
Metabolic Reprogramming and Potential Therapeutic Targets in Lymphoma



Abstract
1. Introduction
2. Metabolic Alterations in Lymphoma
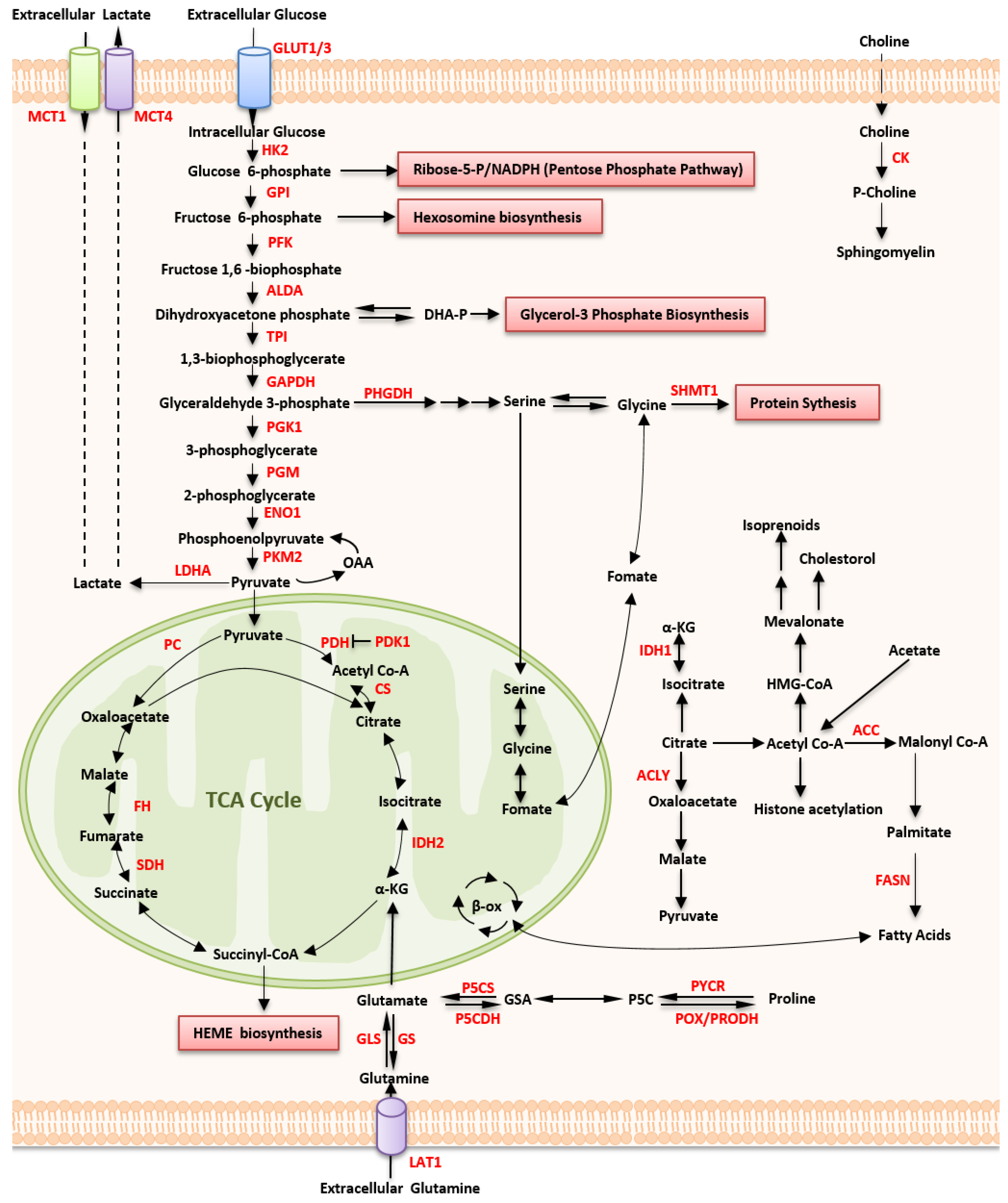
2.1. Altered Glucose Metabolism
2.1.1. Up-Regulated Glucose Uptake
2.1.2. Aerobic Glycolysis

{kind=link}
{kind=link}
{kind=link}
| Enzymes /Metabolites | Subtype of Lymphoma | Alteration | Mechanism | Ref |
|---|---|---|---|---|
| GLUT3 | DLBCL | upregulated | FDG uptake | [17] |
| GLUT3 | PCNSL | upregulated | glucose transporter of FDG accumulation | [18] |
| GLUT1 | cHL | upregulated | better clinical outcomes | [19] |
| HK2 | DLBCL | upregulated | FDG uptake, promote cell growth | [17,24] |
| LDH | PCL | upregulated | aggressive clinical behavior, resistance to chemotherapy, and low survival rates | [25] |
| LDH-5 | NHL | highly upregulated | correlated with HIF-1α cytoplasmic accumulation in NHL cells | [26] |
| MCT1 | DLBCL | highly expressed | an OXPHOS phenotype | [27] |
| SHMT | DLBCL | make glycine for purine synthesis | [32] | |
| IDO 1 | DLBCL | converts tryptophan into kynurenine-pathway metabolites, inhibits T-cell activity, and induces immune tolerance, predict a favorable prognosis | [33] | |
| IDO 1 | HL | converts tryptophan into kynurenine-pathway metabolites, inhibits T-cell activity, and induces immune tolerance, predict dismal clinical outcomes | [34] | |
| FASN | DLBCL | overexpressed | aggressive clinical course and therapeutic resistance, shorter OS and PFS | [35] |
| PI3K/AKT/mTOR | BCL and TCL | activated | related to p53, HIF-1α, and MYC | [36,37,38,39] |
| PKM2 | ALCL | inactivated | induces oncogenesis by phosphorylating nuclear STAT3 and regulating the transcription of genes involved in cell survival and proliferation | [40] |
| POX/PRODH | MYC-driven lymphoma | expressed | mitochondrial tumor suppressor | [41] |
| GLS | BL | upregulated | enhances glutamine uptake and glutamine catabolism | [41] |
| Chokα | TCL | overexpressed | regulation of choline metabolism | [42] |
| SDH | BL | reduced expression | increased the intracellular drug concentration | [43] |
| IDH2 | AITL | mutated | increasing DNA hypermethylation of gene promoters | [44,45] |
| D2HGDH | DLBCL | mutated | affects histone and DNA methylation and HIF-1α hydroxylation | [46] |
2.1.3. Mitochondrial Metabolism
2.2. Altered Amino Acid Metabolism
2.2.1. Glutamine Metabolism
2.2.2. Proline Metabolism
2.2.3. Serine, Glycine, and Serine Hydroxymethyltransferase
2.2.4. Indoleamine 2,3-Dioxygenase 1
2.3. Altered Lipid Metabolism
2.3.1. Fatty Acid Synthase
2.3.2. PPARδ
2.3.3. Cholesterol Synthesis
2.3.4. Choline Metabolism
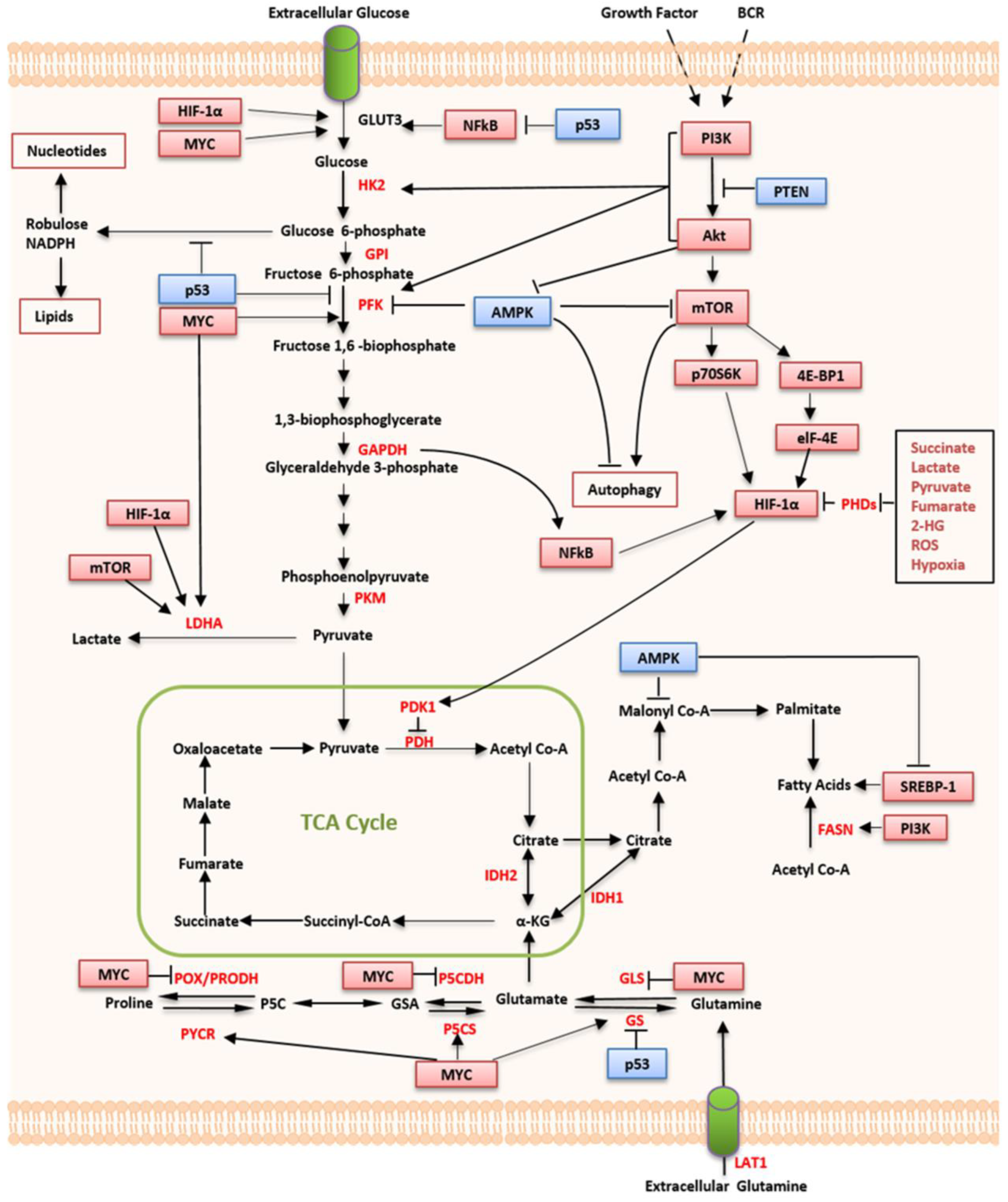
3. Regulation of Altered Metabolism in Lymphoma
3.1. HIF
3.2. MYC
3.3. The PI3K/mTOR Pathway
3.4. p53
3.5. AMPK
3.6. SIRT4
3.7. EBV
4. Oncometabolites in Lymphoma
4.1. Glycolytic Enzymes
4.2. SDH
4.3. IDH2, D2HG, and D2HGDH
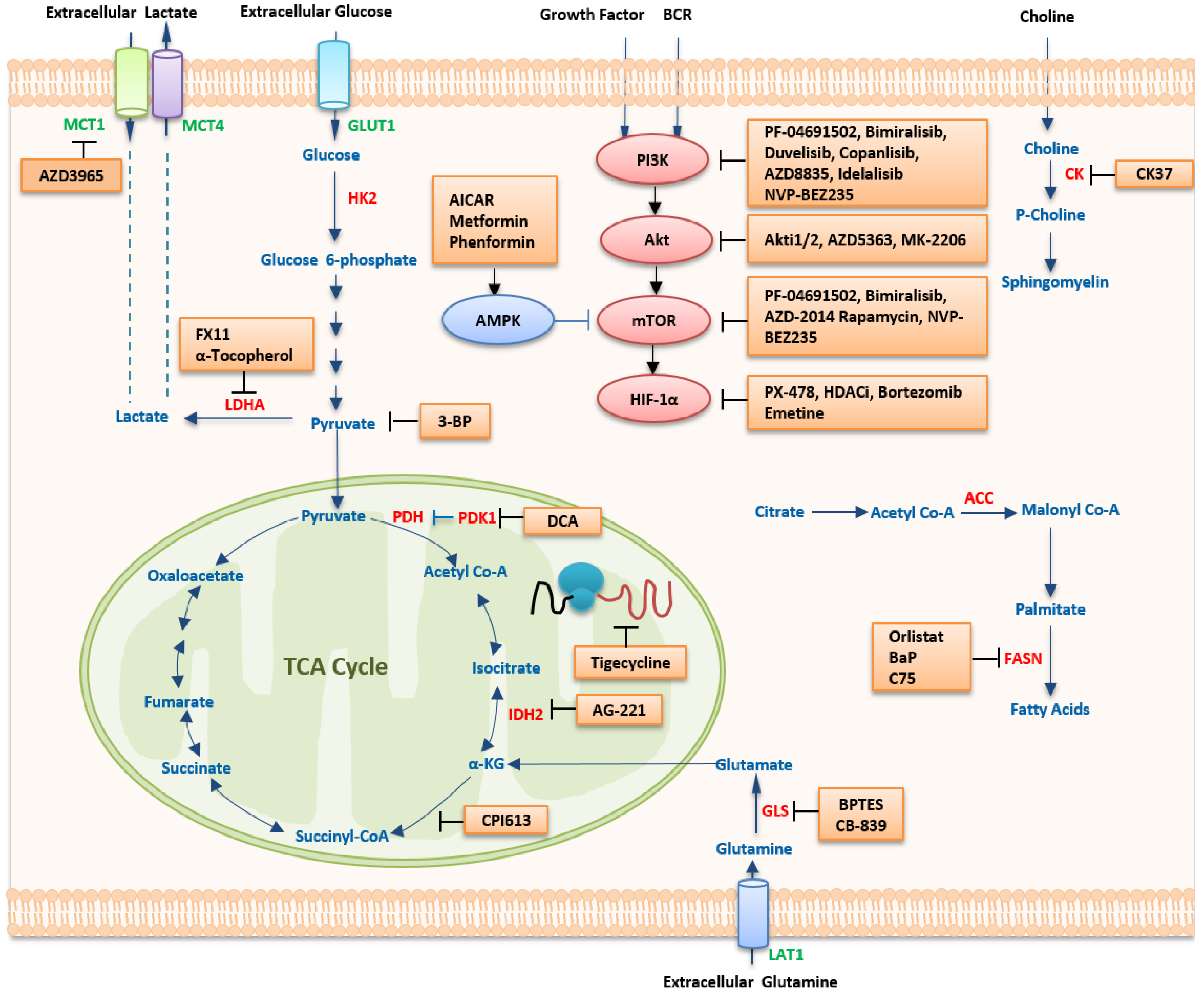
5. Therapeutic Strategies Targeting Metabolism
5.1. Targeting Metabolic Pathways
5.1.1. Targeting Glucose Metabolism
| Pathway | Compound | Application | Development Stage | Ref |
|---|---|---|---|---|
| HK2 | 2-DG | B-NHL | cell lines | [192] |
| 3-BrPA | BL | cell lines, mice | [195,196] | |
| Pyruvate | 3-BP | T-NHL | mice (DL) | [197] |
| LDHA | FX11 | BL | cell line | [198,199] |
| α-Tocopherol | T-NHL | mice (DL) | [199] | |
| PDK | DCA | DL | mice | [202,203] |
| Mitochondrial protein translation | Tigecycline | OxPhos-DLBCL cell lines | FDA-approved | [56] |
| Glutaminase | BPTES | BL | mice | [47] |
| Glutamine uptake | L-asparaginase | NHL | cell lines | [207] |
| SHMT1/2 | SHIN1 | DLBCL | cell lines | [33] |
| FASN | orlistat | MCL | cell lines | [86,208] |
| T-NHL | cell lines, mice | [209] | ||
| C75 | DLBCL, PEL, and B-NHL | cell lines | [84,85] | |
| NA | BaP | BL | patients | [210] |
| PPARα | Fenofibrate | B-NHL | mice | [211] |
| Choline kinase | CK37 | T-NHL | mice | [42] |
| HIF-1α | PX-478 | PEL | cell lines | [115] |
| PCI-24781(HDACi) | DLBCL | Phase 1/2 | [212] | |
| SAHA(HDACi) | B-NHL | cell lines, mice | [213] | |
| MYC | 10058-F4 | DLBCL | cell lines | [214,215,216] |
| PI3K | LY294002 | B-NHL | cell lines | [217] |
| AZD8835 | ABC-DLBCL | cell lines | [218] | |
| AKT | Akti1/2 | PEL | cell lines | [219] |
| AZD-5363 | PTEN-deficient DLBCL | cell lines, mice | [218] | |
| MK-2206 | ABC-DLBCL | mice | [220] | |
| NaB (HDACi) | BL | cell lines | [221] | |
| mTOR | Rapamycin | ALCL, NHL | cell lines | [36,131] |
| mTOR C1/2 | AZD-2014 | MCL | cell lines | [222] |
| Dual inhibitor of PI3K and mTOR | NVP-BEZ235 | PEL | mice | [223] |
| PF-04091502 | PEL | cell lines | [219] | |
| Bimiralisib (PQR309) | DLBCL, MCL, SMZL, CLL, HL, and ALCL | cell lines, mice | [224] | |
| AMPK | AICAR | MCL, SMZL, FL, and CLL | cell lines, mice and patients | [159,225,226,227,228,229] |
| Metformin | B and T-NHL | cell lines, mice | [157,230] | |
| Phenformin | PTEN-deficient T-cell lymphomas | cell lines | [230] |
5.1.2. Targeting Amino Acid Metabolism
5.1.3. Targeting Lipid Metabolism
5.2. Targeting Oncogenic Regulators
5.2.1. HIF-1α Inhibitors
5.2.2. MYC Inhibitors
5.2.3. The PI3K/mTOR Pathway
PI3K Inhibitors
Akt Inhibitors
mTOR Inhibitors
Dual Inhibitors of PI3K and mTOR
5.2.4. AMPK Activators
6. Potential Metabolic Biomarkers of Lymphoma
7. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nabi, K.; Le, A. The Intratumoral Heterogeneity of Cancer Metabolism. Adv. Exp. Med. Biol. 2021, 1311, 149–160. [Google Scholar] [PubMed]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Stine, Z.E.; Schug, Z.T.; Salvino, J.M.; Dang, C.V. Targeting cancer metabolism in the era of precision oncology. Nat. Rev. Drug Discov. 2022, 21, 141–162. [Google Scholar] [CrossRef]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, Metabolism and Cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef]
- Brunner, J.S.; Finley, L.W.S. Metabolic determinants of tumour initiation. Nat. Rev. Endocrinol. 2022, 19, 134–150. [Google Scholar] [CrossRef]
