
Characterization of a PIP Binding Site in the N-Terminal Domain of V-ATPase a4 and Its Role in Plasma Membrane Association


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
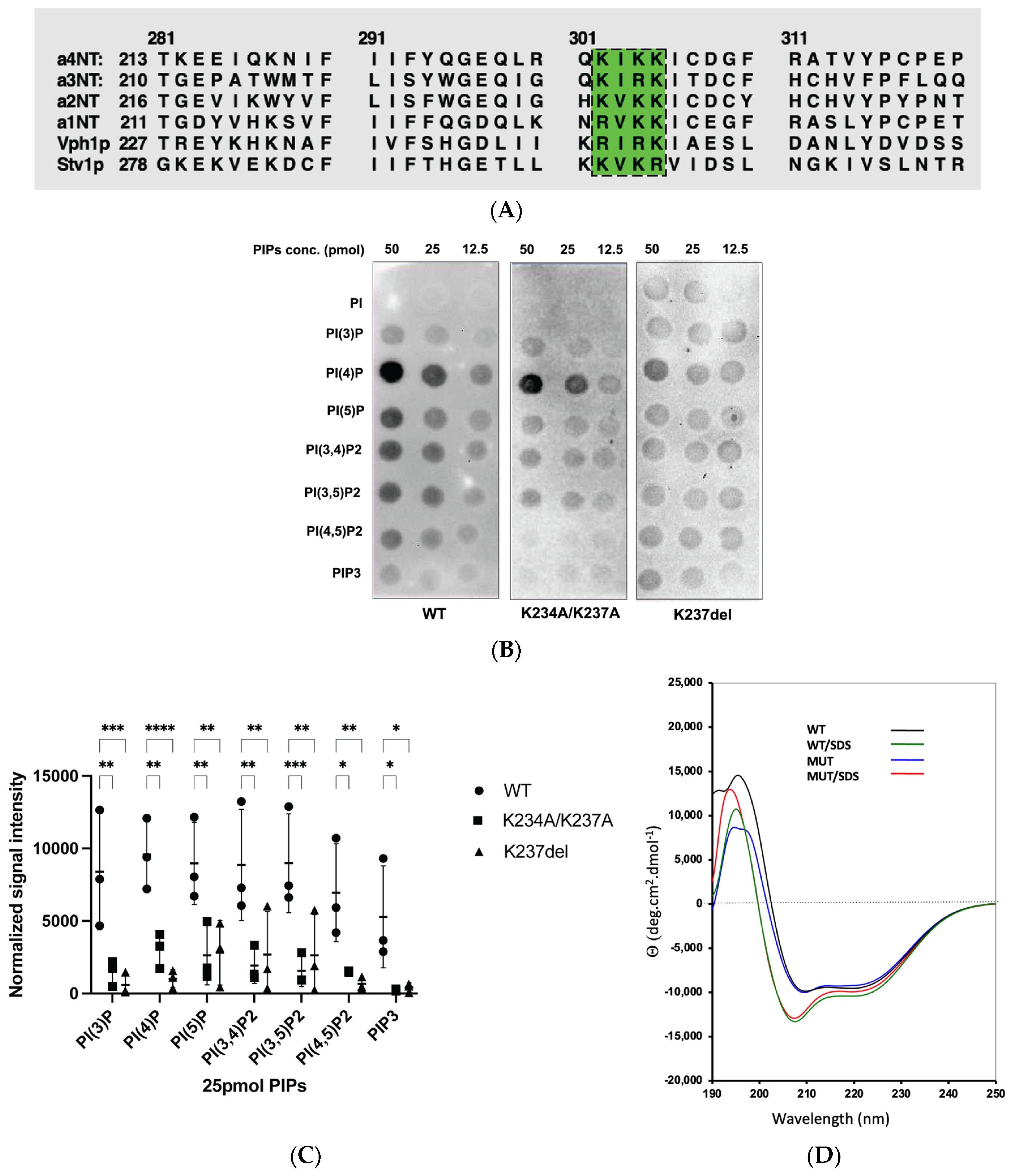
2.1. Homology Model of a4NT Revealed a Putative Lipid Binding Motif K234IKK237 Conserved in Yeast and All Four Mammalian Isoforms
2.2. Mutation within the Putative a4-PIP Binding Motif Reduced In Vitro PIP Binding and Interactions with PI(4,5)P2-Enriched Liposomes
2.3. K234A/K237A and K237del Mutations Reduce a4NT-Membrane Association In Vivo
2.4. Ionomycin Reduces a4NT-Membrane Association In Vivo
3. Discussion
4. Materials and Methods
4.1. Plasmids
4.2. Expression and Purification of Human a4NT Wildtype and Mutants from E.coli
4.3. Protein Structure Prediction
4.4. Protein–Lipid Overlay Assay
4.5. PolyPIPosome Pull-Down Assay
4.6. HEK293 Transfection and Cellular Fractionation
4.7. Immunofluorescence
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kibak, H.; Taiz, L.; Starke, T.; Bernasconi, P.; Gogarten, J.P. Evolution of structure and function of V-ATPases. J. Bioenerg. Biomembr. 1992, 24, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Nelson, N. Structure and function of V-ATPases in endocytic and secretory organelles. J. Exp. Biol. 1992, 172, 149–153. [Google Scholar] [CrossRef]
- Futai, M.; Sun-Wada, G.H.; Wada, Y.; Matsumoto, N.; Nakanishi-Matsui, M. Vacuolar-type ATPase: A proton pump to lysosomal trafficking. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2019, 95, 17. [Google Scholar] [CrossRef] [PubMed]
- Futai, M.; Oka, T.; Sun-Wada, G.; Moriyama, Y.; Kanazawa, H.; Wada, Y. Luminal acidification of diverse organelles by V-ATPase in animal cells. J. Exp. Biol. 2000, 203 Pt 1, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Murata, Y.; Namba, M.; Yoshimizu, T.; Toyomura, T.; Yamamoto, A.; Sun-Wada, G.H.; Hamasaki, N.; Wada, Y.; Futai, M. a4, a unique kidney-specific isoform of mouse vacuolar H+-ATPase subunit a. J. Biol. Chem. 2001, 276, 40050–40054. [Google Scholar] [CrossRef]
- Kartner, N.; Manolson, M.F. Novel techniques in the development of osteoporosis drug therapy: The osteoclast ruffled-border vacuolar H(+)-ATPase as an emerging target. Expert Opin. Drug Discov. 2014, 9, 505–522. [Google Scholar] [CrossRef]
- Nakhoul, N.L.; Hamm, L.L. Vacuolar H(+)-ATPase in the kidney. J. Nephrol. 2002, 15 (Suppl. 5), S22–S31. [Google Scholar]
- Pietrement, C.; Sun-Wada, G.H.; Silva, N.D.; McKee, M.; Marshansky, V.; Brown, D.; Futai, M.; Breton, S. Distinct expression patterns of different subunit isoforms of the V-ATPase in the rat epididymis. Biol. Reprod. 2006, 74, 185–194. [Google Scholar] [CrossRef]
- Hinton, A.; Sennoune, S.R.; Bond, S.; Fang, M.; Reuveni, M.; Sahagian, G.G.; Jay, D.; Martinez-Zaguilan, R.; Forgac, M. Function of a subunit isoforms of the V-ATPase in pH homeostasis and in vitro invasion of MDA-MB231 human breast cancer cells. J. Biol. Chem. 2009, 284, 16400–16408. [Google Scholar] [CrossRef]
