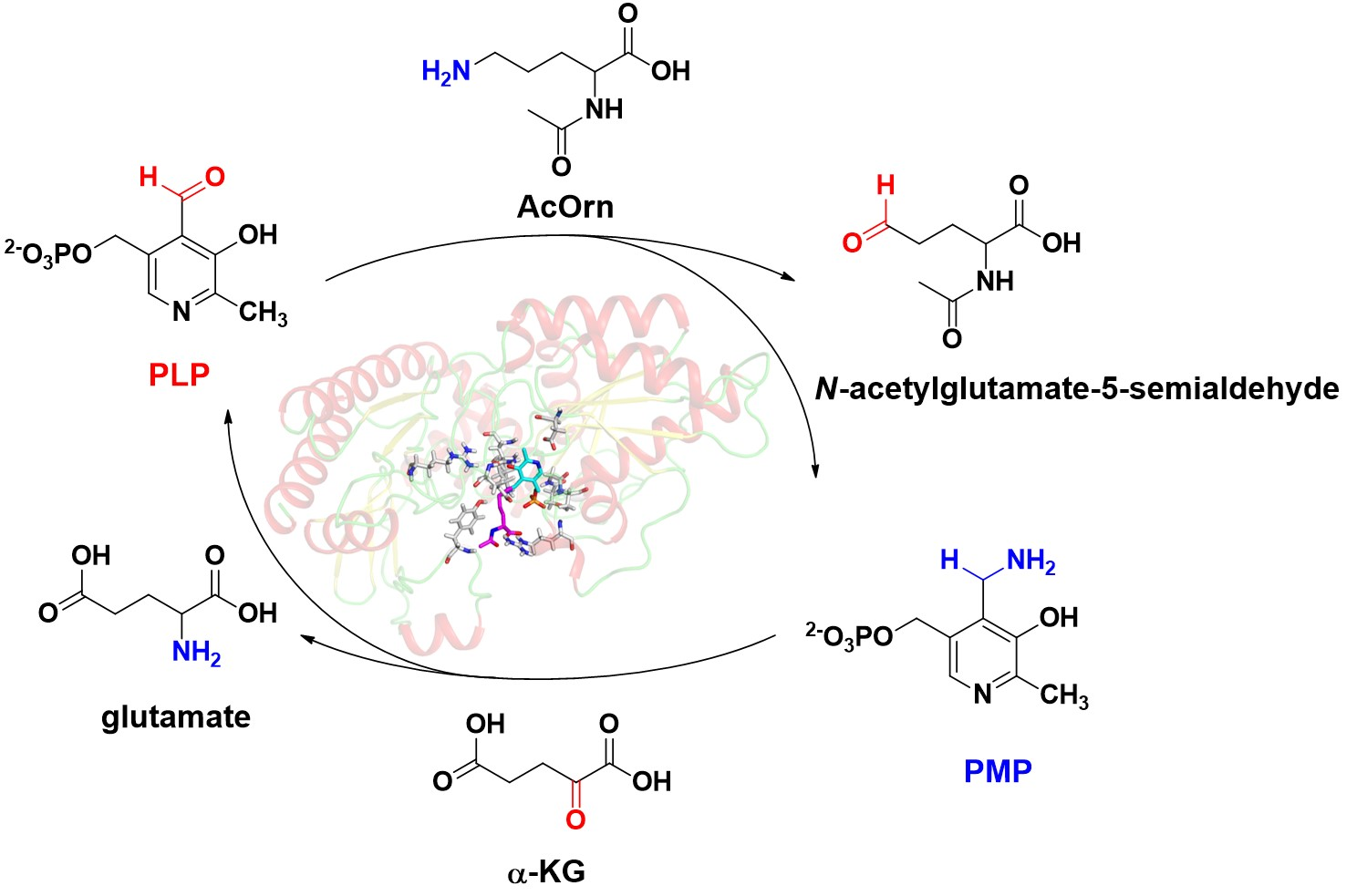
1. Introduction
2. Results
2.1. Oligomer State of Recombinant Slr1022 Protein
2.2. Kinetic Characterization of Recombinant Slr1022 Protein towards Various Substrates
2.3. Effects of Metal Ions and Temperature on Catalytic Activity of Slr1022 as AcOAT
2.4. Structure Modeling and Model Quality Evaluation of Slr1022
2.5. AcOrn Transaminase Activity Assay of Slr1022 Variants
2.6. Spectroscopic Characterization of Slr1022 as AcOAT
2.7. Kinetic and Catalytic Mechanism of Slr1022 as AcOAT
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Expression and Purification of Recombinant Slr1022 Protein
4.3. Oligomer State of Recombinant Slr1022 Protein
4.4. Kinetic Characterization of Slr1022 as AcOAT
4.5. Orn Transaminase Activity Assay of Slr1022
4.6. GABA Transaminase Activity Assay of Slr1022
4.7. Effect of Metal Ions on the AcOrn Transaminase Activity of Slr1022
4.8. Effect of Temperature on the AcOrn Transaminase Activity of Slr1022
4.9. Structure Modeling and Model Quality Evaluation of Slr1022
4.10. Spectroscopic Properties of Slr1022 as AcOAT
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Heyl, D.; Luz, E.; Harris, S.A.; Folker, K. Phosphates of the Vitamin B, Group. I. The Structure of Codecarboxylase. J. Am. Chem. Soc. 1951, 73, 3430–3433. [Google Scholar] [CrossRef]
- Eliot, A.C.; Kirsch, J.F. Pyridoxal phosphate enzymes: Mechanistic, structural, and evolutionary considerations. Ann. Rev. Biochem. 2004, 73, 383–415. [Google Scholar] [CrossRef] [PubMed]
- Tran, J.U.; Brown, B.L. Structural Basis for Allostery In PLP-Dependent Enzymes. Front. Mol. Biosci. 2022, 9, 884281. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Han, Q.; Tan, Y.; Ding, H.; Li, J. Current Advances on Structure-Function Relationships of Pyridoxal 5’-Phosphate-Dependent Enzymes. Front. Mol. Biosci. 2019, 6, 4. [Google Scholar] [CrossRef]
- Koper, K.; Han, S.W.; Pastor, D.C.; Yoshikuni, Y.; Maeda, H.A. Evolutionary origin and functional diversification of aminotransferases. J. Biol. Chem. 2022, 298, 102122. [Google Scholar] [CrossRef] [PubMed]
- Yoon, C.S.; Kim, D.W.; Jang, S.H.; Lee, B.R.; Choi, H.S.; Choi, S.H.; Kim, S.Y.; An, J.J.; Kwon, O.S.; Kang, T.C.; et al. Cysteine-321 of human brain GABA transaminase is involved in intersubunit cross-linking. Mol. Cells 2004, 18, 214–219. [Google Scholar]