- Vaupel, P.; Schmidberger, H.; Mayer, A. The Warburg effect: Essential part of metabolic reprogramming and central contributor to cancer progression. Int. J. Radiat. Biol. 2019, 95, 912–919. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Zhu, J.; Thompson, C.B. The hallmarks of cancer metabolism: Still emerging. Cell Metab. 2022, 34, 355–377. [Google Scholar] [CrossRef]
- Cheson, B.D.; Fisher, R.I.; Barrington, S.F.; Cavalli, F.; Schwartz, L.H.; Zucca, E.; Lister, T.A. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: The Lugano classification. J. Clin. Oncol. 2014, 32, 3059–3068. [Google Scholar] [CrossRef]
- Ricard, F.; Barrington, S.; Korn, R.; Brueggenwerth, G.; Trotman, J.; Cheson, B.; Salles, G.; Schwartz, L.; Goldmacher, G.; Jarecha, R.; et al. Application of the lugano classification for initial evaluation, staging, and response assessment of hodgkin and non-hodgkin lymphoma: The prolog consensus initiative (part 2-technical). J. Nucl. Med. 2022, 64, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Ricard, F.; Cheson, B.; Barrington, S.; Trotman, J.; Schmid, A.; Brueggenwerth, G.; Salles, G.; Schwartz, L.; Goldmacher, G.; Jarecha, R.; et al. Application of the lugano classification for initial evaluation, staging, and response assessment of hodgkin and non-hodgkin lymphoma: The prolog consensus initiative (part 1-clinical). J. Nucl. Med. 2022, 64, 102–108. [Google Scholar] [CrossRef]
- Calvo-Vidal, M.N.; Cerchietti, L. The metabolism of lymphomas. Curr. Opin. Hematol. 2013, 20, 345–354. [Google Scholar] [CrossRef]
- Maxwell, S.A.; Mousavi-Fard, S. Non-Hodgkin’s B-cell lymphoma: Advances in molecular strategies targeting drug resistance. Exp. Biol. Med. 2013, 238, 971–990. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.F.; Chang, C.C.; Liu, Y.C.; Chang, C.S.; Hsiao, H.H.; Liu, T.C.; Huang, C.T.; Lin, S.F. Utilization of 18F-FDG PET/CT as a staging tool in patients with newly diagnosed lymphoma. Kaohsiung J. Med. Sci. 2015, 31, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Chihara, D.; Oki, Y.; Onoda, H.; Taji, H.; Yamamoto, K.; Tamaki, T.; Morishima, Y. High maximum standard uptake value (SUVmax) on PET scan is associated with shorter survival in patients with diffuse large B cell lymphoma. Int. J. Hematol. 2011, 93, 502–508. [Google Scholar] [CrossRef]
- Zaucha, J.M.; Chauvie, S.; Zaucha, R.; Biggii, A.; Gallamini, A. The role of PET/CT in the modern treatment of Hodgkin lymphoma. Cancer Treat. Rev. 2019, 77, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Shim, H.K.; Lee, W.W.; Park, S.Y.; Kim, H.; So, Y.; Kim, S.E. Expressions of glucose transporter Types 1 and 3 and hexokinase-II in diffuse large B-cell lymphoma and other B-cell non-Hodgkin’s lymphomas. Nucl. Med. Biol. 2009, 36, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Akahane, T.; Yamamoto, D.; Nakamura, H.; Sawa, H.; Nitta, K.; Ide, W.; Hashimoto, I.; Kamada, H. Correlation between positron emission tomography findings and glucose transporter 1, 3 and L-type amino acid transporter 1 mRNA expression in primary central nervous system lymphomas. Mol. Clin. Oncol. 2014, 2, 525–529. [Google Scholar] [CrossRef][Green Version]
- Koh, Y.W.; Han, J.H.; Park, S.Y.; Yoon, D.H.; Suh, C.; Huh, J. GLUT1 as a Prognostic Factor for Classical Hodgkin’s Lymphoma: Correlation with PD-L1 and PD-L2 Expression. J. Pathol. Transl. Med. 2017, 51, 152–158. [Google Scholar] [CrossRef]
- Levine, A.J.; Puzio-Kuter, A.M. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 2010, 330, 1340–1344. [Google Scholar] [CrossRef]
- Lee, S.C.; Marzec, M.; Liu, X.; Wehrli, S.; Kantekure, K.; Ragunath, P.N.; Nelson, D.S.; Delikatny, E.J.; Glickson, J.D.; Wasik, M.A. Decreased lactate concentration and glycolytic enzyme expression reflect inhibition of mTOR signal transduction pathway in B-cell lymphoma. NMR Biomed. 2013, 26, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Chiche, J.; Pommier, S.; Beneteau, M.; Mondragón, L.; Meynet, O.; Zunino, B.; Mouchotte, A.; Verhoeyen, E.; Guyot, M.; Pagès, G.; et al. GAPDH enhances the aggressiveness and the vascularization of non-Hodgkin’s B lymphomas via NF-κB-dependent induction of HIF-1α. Leukemia 2015, 29, 1163–1176. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, K.; Kawashima, I.; Koshiisi, M.; Kumagai, T.; Suzuki, M.; Suzuki, J.; Mitsumori, T.; Kirito, K. Glycolytic enzyme hexokinase II is a putative therapeutic target in B-cell malignant lymphoma. Exp. Hematol. 2019, 78, 46–55.e43. [Google Scholar] [CrossRef]
- Bhalla, K.; Jaber, S.; Nahid, M.N.; Underwood, K.; Beheshti, A.; Landon, A.; Bhandary, B.; Bastian, P.; Evens, A.M.; Haley, J.; et al. Role of hypoxia in Diffuse Large B-cell Lymphoma: Metabolic repression and selective translation of HK2 facilitates development of DLBCL. Sci. Rep. 2018, 8, 744. [Google Scholar] [CrossRef]
- Liu, W.P.; Song, Y.Q.; Zheng, W.; Wang, X.P.; Ding, N.; Zhu, J. Aggressive behavior and elevated lactate dehydrogenase at baseline confer inferior prognosis in patients with primary cutaneous lymphoma. Clin. Lymphoma Myeloma Leuk. 2013, 13, 534–540. [Google Scholar] [CrossRef]
- Lu, R.; Jiang, M.; Chen, Z.; Xu, X.; Hu, H.; Zhao, X.; Gao, X.; Guo, L. Lactate dehydrogenase 5 expression in Non-Hodgkin lymphoma is associated with the induced hypoxia regulated protein and poor prognosis. PLoS ONE 2013, 8, e74853. [Google Scholar] [CrossRef]
- Gooptu, M.; Whitaker-Menezes, D.; Sprandio, J.; Domingo-Vidal, M.; Lin, Z.; Uppal, G.; Gong, J.; Fratamico, R.; Leiby, B.; Dulau-Florea, A.; et al. Mitochondrial and glycolytic metabolic compartmentalization in diffuse large B-cell lymphoma. Semin. Oncol. 2017, 44, 204–217. [Google Scholar] [CrossRef]
- Doherty, J.R.; Cleveland, J.L. Targeting lactate metabolism for cancer therapeutics. J. Clin. Investig. 2013, 123, 3685–3692. [Google Scholar] [CrossRef]
- Parks, S.K.; Chiche, J.; Pouysségur, J. Disrupting proton dynamics and energy metabolism for cancer therapy. Nat. Rev. Cancer 2013, 13, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Le Floch, R.; Chiche, J.; Marchiq, I.; Naiken, T.; Ilc, K.; Murray, C.M.; Critchlow, S.E.; Roux, D.; Simon, M.P.; Pouysségur, J. CD147 subunit of lactate/H+ symporters MCT1 and hypoxia-inducible MCT4 is critical for energetics and growth of glycolytic tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 16663–16668. [Google Scholar] [CrossRef] [PubMed]