- Stransky, L.; Cotter, K.; Forgac, M. The Function of V-ATPases in Cancer. Physiol. Rev. 2016, 96, 1071–1091. [Google Scholar] [CrossRef]
- Capecci, J.; Forgac, M. The function of vacuolar ATPase (V-ATPase) a subunit isoforms in invasiveness of MCF10a and MCF10CA1a human breast cancer cells. J. Biol. Chem. 2013, 288, 32731–32741. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Benlekbir, S.; Rubinstein, J.L. Electron cryomicroscopy observation of rotational states in a eukaryotic V-ATPase. Nature 2015, 521, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Kartner, N.; Yao, Y.; Bhargava, A.; Manolson, M.F. Topology, glycosylation and conformational changes in the membrane domain of the vacuolar H+-ATPase a subunit. J. Cell Biochem. 2013, 114, 1474–1487. [Google Scholar] [CrossRef] [PubMed]
- Aoto, K.; Kato, M.; Akita, T.; Nakashima, M.; Mutoh, H.; Akasaka, N.; Tohyama, J.; Nomura, Y.; Hoshino, K.; Ago, Y.; et al. ATP6V0A1 encoding the a1-subunit of the V0 domain of vacuolar H(+)-ATPases is essential for brain development in humans and mice. Nat. Commun. 2021, 12, 2107. [Google Scholar] [CrossRef] [PubMed]
- Fischer, B.; Dimopoulou, A.; Egerer, J.; Gardeitchik, T.; Kidd, A.; Jost, D.; Kayserili, H.; Alanay, Y.; Tantcheva-Poor, I.; Mangold, E.; et al. Further characterization of ATP6V0A2-related autosomal recessive cutis laxa. Hum. Genet. 2012, 131, 1761–1773. [Google Scholar] [CrossRef] [PubMed]
- Kornak, U.; Reynders, E.; Dimopoulou, A.; van Reeuwijk, J.; Fischer, B.; Rajab, A.; Budde, B.; Nurnberg, P.; Foulquier, F.; Lefeber, D.; et al. Impaired glycosylation and cutis laxa caused by mutations in the vesicular H+-ATPase subunit ATP6V0A2. Nat. Genet. 2008, 40, 32–34. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Guitron, L.E.; Ceron-Torres, T.; Sobacchi, C.; Ochoa-Ruiz, E.; Villegas-Huesca, S. Autosomal recessive osteopetrosis type I: Description of pathogenic variant of TCIRG1 gene. Bol. Med. Hosp. Infant. Mex. 2018, 75, 255–259. [Google Scholar]
- Pangrazio, A.; Caldana, M.E.; Lo Iacono, N.; Mantero, S.; Vezzoni, P.; Villa, A.; Sobacchi, C. Autosomal recessive osteopetrosis: Report of 41 novel mutations in the TCIRG1 gene and diagnostic implications. Osteoporos. Int. 2012, 23, 2713–2718. [Google Scholar] [CrossRef]
- Chu, A.; Zirngibl, R.A.; Manolson, M.F. The V-ATPase a3 Subunit: Structure, Function and Therapeutic Potential of an Essential Biomolecule in Osteoclastic Bone Resorption. Int. J. Mol. Sci. 2021, 22, 6934. [Google Scholar] [CrossRef]
- Stehberger, P.A.; Schulz, N.; Finberg, K.E.; Karet, F.E.; Giebisch, G.; Lifton, R.P.; Geibel, J.P.; Wagner, C.A. Localization and regulation of the ATP6V0A4 (a4) vacuolar H+-ATPase subunit defective in an inherited form of distal renal tubular acidosis. J. Am. Soc. Nephrol. 2003, 14, 3027–3038. [Google Scholar] [CrossRef]