- Ginguay, A.; Cynober, L.; Curis, E.; Nicolis, I. Ornithine Aminotransferase, an Important Glutamate-Metabolizing Enzyme at the Crossroads of Multiple Metabolic Pathways. Biology 2017, 6, 18. [Google Scholar] [CrossRef]
- Slocum, R.D. Genes, enzymes and regulation of arginine biosynthesis in plants. Plant Physiol. Biochem. 2005, 43, 729–745. [Google Scholar] [CrossRef]
- Toney, M.D. Aspartate aminotransferase: An old dog teaches new tricks. Arch. Biochem. Biophys. 2014, 544, 119–127. [Google Scholar] [CrossRef]
- Voellmy, R.; Leisinger, T. Dual role for N-2-acetylornithine 5-aminotransferase from Pseudomonas aeruginosa in arginine biosynthesis and arginine catabolism. J. Bacteriol. 1975, 122, 799–809. [Google Scholar] [CrossRef]
- Billheimer, J.T.; Shen, M.Y.; Carnevale, H.N.; Horton, H.R.; Jones, E.E. Isolation and characterization of acetylornithine delta-transaminase of wild-type Escherichia coli W. Comparison with arginine-inducible acetylornithine delta-transaminase. Arch. Biochem. Biophys. 1979, 195, 401–413. [Google Scholar] [CrossRef]
- Rajaram, V.; Ratna Prasuna, P.; Savithri, H.S.; Murthy, M.R. Structure of biosynthetic N-acetylornithine aminotransferase from Salmonella typhimurium: Studies on substrate specificity and inhibitor binding. Proteins 2008, 70, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Quintero, M.J.; Muro-Pastor, A.M.; Herrero, A.; Flores, E. Arginine catabolism in the cyanobacterium Synechocystis sp. Strain PCC 6803 involves the urea cycle and arginase pathway. J. Bacteriol. 2000, 182, 1008–1015. [Google Scholar] [CrossRef]
- Schriek, S.; Ruckert, C.; Staiger, D.; Pistorius, E.K.; Michel, K.P. Bioinformatic evaluation of L-arginine catabolic pathways in 24 cyanobacteria and transcriptional analysis of genes encoding enzymes of L-arginine catabolism in the cyanobacterium Synechocystis sp. PCC 6803. BMC Genom. 2007, 8, 437. [Google Scholar] [CrossRef]
- Xiong, W.; Brune, D.; Vermaas, W.F. The γ-aminobutyric acid shunt contributes to closing the tricarboxylic acid cycle in Synechocystis sp. PCC 6803. Mol. Microbiol. 2014, 93, 786–796. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Qian, X.; Chang, S.; Dismukes, G.C.; Bryant, D.A. Natural and synthetic variants of the tricarboxylic acid cycle in cyanobacteria: Introduction of the GABA shunt into Synechococcus sp. PCC 7002. Front. Microbiol. 2016, 7, 1972. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.; Li, Z.-M.; Wang, X.; Hu, Z.; Bao, L.; Li, Z. Biochemical Characterization and Structural Analysis of N-Acetylornithine Transaminase from Synechocystis Sp. PCC6803. Biotechnol. Bull. 2021, 37, 98–107. [Google Scholar]