- Baenke, F.; Dubuis, S.; Brault, C.; Weigelt, B.; Dankworth, B.; Griffiths, B.; Jiang, M.; Mackay, A.; Saunders, B.; Spencer-Dene, B.; et al. Functional screening identifies MCT4 as a key regulator of breast cancer cell metabolism and survival. J. Pathol. 2015, 237, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Ducker, G.S.; Ghergurovich, J.M.; Mainolfi, N.; Suri, V.; Jeong, S.K.; Hsin-Jung Li, S.; Friedman, A.; Manfredi, M.G.; Gitai, Z.; Kim, H.; et al. Human SHMT inhibitors reveal defective glycine import as a targetable metabolic vulnerability of diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. USA 2017, 114, 11404–11409. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.J.; Kim, S.; Paik, J.H.; Kim, T.M.; Heo, D.S.; Kim, C.W.; Jeon, Y.K. An increase in indoleamine 2,3-dioxygenase-positive cells in the tumor microenvironment predicts favorable prognosis in patients with diffuse large B-cell lymphoma treated with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisolone. Leuk. Lymphoma 2016, 57, 1956–1960. [Google Scholar] [CrossRef]
- Choe, J.Y.; Yun, J.Y.; Jeon, Y.K.; Kim, S.H.; Park, G.; Huh, J.R.; Oh, S.; Kim, J.E. Indoleamine 2,3-dioxygenase (IDO) is frequently expressed in stromal cells of Hodgkin lymphoma and is associated with adverse clinical features: A retrospective cohort study. BMC Cancer 2014, 14, 335. [Google Scholar] [CrossRef] [PubMed]
- Dashnamoorthy, R.; Abermil, N.; Behesti, A.; Kozlowski, P.; Lansigan, F.; Kinlaw, W.B.; Gartenhaus, R.; Jones, G.; Hlatky, L.; Evens, A.M. The Lipid Addiction of Diffuse Large B-Cell Lymphoma (DLBCL) and Potential Treatment Strategies with Novel Fatty Acid Synthase (FASN) Small Molecule Inhibitors. Blood 2014, 124, 4490. [Google Scholar] [CrossRef]
- Argyriou, P.; Papageorgiou, S.G.; Panteleon, V.; Psyrri, A.; Bakou, V.; Pappa, V.; Spathis, A.; Economopoulou, P.; Papageorgiou, E.; Economopoulos, T.; et al. Hypoxia-inducible factors in mantle cell lymphoma: Implication for an activated mTORC1→HIF-1α pathway. Ann. Hematol. 2011, 90, 315–322. [Google Scholar] [CrossRef]
- Sander, S.; Calado, D.P.; Srinivasan, L.; Köchert, K.; Zhang, B.; Rosolowski, M.; Rodig, S.J.; Holzmann, K.; Stilgenbauer, S.; Siebert, R.; et al. Synergy between PI3K signaling and MYC in Burkitt lymphomagenesis. Cancer Cell 2012, 22, 167–179. [Google Scholar] [CrossRef]
- Schmitz, R.; Young, R.M.; Ceribelli, M.; Jhavar, S.; Xiao, W.; Zhang, M.; Wright, G.; Shaffer, A.L.; Hodson, D.J.; Buras, E.; et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 2012, 490, 116–120. [Google Scholar] [CrossRef]
- Yamamoto-Sugitani, M.; Kuroda, J.; Shimura, Y.; Nagoshi, H.; Chinen, Y.; Ohshiro, M.; Mizutani, S.; Kiyota, M.; Nakayama, R.; Kobayashi, T.; et al. Comprehensive cytogenetic study of primary cutaneous gamma-delta T-cell lymphoma by means of spectral karyotyping and genome-wide single nucleotide polymorphism array. Cancer Genet. 2012, 205, 459–464. [Google Scholar] [CrossRef]
- Hwang, S.R.; Murga-Zamalloa, C.; Brown, N.; Basappa, J.; McDonnell, S.R.; Mendoza-Reinoso, V.; Basrur, V.; Wilcox, R.; Elenitoba-Johnson, K.; Lim, M.S. Pyrimidine tract-binding protein 1 mediates pyruvate kinase M2-dependent phosphorylation of signal transducer and activator of transcription 3 and oncogenesis in anaplastic large cell lymphoma. Lab. Investig. 2017, 97, 962–970. [Google Scholar] [CrossRef][Green Version]
- Liu, W.; Le, A.; Hancock, C.; Lane, A.N.; Dang, C.V.; Fan, T.W.; Phang, J.M. Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proc. Natl. Acad. Sci. USA 2012, 109, 8983–8988. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Bian, J.; Wang, L.; Zhou, J.Y.; Wang, Y.; Zhao, Y.; Wu, L.L.; Hu, J.J.; Li, B.; Chen, S.J.; et al. Dysregulated choline metabolism in T-cell lymphoma: Role of choline kinase-α and therapeutic targeting. Blood Cancer J. 2015, 5, 287. [Google Scholar] [CrossRef] [PubMed]
- Kusao, I.; Troelstrup, D.; Shiramizu, B. Possible Mitochondria-Associated Enzymatic Role in Non-Hodgkin Lymphoma Residual Disease. Cancer Growth Metastasis 2008, 1, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Lemonnier, F.; Cairns, R.A.; Inoue, S.; Li, W.Y.; Dupuy, A.; Broutin, S.; Martin, N.; Fataccioli, V.; Pelletier, R.; Wakeham, A.; et al. The IDH2 R172K mutation associated with angioimmunoblastic T-cell lymphoma produces 2HG in T cells and impacts lymphoid development. Proc. Natl. Acad. Sci. USA 2016, 113, 15084–15089. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; McKeithan, T.W.; Gong, Q.; Zhang, W.; Bouska, A.; Rosenwald, A.; Gascoyne, R.D.; Wu, X.; Wang, J.; Muhammad, Z.; et al. IDH2R172 mutations define a unique subgroup of patients with angioimmunoblastic T-cell lymphoma. Blood 2015, 126, 1741–1752. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.P.; Abbas, S.; Kim, S.W.; Ortega, M.; Bouamar, H.; Escobedo, Y.; Varadarajan, P.; Qin, Y.; Sudderth, J.; Schulz, E.; et al. D2HGDH regulates alpha-ketoglutarate levels and dioxygenase function by modulating IDH2. Nat. Commun. 2015, 6, 7768. [Google Scholar] [CrossRef] [PubMed]
- Le, A.; Lane, A.N.; Hamaker, M.; Bose, S.; Gouw, A.; Barbi, J.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Zhang, H.; et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012, 15, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, R.; Gilkerson, R.; Aggeler, R.; Yamagata, K.; Remington, S.J.; Capaldi, R.A. Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res. 2004, 64, 985–993. [Google Scholar] [CrossRef]
- Zaugg, K.; Yao, Y.; Reilly, P.T.; Kannan, K.; Kiarash, R.; Mason, J.; Huang, P.; Sawyer, S.K.; Fuerth, B.; Faubert, B.; et al. Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev. 2011, 25, 1041–1051. [Google Scholar] [CrossRef]
- Marin-Valencia, I.; Yang, C.; Mashimo, T.; Cho, S.; Baek, H.; Yang, X.L.; Rajagopalan, K.N.; Maddie, M.; Vemireddy, V.; Zhao, Z.; et al. Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab. 2012, 15, 827–837. [Google Scholar] [CrossRef]