- Finnigan, G.C.; Cronan, G.E.; Park, H.J.; Srinivasan, S.; Quiocho, F.A.; Stevens, T.H. Sorting of the yeast vacuolar-type, proton-translocating ATPase enzyme complex (V-ATPase): Identification of a necessary and sufficient Golgi/endosomal retention signal in Stv1p. J. Biol. Chem. 2012, 287, 19487–19500. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Kane, P.M. Direct interaction of the Golgi V-ATPase a-subunit isoform with PI(4)P drives localization of Golgi V-ATPases in yeast. Mol. Biol. Cell 2017, 28, 2518–2530. [Google Scholar] [CrossRef] [PubMed]
- Li, S.C.; Diakov, T.T.; Xu, T.; Tarsio, M.; Zhu, W.; Couoh-Cardel, S.; Weisman, L.S.; Kane, P.M. The signaling lipid PI(3,5)P(2) stabilizes V(1)-V(o) sector interactions and activates the V-ATPase. Mol. Biol. Cell 2014, 25, 1251–1262. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.G. Phosphoinositides in constitutive membrane traffic. Physiol. Rev. 2004, 84, 699–730. [Google Scholar] [CrossRef] [PubMed]
- Kanaho, Y.; Suzuki, T. Phosphoinositide kinases as enzymes that produce versatile signaling lipids, phosphoinositides. J. Biochem. 2002, 131, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Payrastre, B. Phosphoinositides: Lipid kinases and phosphatases. Methods Mol. Biol. 2004, 273, 201–212. [Google Scholar]
- Posor, Y.; Jang, W.; Haucke, V. Phosphoinositides as membrane organizers. Nat. Rev. Mol. Cell Biol. 2022, 23, 797–816. [Google Scholar] [CrossRef]
- Li, S.; Ghosh, C.; Xing, Y.; Sun, Y. Phosphatidylinositol 4,5-bisphosphate in the Control of Membrane Trafficking. Int. J. Biol. Sci. 2020, 16, 2761–2774. [Google Scholar] [CrossRef]
- Yoneda, A.; Kanemaru, K.; Matsubara, A.; Takai, E.; Shimozawa, M.; Satow, R.; Yamaguchi, H.; Nakamura, Y.; Fukami, K. Phosphatidylinositol 4,5-bisphosphate is localized in the plasma membrane outer leaflet and regulates cell adhesion and motility. Biochem. Biophys. Res. Commun. 2020, 527, 1050–1056. [Google Scholar] [CrossRef]
- Katan, M.; Cockcroft, S. Phosphatidylinositol(4,5)bisphosphate: Diverse functions at the plasma membrane. Essays Biochem. 2020, 64, 513–531. [Google Scholar]
- Ju, M.; Shi, J.; Saleh, S.N.; Albert, A.P.; Large, W.A. Ins(1,4,5)P3 interacts with PIP2 to regulate activation of TRPC6/C7 channels by diacylglycerol in native vascular myocytes. J. Physiol. 2010, 588 Pt 9, 1419–1433. [Google Scholar] [CrossRef]
- Kanemaru, K.; Shimozawa, M.; Kitamata, M.; Furuishi, R.; Kayano, H.; Sukawa, Y.; Chiba, Y.; Fukuyama, T.; Hasegawa, J.; Nakanishi, H.; et al. Plasma membrane phosphatidylinositol (4,5)-bisphosphate is critical for determination of epithelial characteristics. Nat. Commun. 2022, 13, 2347. [Google Scholar] [CrossRef]
- Gada, K.D.; Logothetis, D.E. Protein Kinase C regulation of ion channels: The involvement of PIP2. J. Biol. Chem. 2022, 298, 102035. [Google Scholar] [CrossRef]