- Gupta, P.; Thomas, S.E.; Zaidan, S.A.; Pasillas, M.A.; Cory-Wright, J.; Sebastian-Perez, V.; Burgess, A.; Cattermole, E.; Meghir, C.; Abell, C.; et al. A fragment-based approach to assess the ligandability of ArgB, ArgC, ArgD and ArgF in the L-arginine biosynthetic pathway of Mycobacterium tuberculosis. Comput. Struct. Biotechnol. J. 2021, 19, 3491–3506. [Google Scholar] [CrossRef]
- Mehta, P.K.; Hale, T.I.; Christen, P. Aminotransferases: Demonstration of homology and division into evolutionary subgroups. Eur. J. Biochem. 1993, 214, 549–561. [Google Scholar] [CrossRef]
- Lee, H.; Juncosa, J.I.; Silverman, R.B. Ornithine aminotransferase versus GABA aminotransferase: Implications for the design of new anticancer drugs. Med. Res. Rev. 2015, 35, 286–305. [Google Scholar] [CrossRef]
- Butrin, A.; Butrin, A.; Wawrzak, Z.; Moran, G.R.; Liu, D. Determination of the pH dependence, substrate specificity, and turnovers of alternative substrates for human ornithine aminotransferase. J. Biol. Chem. 2022, 298, 101969. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Tang, X.; Zhu, Y.; Yu, Z.; Su, K.; Zhang, Y.; Dong, Y.; Zhu, W.; Zhang, C.; Wu, R.; et al. Structural studies reveal flexible roof of active site responsible for omega-transaminase CrmG overcoming by-product inhibition. Commun. Biol. 2020, 3, 455. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Peterson, P.E.; Langston, J.A.; Jin, X.; Zhou, X.; Fisher, A.J.; Toney, M.D. Kinetic and Crystallographic Analysis of Active Site Mutants of Escherichia coli γ-Aminobutyrate Aminotransferase. Biochemistry 2005, 44, 2982–2992. [Google Scholar] [CrossRef]
- Storici, P.; Capitani, G.; Muller, R.; Schirmer, T.; Jansonius, J.N. Crystal Structure of Human Ornithine Aminotransferase Complexed with the Highly Specific and Potent Inhibitor 5-Fluoromethylornithine. J. Mol. Biol. 1999, 285, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Ledwidge, R.; Blanchard, J.S. The dual biosynthetic capability of N-acetylornithine aminotransferase in arginine and lysine biosynthesis. Biochemistry 1999, 38, 3019–3024. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Zhang, X.; Rao, Z.; Yang, J.; Dou, W.; Jin, J.; Xu, Z. Cloning, expression and characterization of N-acetylornithine aminotransferase from Corynebacterium crenatum and its effects on L-arginine fermentation. Chin. J. Biotechnol. 2011, 27, 1013–1023. [Google Scholar]