- Moreno-Sánchez, R.; Rodríguez-Enríquez, S.; Saavedra, E.; Marín-Hernández, A.; Gallardo-Pérez, J.C. The bioenergetics of cancer: Is glycolysis the main ATP supplier in all tumor cells? Biofactors 2009, 35, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Meynet, O.; Bénéteau, M.; Jacquin, M.A.; Pradelli, L.A.; Cornille, A.; Carles, M.; Ricci, J.E. Glycolysis inhibition targets Mcl-1 to restore sensitivity of lymphoma cells to ABT-737-induced apoptosis. Leukemia 2012, 26, 1145–1147. [Google Scholar] [CrossRef][Green Version]
- Monti, S.; Savage, K.J.; Kutok, J.L.; Feuerhake, F.; Kurtin, P.; Mihm, M.; Wu, B.; Pasqualucci, L.; Neuberg, D.; Aguiar, R.C.; et al. Molecular profiling of diffuse large B-cell lymphoma identifies robust subtypes including one characterized by host inflammatory response. Blood 2005, 105, 1851–1861. [Google Scholar] [CrossRef]
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Sha, C.; Barrans, S.; Cucco, F.; Bentley, M.A.; Care, M.A.; Cummin, T.; Kennedy, H.; Thompson, J.S.; Uddin, R.; Worrillow, L.; et al. Molecular High-Grade B-Cell Lymphoma: Defining a Poor-Risk Group That Requires Different Approaches to Therapy. J. Clin. Oncol. 2019, 37, 202–212. [Google Scholar] [CrossRef]
- Norberg, E.; Lako, A.; Chen, P.H.; Stanley, I.A.; Zhou, F.; Ficarro, S.B.; Chapuy, B.; Chen, L.; Rodig, S.; Shin, D.; et al. Differential contribution of the mitochondrial translation pathway to the survival of diffuse large B-cell lymphoma subsets. Cell Death Differ. 2017, 24, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Caro, P.; Kishan, A.U.; Norberg, E.; Stanley, I.A.; Chapuy, B.; Ficarro, S.B.; Polak, K.; Tondera, D.; Gounarides, J.; Yin, H.; et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell 2012, 22, 547–560. [Google Scholar] [CrossRef]
- Monti, S.; Chapuy, B.; Takeyama, K.; Rodig, S.J.; Hao, Y.; Yeda, K.T.; Inguilizian, H.; Mermel, C.; Currie, T.; Dogan, A.; et al. Integrative analysis reveals an outcome-associated and targetable pattern of p53 and cell cycle deregulation in diffuse large B cell lymphoma. Cancer Cell 2012, 22, 359–372. [Google Scholar] [CrossRef]
- Chen, L.; Monti, S.; Juszczynski, P.; Daley, J.; Chen, W.; Witzig, T.E.; Habermann, T.M.; Kutok, J.L.; Shipp, M.A. SYK-dependent tonic B-cell receptor signaling is a rational treatment target in diffuse large B-cell lymphoma. Blood 2008, 111, 2230–2237. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef]
- Wishart, D.S. Metabolomics for Investigating Physiological and Pathophysiological Processes. Physiol. Rev. 2019, 99, 1819–1875. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Fan, T.W.; Lane, A.N.; Higashi, R.M. Chloroformate derivatization for tracing the fate of Amino acids in cells and tissues by multiple stable isotope resolved metabolomics (mSIRM). Anal. Chim. Acta 2017, 976, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. C-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Cheng, T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef]
- Wei, P.; Bott, A.J.; Cluntun, A.A.; Morgan, J.T.; Cunningham, C.N.; Schell, J.C.; Ouyang, Y.; Ficarro, S.B.; Marto, J.A.; Danial, N.N.; et al. Mitochondrial pyruvate supports lymphoma proliferation by fueling a glutamate pyruvate transaminase 2-dependent glutaminolysis pathway. Sci. Adv. 2022, 8, eabq0117. [Google Scholar] [CrossRef]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef]
- Cruzat, V.; Macedo Rogero, M.; Noel Keane, K.; Curi, R.; Newsholme, P. Glutamine: Metabolism and Immune Function, Supplementation and Clinical Translation. Nutrients 2018, 10, 1564. [Google Scholar] [CrossRef]
- Li, L.; Nie, L.; Jordan, A.; Cai, Q.; Liu, Y.; Li, Y.; Che, Y.; Vargas, J.; Chen, Z.; Leeming, A.; et al. Targeting glutaminase is therapeutically effective in ibrutinib-resistant mantle cell lymphoma. Haematologica 2022. [Google Scholar] [CrossRef]
- Zimta, A.A.; Cenariu, D.; Irimie, A.; Magdo, L.; Nabavi, S.M.; Atanasov, A.G.; Berindan-Neagoe, I. The Role of Nrf2 Activity in Cancer Development and Progression. Cancers 2019, 11, 1755. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Zhao, Y.; Xue, L.; Zhang, J.; Qiao, Y.; Jin, Q.; Li, H. Expression of Keap1 and Nrf2 in diffuse large B-cell lymphoma and its clinical significance. Exp. Ther. Med. 2018, 16, 573–578. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Goswami, D.; Adiseshaiah, P.P.; Burgan, W.; Yi, M.; Guerin, T.M.; Kozlov, S.V.; Nissley, D.V.; McCormick, F. Undermining Glutaminolysis Bolsters Chemotherapy While NRF2 Promotes Chemoresistance in KRAS-Driven Pancreatic Cancers. Cancer Res. 2020, 80, 1630–1643. [Google Scholar] [CrossRef]
- Rushworth, S.A.; Macewan, D.J. The role of nrf2 and cytoprotection in regulating chemotherapy resistance of human leukemia cells. Cancers 2011, 3, 1605–1621. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Stine, Z.E.; Xia, J.; Lu, Y.; O’Connor, R.S.; Altman, B.J.; Hsieh, A.L.; Gouw, A.M.; Thomas, A.G.; Gao, P.; et al. Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. J. Clin. Investig. 2015, 125, 2293–2306. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, K.; Bhatia, R.; Rauth, S.; Kisling, A.; Atri, P.; Thompson, C.; Vengoji, R.; Ram Krishn, S.; Shinde, D.; Thomas, V.; et al. Mucin 5AC Serves as the Nexus for β-Catenin/c-Myc Interplay to Promote Glutamine Dependency During Pancreatic Cancer Chemoresistance. Gastroenterology 2022, 162, 253–268.e213. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W. Serine, glycine and one-carbon units: Cancer metabolism in full circle. Nat. Rev. Cancer 2013, 13, 572–583. [Google Scholar] [CrossRef]
- Maddocks, O.D.K.; Athineos, D.; Cheung, E.C.; Lee, P.; Zhang, T.; van den Broek, N.J.F.; Mackay, G.M.; Labuschagne, C.F.; Gay, D.; Kruiswijk, F.; et al. Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 2017, 544, 372–376. [Google Scholar] [CrossRef]
- Maliniemi, P.; Laukkanen, K.; Väkevä, L.; Dettmer, K.; Lipsanen, T.; Jeskanen, L.; Bessede, A.; Oefner, P.J.; Kadin, M.E.; Ranki, A. Biological and clinical significance of tryptophan-catabolizing enzymes in cutaneous T-cell lymphomas. Oncoimmunology 2017, 6, e1273310. [Google Scholar] [CrossRef]