- Zhang, L.; Huang, W.; Tanimura, A.; Morita, T.; Harihar, S.; Dewald, D.B.; Prestwich, G.D. Synthesis and biological activity of metabolically stabilized cyclopentyl trisphosphate analogues of D-myo-Ins(1,4,5)P3. ChemMedChem 2007, 2, 1281–1289. [Google Scholar] [CrossRef] [PubMed]
- Choudhry, H.; Aggarwal, M.; Pan, P.Y. Mini-review: Synaptojanin 1 and its implications in membrane trafficking. Neurosci. Lett. 2021, 765, 136288. [Google Scholar] [CrossRef] [PubMed]
- Ahaley, S.S. Synaptojanin regulates Hedgehog signalling by modulating phosphatidylinositol 4-phosphate levels. J. Biosci. 2018, 43, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Sengelaub, C.A.; Navrazhina, K.; Ross, J.B.; Halberg, N.; Tavazoie, S.F. PTPRN2 and PLCbeta1 promote metastatic breast cancer cell migration through PI(4,5)P2-dependent actin remodeling. EMBO J. 2016, 35, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Senju, Y.; Lappalainen, P. Regulation of actin dynamics by PI(4,5)P2 in cell migration and endocytosis. Curr. Opin. Cell Biol. 2019, 56, 7–13. [Google Scholar] [CrossRef]
- Hilpela, P.; Vartiainen, M.K.; Lappalainen, P. Regulation of the actin cytoskeleton by PI(4,5)P2 and PI(3,4,5)P3. Curr. Top. Microbiol. Immunol. 2004, 282, 117–163. [Google Scholar]
- Manolson, M.F.; Wu, B.; Proteau, D.; Taillon, B.E.; Roberts, B.T.; Hoyt, M.A.; Jones, E.W. STV1 gene encodes functional homologue of 95-kDa yeast vacuolar H(+)-ATPase subunit Vph1p. J. Biol. Chem. 1994, 269, 14064–14074. [Google Scholar] [CrossRef]
- McGuire, C.M.; Collins, M.P.; Sun-Wada, G.; Wada, Y.; Forgac, M. Isoform-specific gene disruptions reveal a role for the V-ATPase subunit a4 isoform in the invasiveness of 4T1-12B breast cancer cells. J. Biol. Chem. 2019, 294, 11248–11258. [Google Scholar] [CrossRef] [PubMed]
- Ochotny, N.; Van Vliet, A.; Chan, N.; Yao, Y.; Morel, M.; Kartner, N.; von Schroeder, H.P.; Heersche, J.N.; Manolson, M.F. Effects of human a3 and a4 mutations that result in osteopetrosis and distal renal tubular acidosis on yeast V-ATPase expression and activity. J. Biol. Chem. 2006, 281, 26102–26111. [Google Scholar] [CrossRef] [PubMed]
- Esmail, S.; Kartner, N.; Yao, Y.; Kim, J.W.; Reithmeier, R.A.F.; Manolson, M.F. Molecular mechanisms of cutis laxa- and distal renal tubular acidosis-causing mutations in V-ATPase a subunits, ATP6V0A2 and ATP6V0A4. J. Biol. Chem. 2018, 293, 2787–2800. [Google Scholar] [CrossRef]
- Imai, E.; Kaneko, S.; Mori, T.; Okado, T.; Uchida, S.; Tsukamoto, Y. A novel heterozygous mutation in the ATP6V0A4 gene encoding the V-ATPase a4 subunit in an adult patient with incomplete distal renal tubular acidosis. Clin. Kidney J. 2016, 9, 424–428. [Google Scholar] [CrossRef]
- Stover, E.H.; Borthwick, K.J.; Bavalia, C.; Eady, N.; Fritz, D.M.; Rungroj, N.; Giersch, A.B.; Morton, C.C.; Axon, P.R.; Akil, I.; et al. Novel ATP6V1B1 and ATP6V0A4 mutations in autosomal recessive distal renal tubular acidosis with new evidence for hearing loss. J. Med. Genet. 2002, 39, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Yang, H.; Yang, H.; Eom, S.H.; Im, Y.J. Structure of Osh3 reveals a conserved mode of phosphoinositide binding in oxysterol-binding proteins. Structure 2013, 21, 1203–1213. [Google Scholar] [CrossRef]