- Baek, M.; Dimaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Yano, T.; Kuramitsu, S.; Tanase, S.; Morino, Y.; Kagamiyama, H. Role of Asp222 in the Catalytic Mechanism of Escherichia coli Aspartate Enzyme-Bound Coenzyme Pyridoxal 5’-Phosphate Aminotransferase: The Amino Acid Residue Which Enhances the Function Pyridoxal 5’-Phosphate. Biochemistry 1992, 31, 5878–5887. [Google Scholar] [CrossRef] [PubMed]
- Onuffer, J.J.; Kirsch, J.F. Redesign of the substrate specificity of Escherichia coli aspartate aminotransferase to that of Escherichia m coli tyrosine aminotransferase by homology modeling and site-directed mutagenesis. Protein Sci. 1995, 4, 1750–1757. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Yoon, C.S.; Eum, W.S.; Lee, B.R.; An, J.J.; Lee, S.H.; Lee, S.R.; Ahn, J.-Y.; Kwon, O.-S.; Kang, T.-C.; et al. Site-directed Mutagenesis of Human Brain GABA Transaminase: Lysine-357 Is Involved in Cofactor Binding at the Active Site. Mol. Cells 2004, 18, 314–319. [Google Scholar] [PubMed]
- Wang, L.; Qian, H.; Nian, Y.; Han, Y.; Ren, Z.; Zhang, H.; Hu, L.; Prasad, B.V.V.; Laganowsky, A.; Yan, N.; et al. Structure and mechanism of human diacylglycerol O-acyltransferase 1. Nature 2020, 581, 329–332. [Google Scholar] [CrossRef]
- Wang, X.; Lai, C.; Lei, G.; Wang, F.; Long, H.; Wu, X.; Chen, J.; Huo, G.; Li, Z. Kinetic characterization and structural modeling of an NADP(+)-dependent succinic semialdehyde dehydrogenase from Anabaena sp. PCC7120. Int. J. Biol. Macromol. 2018, 108, 615–624. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.L.; Macarthur, M.W.; Hutchinson, E.G.; Thornton, J.M. Stereochemical quality of protein structure coordinates. Proteins 1992, 12, 345–364. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.; Luthy, R.; Bowie, J.U. VERIFY3D: Assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997, 277, 396–404. [Google Scholar] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzymatic Function (Substrate) | Respective Substrate | α-KG | ||||
|---|---|---|---|---|---|---|
| KM (mmol/L) | kcat (s−1) | kcat/KM (L/(mol·s)) | KM (mmol/L) | kcat (s−1) | kcat/KM (L/(mol·s)) | |
| AcOAT (AcOrn) [17] | 0.12 ± 0.01 | 2.31 ± 0.06 | 1.93 × 104 | 0.039 ± 0.004 | 2.50 ± 0.08 | 6.41 × 104 |