- Masaki, A.; Ishida, T.; Maeda, Y.; Suzuki, S.; Ito, A.; Takino, H.; Ogura, H.; Totani, H.; Yoshida, T.; Kinoshita, S.; et al. Prognostic Significance of Tryptophan Catabolism in Adult T-cell Leukemia/Lymphoma. Clin. Cancer Res. 2015, 21, 2830–2839. [Google Scholar] [CrossRef] [PubMed]
- Ninomiya, S.; Narala, N.; Huye, L.; Yagyu, S.; Savoldo, B.; Dotti, G.; Heslop, H.E.; Brenner, M.K.; Rooney, C.M.; Ramos, C.A. Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood 2015, 125, 3905–3916. [Google Scholar] [CrossRef] [PubMed]
- Kapadia, B.; Nanaji, N.M.; Bhalla, K.; Bhandary, B.; Lapidus, R.; Beheshti, A.; Evens, A.M.; Gartenhaus, R.B. Fatty Acid Synthase induced S6Kinase facilitates USP11-eIF4B complex formation for sustained oncogenic translation in DLBCL. Nat. Commun. 2018, 9, 829. [Google Scholar] [CrossRef] [PubMed]
- Uddin, S.; Hussain, A.R.; Ahmed, M.; Bu, R.; Ahmed, S.O.; Ajarim, D.; Al-Dayel, F.; Bavi, P.; Al-Kuraya, K.S. Inhibition of fatty acid synthase suppresses c-Met receptor kinase and induces apoptosis in diffuse large B-cell lymphoma. Mol. Cancer Ther. 2010, 9, 1244–1255. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, A.P.; Jacobs, S.R.; Freemerman, A.J.; Makowski, L.; Rathmell, J.C.; Dittmer, D.P.; Damania, B. Dysregulation of fatty acid synthesis and glycolysis in non-Hodgkin lymphoma. Proc. Natl. Acad. Sci. USA 2012, 109, 11818–11823. [Google Scholar] [CrossRef]
- Gelebart, P.; Zak, Z.; Anand, M.; Belch, A.; Lai, R. Blockade of fatty acid synthase triggers significant apoptosis in mantle cell lymphoma. PLoS ONE 2012, 7, e33738. [Google Scholar] [CrossRef]
- Dashnamoorthy, R.; Lansigan, F.; Davis, W.L.; Kinlaw, W.B., III; Gartenhaus, R., III; Evens, A.M. Targeting The Interactions Of Fatty Acid Metabolism With PI3K/mTOR and MAPK As a Novel Therapeutic Strategy In Diffuse Large B-Cell Lymphoma (DLBCL). Blood 2013, 122, 5133. [Google Scholar] [CrossRef]
- Röhrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef]
- Spaner, D.E.; Lee, E.; Shi, Y.; Wen, F.; Li, Y.; Tung, S.; McCaw, L.; Wong, K.; Gary-Gouy, H.; Dalloul, A.; et al. PPAR-alpha is a therapeutic target for chronic lymphocytic leukemia. Leukemia 2013, 27, 1090–1099. [Google Scholar] [CrossRef]
- Harmon, G.S.; Lam, M.T.; Glass, C.K. PPARs and lipid ligands in inflammation and metabolism. Chem. Rev. 2011, 111, 6321–6340. [Google Scholar] [CrossRef]
- Wang, X.; Wang, G.; Shi, Y.; Sun, L.; Gorczynski, R.; Li, Y.J.; Xu, Z.; Spaner, D.E. PPAR-delta promotes survival of breast cancer cells in harsh metabolic conditions. Oncogenesis 2016, 5, e232. [Google Scholar] [CrossRef] [PubMed]
- Khozoie, C.; Borland, M.G.; Zhu, B.; Baek, S.; John, S.; Hager, G.L.; Shah, Y.M.; Gonzalez, F.J.; Peters, J.M. Analysis of the peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) cistrome reveals novel co-regulatory role of ATF4. BMC Genom. 2012, 13, 665. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Shi, Y.; Wang, G.; Wang, X.; Zeng, S.; Dunn, S.E.; Fairn, G.D.; Li, Y.J.; Spaner, D.E. PPAR-delta modulates membrane cholesterol and cytokine signaling in malignant B cells. Leukemia 2018, 32, 184–193. [Google Scholar] [CrossRef]
- Rink, J.S.; Yang, S.; Cen, O.; Taxter, T.; McMahon, K.M.; Misener, S.; Behdad, A.; Longnecker, R.; Gordon, L.I.; Thaxton, C.S. Rational Targeting of Cellular Cholesterol in Diffuse Large B-Cell Lymphoma (DLBCL) Enabled by Functional Lipoprotein Nanoparticles: A Therapeutic Strategy Dependent on Cell of Origin. Mol. Pharm. 2017, 14, 4042–4051. [Google Scholar] [CrossRef] [PubMed]
- Glunde, K.; Bhujwalla, Z.M.; Ronen, S.M. Choline metabolism in malignant transformation. Nat. Rev. Cancer 2011, 11, 835–848. [Google Scholar] [CrossRef] [PubMed]
- Arlauckas, S.P.; Popov, A.V.; Delikatny, E.J. Choline kinase alpha-Putting the ChoK-hold on tumor metabolism. Prog. Lipid Res. 2016, 63, 28–40. [Google Scholar] [CrossRef]
- Kierans, S.J.; Taylor, C.T. Regulation of glycolysis by the hypoxia-inducible factor (HIF): Implications for cellular physiology. J. Physiol. 2021, 599, 23–37. [Google Scholar] [CrossRef]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef]
- Gonzalez-King, H.; García, N.A.; Ontoria-Oviedo, I.; Ciria, M.; Montero, J.A.; Sepúlveda, P. Hypoxia Inducible Factor-1α Potentiates Jagged 1-Mediated Angiogenesis by Mesenchymal Stem Cell-Derived Exosomes. Stem Cells 2017, 35, 1747–1759. [Google Scholar] [CrossRef]
- Eyrich, N.W.; Potts, C.R.; Robinson, M.H.; Maximov, V.; Kenney, A.M. Reactive Oxygen Species Signaling Promotes Hypoxia-Inducible Factor 1α Stabilization in Sonic Hedgehog-Driven Cerebellar Progenitor Cell Proliferation. Mol. Cell Biol. 2019, 39, e00268-18. [Google Scholar] [CrossRef]
- Majumder, P.K.; Febbo, P.G.; Bikoff, R.; Berger, R.; Xue, Q.; McMahon, L.M.; Manola, J.; Brugarolas, J.; McDonnell, T.J.; Golub, T.R.; et al. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat. Med. 2004, 10, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Laughner, E.; Taghavi, P.; Chiles, K.; Mahon, P.C.; Semenza, G.L. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: Novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol. Cell Biol. 2001, 21, 3995–4004. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, R.; Hirota, K.; Fan, F.; Jung, Y.D.; Ellis, L.M.; Semenza, G.L. Insulin-like growth factor 1 induces hypoxia-inducible factor 1-mediated vascular endothelial growth factor expression, which is dependent on MAP kinase and phosphatidylinositol 3-kinase signaling in colon cancer cells. J. Biol. Chem. 2002, 277, 38205–38211. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Zhang, H.; Dinavahi, R.; Li, F.; Xiang, Y.; Raman, V.; Bhujwalla, Z.M.; Felsher, D.W.; Cheng, L.; Pevsner, J.; et al. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell 2007, 12, 230–238. [Google Scholar] [CrossRef]
- Gupta, D.; Crosby, M.E.; Almasan, A.; Macklis, R.M. Regulation of CD20 expression by radiation-induced changes in intracellular redox status. Free Radic Biol. Med. 2008, 44, 614–623. [Google Scholar] [CrossRef][Green Version]
- Evens, A.M.; Schumacker, P.T.; Helenowski, I.B.; Singh, A.T.; Dokic, D.; Keswani, A.; Kordeluk, E.; Raji, A.; Winter, J.N.; Jovanovic, B.D.; et al. Hypoxia inducible factor-alpha activation in lymphoma and relationship to the thioredoxin family. Br. J. Haematol. 2008, 141, 676–680. [Google Scholar] [CrossRef]