- Brown, M.F. Curvature forces in membrane lipid-protein interactions. Biochemistry 2012, 51, 9782–9795. [Google Scholar] [CrossRef]
- Bigay, J.; Antonny, B. Curvature, lipid packing, and electrostatics of membrane organelles: Defining cellular territories in determining specificity. Dev. Cell 2012, 23, 886–895. [Google Scholar] [CrossRef]
- Dickson, E.J.; Jensen, J.B.; Hille, B. Golgi and plasma membrane pools of PI(4)P contribute to plasma membrane PI(4,5)P2 and maintenance of KCNQ2/3 ion channel current. Proc. Natl. Acad. Sci. USA 2014, 111, E2281–E2290. [Google Scholar] [CrossRef]
- Feng, J.; He, L.; Li, Y.; Xiao, F.; Hu, G. Modeling of PH Domains and Phosphoinositides Interactions and Beyond. Adv. Exp. Med. Biol. 2019, 1111, 19–32. [Google Scholar]
- Rosenhouse-Dantsker, A.; Logothetis, D.E. Molecular characteristics of phosphoinositide binding. Pflug. Arch. 2007, 455, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Yoshida, S.; Muroi, E.; Kawamura, M.; Kouchi, Z.; Nakamura, Y.; Sakai, R.; Fukami, K. Phosphatidylinositol 4,5-bisphosphate and PIP5-kinase Ialpha are required for invadopodia formation in human breast cancer cells. Cancer Sci. 2010, 101, 1632–1638. [Google Scholar] [CrossRef] [PubMed]
- Pike, J.A.; Styles, I.B.; Rappoport, J.Z.; Heath, J.K. Quantifying receptor trafficking and colocalization with confocal microscopy. Methods 2017, 115, 42–54. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chu, A.; Yao, Y.; Saffi, G.T.; Chung, J.H.; Botelho, R.J.; Glibowicka, M.; Deber, C.M.; Manolson, M.F. Characterization of a PIP Binding Site in the N-Terminal Domain of V-ATPase a4 and Its Role in Plasma Membrane Association. Int. J. Mol. Sci. 2023, 24, 4867. https://doi.org/10.3390/ijms24054867
Chu A, Yao Y, Saffi GT, Chung JH, Botelho RJ, Glibowicka M, Deber CM, Manolson MF. Characterization of a PIP Binding Site in the N-Terminal Domain of V-ATPase a4 and Its Role in Plasma Membrane Association. International Journal of Molecular Sciences. 2023; 24(5):4867. https://doi.org/10.3390/ijms24054867
Chicago/Turabian StyleChu, Anh, Yeqi Yao, Golam T. Saffi, Ji Hyun Chung, Roberto J. Botelho, Miroslawa Glibowicka, Charles M. Deber, and Morris F. Manolson. 2023. "Characterization of a PIP Binding Site in the N-Terminal Domain of V-ATPase a4 and Its Role in Plasma Membrane Association" International Journal of Molecular Sciences 24, no. 5: 4867. https://doi.org/10.3390/ijms24054867
APA StyleChu, A., Yao, Y., Saffi, G. T., Chung, J. H., Botelho, R. J., Glibowicka, M., Deber, C. M., & Manolson, M. F. (2023). Characterization of a PIP Binding Site in the N-Terminal Domain of V-ATPase a4 and Its Role in Plasma Membrane Association. International Journal of Molecular Sciences, 24(5), 4867. https://doi.org/10.3390/ijms24054867