| OAT (Orn) | 20.9 ± 3.5 | 0.16 ± 0.01 | 7.7 | 0.13 ± 0.03 | 0.11 ± 0.01 | 8.46 × 102 |
| GABA-AT (GABA) | 78.1 ± 18.7 | 0.14 ± 0.01 | 1.8 | 0.04 ± 0.01 | 0.056 ± 0.003 | 1.4 × 103 |
| Proteins | KMAcOrn (mmol/L) | KMα-KG (mmol/L) | kcat (s−1) | Relative Catalytic Rate (% of Wild-Type) |
|---|---|---|---|---|
| wild-type [17] | 0.12 ± 0.01 | 0.039 ± 0.004 | 2.31 ± 0.06 | 100 |
| T308A | 0.14 ± 0.02 | 0.025 ± 0.003 | 0.007 ± 0.002 | 0.3 |
| Q254A | 0.11 ± 0.01 | 0.04 ± 0.01 | 0.02 ± 0.0005 | 0.9 |
| S125A | 0.23 ± 0.02 | 0.065 ± 0.008 | 0.32 ± 0.01 | 14 |
| A127S | 0.59 ± 0.12 | 0.09 ± 0.02 | 0.21 ± 0.01 | 9.1 |
| D251E | 1.39 ± 0.22 | 0.039 ± 0.006 | 0.47 ± 0.03 | 20 |
| D251A | NA | NA | NA | NA |
| K280A | NA | NA | NA | NA |
| G126A | NA | NA | NA | NA |
| E223S | 8.81 ± 2.96 | 0.015 ± 0.004 | 3.67 ± 0.43 | 160 |
| E223A | 7.77 ± 1.76 | 0.15 ± 0.02 | 1.17 ± 0.09 | 50 |
| R163A | 493 ± 108 | 0.92 ± 0.19 | 0.22 ± 0.02 | 9.5 |
| R402A | 250 ± 32 | 0.87 ± 0.14 | 0.41 ± 0.02 | 18 |
| Y39F | 0.47 ± 0.05 | 0.08 ± 0.03 | 0.17 ± 0.01 | 7.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.-M.; Bai, F.; Wang, X.; Xie, C.; Wan, Y.; Li, Y.; Liu, J.; Li, Z. Kinetic Characterization and Catalytic Mechanism of N-Acetylornithine Aminotransferase Encoded by slr1022 Gene from Synechocystis sp. PCC6803. Int. J. Mol. Sci. 2023, 24, 5853. https://doi.org/10.3390/ijms24065853
Li Z-M, Bai F, Wang X, Xie C, Wan Y, Li Y, Liu J, Li Z. Kinetic Characterization and Catalytic Mechanism of N-Acetylornithine Aminotransferase Encoded by slr1022 Gene from Synechocystis sp. PCC6803. International Journal of Molecular Sciences. 2023; 24(6):5853. https://doi.org/10.3390/ijms24065853
Chicago/Turabian StyleLi, Zhi-Min, Fumei Bai, Xiaoqin Wang, Congcong Xie, Yuting Wan, Yating Li, Jianping Liu, and Zhimin Li. 2023. "Kinetic Characterization and Catalytic Mechanism of N-Acetylornithine Aminotransferase Encoded by slr1022 Gene from Synechocystis sp. PCC6803" International Journal of Molecular Sciences 24, no. 6: 5853. https://doi.org/10.3390/ijms24065853
APA StyleLi, Z.-M., Bai, F., Wang, X., Xie, C., Wan, Y., Li, Y., Liu, J., & Li, Z. (2023). Kinetic Characterization and Catalytic Mechanism of N-Acetylornithine Aminotransferase Encoded by slr1022 Gene from Synechocystis sp. PCC6803. International Journal of Molecular Sciences, 24(6), 5853. https://doi.org/10.3390/ijms24065853