- Evens, A.M.; Sehn, L.H.; Farinha, P.; Nelson, B.P.; Raji, A.; Lu, Y.; Brakman, A.; Parimi, V.; Winter, J.N.; Schumacker, P.T.; et al. Hypoxia-inducible factor-1 {alpha} expression predicts superior survival in patients with diffuse large B-cell lymphoma treated with R-CHOP. J. Clin. Oncol. 2010, 28, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.R.; Dojcinov, S.; King, L.; Wosniak, S.; Gerry, S.; Casbard, A.; Bailey, H.; Gallop-Evans, E.; Maughan, T. Prognostic significance of hypoxia inducible factor-1α and vascular endothelial growth factor expression in patients with diffuse large B-cell lymphoma treated with rituximab. Leuk. Lymphoma 2013, 54, 959–966. [Google Scholar] [CrossRef]
- Darekar, S.; Georgiou, K.; Yurchenko, M.; Yenamandra, S.P.; Chachami, G.; Simos, G.; Klein, G.; Kashuba, E. Epstein-Barr virus immortalization of human B-cells leads to stabilization of hypoxia-induced factor 1 alpha, congruent with the Warburg effect. PLoS ONE 2012, 7, e42072. [Google Scholar] [CrossRef]
- Dang, C.V. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb Perspect. Med. 2013, 3, a014217. [Google Scholar] [CrossRef]
- Lewis, B.C.; Prescott, J.E.; Campbell, S.E.; Shim, H.; Orlowski, R.Z.; Dang, C.V. Tumor induction by the c-Myc target genes rcl and lactate dehydrogenase A. Cancer Res. 2000, 60, 6178–6183. [Google Scholar]
- Mushtaq, M.; Darekar, S.; Klein, G.; Kashuba, E. Different Mechanisms of Regulation of the Warburg Effect in Lymphoblastoid and Burkitt Lymphoma Cells. PLoS ONE 2015, 10, e0136142. [Google Scholar] [CrossRef] [PubMed]
- Martinengo, C.; Poggio, T.; Menotti, M.; Scalzo, M.S.; Mastini, C.; Ambrogio, C.; Pellegrino, E.; Riera, L.; Piva, R.; Ribatti, D.; et al. ALK-dependent control of hypoxia-inducible factors mediates tumor growth and metastasis. Cancer Res. 2014, 74, 6094–6106. [Google Scholar] [CrossRef] [PubMed]
- Marzec, M.; Liu, X.; Wong, W.; Yang, Y.; Pasha, T.; Kantekure, K.; Zhang, P.; Woetmann, A.; Cheng, M.; Odum, N.; et al. Oncogenic kinase NPM/ALK induces expression of HIF1α mRNA. Oncogene 2011, 30, 1372–1378. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, P.; Davis, D.A.; Veeranna, R.P.; Carey, R.F.; Viollet, C.; Yarchoan, R. Hypoxia-inducible factor-1 alpha as a therapeutic target for primary effusion lymphoma. PLoS Pathog. 2017, 13, e1006628. [Google Scholar] [CrossRef]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef]
- Sabò, A.; Kress, T.R.; Pelizzola, M.; de Pretis, S.; Gorski, M.M.; Tesi, A.; Morelli, M.J.; Bora, P.; Doni, M.; Verrecchia, A.; et al. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature 2014, 511, 488–492. [Google Scholar] [CrossRef]
- Ott, G.; Rosenwald, A.; Campo, E. Understanding MYC-driven aggressive B-cell lymphomas: Pathogenesis and classification. Blood 2013, 122, 3884–3891. [Google Scholar] [CrossRef]
- Leucci, E.; Cocco, M.; Onnis, A.; De Falco, G.; van Cleef, P.; Bellan, C.; van Rijk, A.; Nyagol, J.; Byakika, B.; Lazzi, S.; et al. MYC translocation-negative classical Burkitt lymphoma cases: An alternative pathogenetic mechanism involving miRNA deregulation. J. Pathol. 2008, 216, 440–450. [Google Scholar] [CrossRef]
- Ruzinova, M.B.; Caron, T.; Rodig, S.J. Altered subcellular localization of c-Myc protein identifies aggressive B-cell lymphomas harboring a c-MYC translocation. Am. J. Surg. Pathol. 2010, 34, 882–891. [Google Scholar] [CrossRef]
- Calvo-Vidal, M.N.; Zamponi, N.; Krumsiek, J.; Stockslager, M.A.; Revuelta, M.V.; Phillip, J.M.; Marullo, R.; Tikhonova, E.; Kotlov, N.; Patel, J.; et al. Oncogenic HSP90 Facilitates Metabolic Alterations in Aggressive B-cell Lymphomas. Cancer Res. 2021, 81, 5202–5216. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, D.; Zhang, L.; Jiang, S.; Liang, J.; Narita, Y.; Hou, I.; Zhong, Q.; Zheng, Z.; Xiao, H.; et al. RNA Sequencing Analyses of Gene Expression during Epstein-Barr Virus Infection of Primary B Lymphocytes. J. Virol. 2019, 93, e00226-19. [Google Scholar] [CrossRef] [PubMed]
- Klein, G. Burkitt lymphoma—A stalking horse for cancer research? Semin. Cancer Biol. 2009, 19, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Elenitoba-Johnson, K.S.; Jenson, S.D.; Abbott, R.T.; Palais, R.A.; Bohling, S.D.; Lin, Z.; Tripp, S.; Shami, P.J.; Wang, L.Y.; Coupland, R.W.; et al. Involvement of multiple signaling pathways in follicular lymphoma transformation: p38-mitogen-activated protein kinase as a target for therapy. Proc. Natl. Acad. Sci. USA 2003, 100, 7259–7264. [Google Scholar] [CrossRef]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef]
- Xiong, J.; Wang, L.; Fei, X.C.; Jiang, X.F.; Zheng, Z.; Zhao, Y.; Wang, C.F.; Li, B.; Chen, S.J.; Janin, A.; et al. MYC is a positive regulator of choline metabolism and impedes mitophagy-dependent necroptosis in diffuse large B-cell lymphoma. Blood Cancer J. 2017, 7, e582. [Google Scholar] [CrossRef]
- Wong, K.K.; Engelman, J.A.; Cantley, L.C. Targeting the PI3K signaling pathway in cancer. Curr. Opin. Genet. Dev. 2010, 20, 87–90. [Google Scholar] [CrossRef]
- Westin, J.R. Status of PI3K/Akt/mTOR pathway inhibitors in lymphoma. Clin. Lymphoma Myeloma Leuk. 2014, 14, 335–342. [Google Scholar] [CrossRef]
- Carnero, A. The PKB/AKT pathway in cancer. Curr. Pharm. Des 2010, 16, 34–44. [Google Scholar] [CrossRef]
- Kittipongdaja, W.; Wu, X.; Garner, J.; Liu, X.; Komas, S.M.; Hwang, S.T.; Schieke, S.M. Rapamycin Suppresses Tumor Growth and Alters the Metabolic Phenotype in T-Cell Lymphoma. J. Investig. Dermatol 2015, 135, 2301–2308. [Google Scholar] [CrossRef] [PubMed]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.C.; Avraamides, C.J.; Dippold, H.C.; Franco, I.; Foubert, P.; Ellies, L.G.; Acevedo, L.M.; Manglicmot, J.R.; Song, X.; Wrasidlo, W.; et al. Receptor tyrosine kinases and TLR/IL1Rs unexpectedly activate myeloid cell PI3kγ, a single convergent point promoting tumor inflammation and progression. Cancer Cell 2011, 19, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef]
- Fan, Y.; Dickman, K.G.; Zong, W.X. Akt and c-Myc differentially activate cellular metabolic programs and prime cells to bioenergetic inhibition. J. Biol. Chem. 2010, 285, 7324–7333. [Google Scholar] [CrossRef]
- Robey, R.B.; Hay, N. Is Akt the “Warburg kinase”?-Akt-energy metabolism interactions and oncogenesis. Semin. Cancer Biol. 2009, 19, 25–31. [Google Scholar] [CrossRef]
- Inoki, K.; Corradetti, M.N.; Guan, K.L. Dysregulation of the TSC-mTOR pathway in human disease. Nat. Genet. 2005, 37, 19–24. [Google Scholar] [CrossRef]
- Duvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. Regulation of mTORC1 and its impact on gene expression at a glance. J. Cell Sci. 2013, 126 Pt 8, 1713–1719. [Google Scholar] [CrossRef]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef]
- Blachly, J.S.; Baiocchi, R.A. Targeting PI3-kinase (PI3K), AKT and mTOR axis in lymphoma. Br. J. Haematol. 2014, 167, 19–32. [Google Scholar] [CrossRef]
- Rasul, A.; Ding, C.; Li, X.; Khan, M.; Yi, F.; Ali, M.; Ma, T. Dracorhodin perchlorate inhibits PI3K/Akt and NF-κB activation, up-regulates the expression of p53, and enhances apoptosis. Apoptosis 2012, 17, 1104–1119. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Levine, A.J. The regulation of energy metabolism and the IGF-1/mTOR pathways by the p53 protein. Trends Cell Biol. 2010, 20, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Zhang, Y.; Pan, J.; Chen, S.; Lu, S.; Shen, F.; Huang, Z. Triptolide dysregulates glucose uptake via inhibition of IKKβ-NF-κB pathway by p53 activation in cardiomyocytes. Toxicol. Lett. 2020, 318, 1–11. [Google Scholar] [CrossRef]
- Yeung, S.J.; Pan, J.; Lee, M.H. Roles of p53, MYC and HIF-1 in regulating glycolysis-the seventh hallmark of cancer. Cell Mol. Life Sci. 2008, 65, 3981–3999. [Google Scholar] [CrossRef]
- Xu-Monette, Z.Y.; Medeiros, L.J.; Li, Y.; Orlowski, R.Z.; Andreeff, M.; Bueso-Ramos, C.E.; Greiner, T.C.; McDonnell, T.J.; Young, K.H. Dysfunction of the TP53 tumor suppressor gene in lymphoid malignancies. Blood 2012, 119, 3668–3683. [Google Scholar] [CrossRef]
- Xu-Monette, Z.Y.; Wu, L.; Visco, C.; Tai, Y.C.; Tzankov, A.; Liu, W.M.; Montes-Moreno, S.; Dybkaer, K.; Chiu, A.; Orazi, A.; et al. Mutational profile and prognostic significance of TP53 in diffuse large B-cell lymphoma patients treated with R-CHOP: Report from an International DLBCL Rituximab-CHOP Consortium Program Study. Blood 2012, 120, 3986–3996. [Google Scholar] [CrossRef]
- Wang, X.J.; Medeiros, L.J.; Bueso-Ramos, C.E.; Tang, G.; Wang, S.; Oki, Y.; Desai, P.; Khoury, J.D.; Miranda, R.N.; Tang, Z.; et al. P53 expression correlates with poorer survival and augments the negative prognostic effect of MYC rearrangement, expression or concurrent MYC/BCL2 expression in diffuse large B-cell lymphoma. Mod. Pathol. 2017, 30, 194–203. [Google Scholar] [CrossRef]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef]
- Faubert, B.; Boily, G.; Izreig, S.; Griss, T.; Samborska, B.; Dong, Z.; Dupuy, F.; Chambers, C.; Fuerth, B.J.; Viollet, B.; et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 2013, 17, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. AMP-activated protein kinase: An energy sensor that regulates all aspects of cell function. Genes Dev. 2011, 25, 1895–1908. [Google Scholar] [CrossRef]
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, A.E.; Demanelis, K.; Fu, A.; Zheng, T.; Zhu, Y. Association of AMP-activated protein kinase with risk and progression of non-Hodgkin lymphoma. Cancer Epidemiol. Biomark. Prev. 2013, 22, 736–744. [Google Scholar] [CrossRef] [PubMed]
- Houde, V.P.; Donzelli, S.; Sacconi, A.; Galic, S.; Hammill, J.A.; Bramson, J.L.; Foster, R.A.; Tsakiridis, T.; Kemp, B.E.; Grasso, G.; et al. AMPK β1 reduces tumor progression and improves survival in p53 null mice. Mol. Oncol. 2017, 11, 1143–1155. [Google Scholar] [CrossRef]
- Ying, H.; Wang, Z.; Zhang, Y.; Yang, T.Y.; Ding, Z.H.; Liu, S.Y.; Shao, J.; Liu, Y.; Fan, X.B. Capsaicin induces apoptosis in human osteosarcoma cells through AMPK-dependent and AMPK-independent signaling pathways. Mol. Cell Biochem. 2013, 384, 229–237. [Google Scholar] [CrossRef]
- Shi, W.Y.; Xiao, D.; Wang, L.; Dong, L.H.; Yan, Z.X.; Shen, Z.X.; Chen, S.J.; Chen, Y.; Zhao, W.L. Therapeutic metformin/AMPK activation blocked lymphoma cell growth via inhibition of mTOR pathway and induction of autophagy. Cell Death Dis. 2012, 3, e275. [Google Scholar] [CrossRef]
- Singh, A.R.; Gu, J.J.; Zhang, Q.; Torka, P.; Sundaram, S.; Mavis, C.; Hernandez-Ilizaliturri, F.J. Metformin sensitizes therapeutic agents and improves outcome in pre-clinical and clinical diffuse large B-cell lymphoma. Cancer Metab. 2020, 8, 10. [Google Scholar] [CrossRef]
- Montraveta, A.; Xargay-Torrent, S.; López-Guerra, M.; Rosich, L.; Pérez-Galán, P.; Salaverria, I.; Beà, S.; Kalko, S.G.; de Frias, M.; Campàs, C.; et al. Synergistic anti-tumor activity of acadesine (AICAR) in combination with the anti-CD20 monoclonal antibody rituximab in in vivo and in vitro models of mantle cell lymphoma. Oncotarget 2014, 5, 726–739. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Chatterjee, A.; Kogan, D.; Patel, D.; Foster, D.A. 5-Aminoimidazole-4-carboxamide-1-β-4-ribofuranoside (AICAR) enhances the efficacy of rapamycin in human cancer cells. Cell Cycle 2015, 14, 3331–3339. [Google Scholar] [CrossRef]
- Liang, S.; Medina, E.A.; Li, B.; Habib, S.L. Preclinical evidence of the enhanced effectiveness of combined rapamycin and AICAR in reducing kidney cancer. Mol. Oncol. 2018, 12, 1917–1934. [Google Scholar] [CrossRef]
- Jeong, S.M.; Lee, A.; Lee, J.; Haigis, M.C. SIRT4 protein suppresses tumor formation in genetic models of Myc-induced B cell lymphoma. J. Biol. Chem. 2014, 289, 4135–4144. [Google Scholar] [CrossRef]
- Bonglack, E.N.; Messinger, J.E.; Cable, J.M.; Ch’ng, J.; Parnell, K.M.; Reinoso-Vizcaíno, N.M.; Barry, A.P.; Russell, V.S.; Dave, S.S.; Christofk, H.R.; et al. Monocarboxylate transporter antagonism reveals metabolic vulnerabilities of viral-driven lymphomas. Proc. Natl. Acad. Sci. USA 2021, 118, e2022495118. [Google Scholar] [CrossRef] [PubMed]
- Piccaluga, P.P.; Weber, A.; Ambrosio, M.R.; Ahmed, Y.; Leoncini, L. Epstein-Barr Virus-Induced Metabolic Rearrangements in Human B-Cell Lymphomas. Front. MicroBiol. 2018, 9, 1233. [Google Scholar] [CrossRef]
- Portis, T.; Dyck, P.; Longnecker, R. Epstein-Barr Virus (EBV) LMP2A induces alterations in gene transcription similar to those observed in Reed-Sternberg cells of Hodgkin lymphoma. Blood 2003, 102, 4166–4178. [Google Scholar] [CrossRef] [PubMed]
- Burton, E.M.; Gewurz, B.E. Epstein-Barr virus oncoprotein-driven B cell metabolism remodeling. PLoS Pathog. 2022, 18, e1010254. [Google Scholar]
- Mrozek-Gorska, P.; Buschle, A.; Pich, D.; Schwarzmayr, T.; Fechtner, R.; Scialdone, A.; Hammerschmidt, W. Epstein-Barr virus reprograms human B lymphocytes immediately in the prelatent phase of infection. Proc. Natl. Acad. Sci. USA 2019, 116, 16046–16055. [Google Scholar] [CrossRef] [PubMed]
- Mawson, A.R.; Majumdar, S. Malaria, Epstein-Barr virus infection and the pathogenesis of Burkitt’s lymphoma. Int. J. Cancer 2017, 141, 1849–1855. [Google Scholar] [CrossRef] [PubMed]
- Hulse, M.; Johnson, S.M.; Boyle, S.; Caruso, L.B.; Tempera, I. Epstein-Barr Virus-Encoded Latent Membrane Protein 1 and B-Cell Growth Transformation Induce Lipogenesis through Fatty Acid Synthase. J. Virol. 2021, 9, e01857-20. [Google Scholar] [CrossRef]
- Pujals, A.; Favre, L.; Pioche-Durieu, C.; Robert, A.; Meurice, G.; Le Gentil, M.; Chelouah, S.; Martin-Garcia, N.; Le Cam, E.; Guettier, C.; et al. Constitutive autophagy contributes to resistance to TP53-mediated apoptosis in Epstein-Barr virus-positive latency III B-cell lymphoproliferations. Autophagy 2015, 11, 2275–2287. [Google Scholar] [CrossRef]
- Nowicki, S.; Gottlieb, E. Oncometabolites: Tailoring our genes. FEBS J. 2015, 282, 2796–2805. [Google Scholar] [CrossRef] [PubMed]
- Zong, W.X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Cardaci, S.; Zheng, L.; MacKay, G.; van den Broek, N.J.; MacKenzie, E.D.; Nixon, C.; Stevenson, D.; Tumanov, S.; Bulusu, V.; Kamphorst, J.J.; et al. Pyruvate carboxylation enables growth of SDH-deficient cells by supporting aspartate biosynthesis. Nat. Cell Biol. 2015, 17, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Lussey-Lepoutre, C.; Hollinshead, K.E.; Ludwig, C.; Menara, M.; Morin, A.; Castro-Vega, L.J.; Parker, S.J.; Janin, M.; Martinelli, C.; Ottolenghi, C.; et al. Loss of succinate dehydrogenase activity results in dependency on pyruvate carboxylation for cellular anabolism. Nat. Commun. 2015, 6, 8784. [Google Scholar] [CrossRef]
- Carey, B.W.; Finley, L.W.; Cross, J.R.; Allis, C.D.; Thompson, C.B. Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 2015, 518, 413–416. [Google Scholar] [CrossRef]
- Chin, R.M.; Fu, X.; Pai, M.Y.; Vergnes, L.; Hwang, H.; Deng, G.; Diep, S.; Lomenick, B.; Meli, V.S.; Monsalve, G.C.; et al. The metabolite α-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature 2014, 510, 397–401. [Google Scholar] [CrossRef]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2011, 481, 385–388. [Google Scholar] [CrossRef]
- Wang, F.; Travins, J.; DeLaBarre, B.; Penard-Lacronique, V.; Schalm, S.; Hansen, E.; Straley, K.; Kernytsky, A.; Liu, W.; Gliser, C.; et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 2013, 340, 622–626. [Google Scholar] [CrossRef]
- Losman, J.A.; Looper, R.E.; Koivunen, P.; Lee, S.; Schneider, R.K.; McMahon, C.; Cowley, G.S.; Root, D.E.; Ebert, B.L.; Kaelin, W.G., Jr. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 2013, 339, 1621–1625. [Google Scholar] [CrossRef]
- Dang, L.; Yen, K.; Attar, E.C. IDH mutations in cancer and progress toward development of targeted therapeutics. Ann. Oncol. 2016, 27, 599–608. [Google Scholar] [CrossRef]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.D.; et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 2009, 361, 1058–1066. [Google Scholar] [CrossRef]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.; Li, X.S.; Woon, E.C.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469. [Google Scholar] [CrossRef]
- Janke, R.; Iavarone, A.T.; Rine, J. Oncometabolite D-2-Hydroxyglutarate enhances gene silencing through inhibition of specific H3K36 histone demethylases. Elife 2017, 6, e22451. [Google Scholar] [CrossRef] [PubMed]
- Koivunen, P.; Lee, S.; Duncan, C.G.; Lopez, G.; Lu, G.; Ramkissoon, S.; Losman, J.A.; Joensuu, P.; Bergmann, U.; Gross, S.; et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 2012, 483, 484–488. [Google Scholar] [CrossRef]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef]
- Turcan, S.; Fabius, A.W.; Borodovsky, A.; Pedraza, A.; Brennan, C.; Huse, J.; Viale, A.; Riggins, G.J.; Chan, T.A. Efficient induction of differentiation and growth inhibition in IDH1 mutant glioma cells by the DNMT Inhibitor Decitabine. Oncotarget 2013, 4, 1729–1736. [Google Scholar] [CrossRef]
- Geschwind, J.F.; Georgiades, C.S.; Ko, Y.H.; Pedersen, P.L. Recently elucidated energy catabolism pathways provide opportunities for novel treatments in hepatocellular carcinoma. Expert Rev. Anticancer Ther. 2004, 4, 449–457. [Google Scholar] [CrossRef]
- Pang, Y.Y.; Wang, T.; Chen, F.Y.; Wu, Y.L.; Shao, X.; Xiao, F.; Huang, H.H.; Zhong, H.; Zhong, J.H. Glycolytic inhibitor 2-deoxy-d-glucose suppresses cell proliferation and enhances methylprednisolone sensitivity in non-Hodgkin lymphoma cells through down-regulation of HIF-1α and c-MYC. Leuk. Lymphoma 2015, 56, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Zagorodna, O.; Martin, S.M.; Rutkowski, D.T.; Kuwana, T.; Spitz, D.R.; Knudson, C.M. 2-deoxyglucose-induced toxicity is regulated by Bcl-2 family members and is enhanced by antagonizing Bcl-2 in lymphoma cell lines. Oncogene 2012, 31, 2738–2749. [Google Scholar] [CrossRef]
- Robinson, G.L.; Dinsdale, D.; Macfarlane, M.; Cain, K. Switching from aerobic glycolysis to oxidative phosphorylation modulates the sensitivity of mantle cell lymphoma cells to TRAIL. Oncogene 2012, 31, 4996–5006. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.H.; Pedersen, P.L.; Geschwind, J.F. Glucose catabolism in the rabbit VX2 tumor model for liver cancer: Characterization and targeting hexokinase. Cancer Lett. 2001, 173, 83–91. [Google Scholar] [CrossRef]
- Schaefer, N.G.; Geschwind, J.F.; Engles, J.; Buchanan, J.W.; Wahl, R.L. Systemic administration of 3-bromopyruvate in treating disseminated aggressive lymphoma. Transl. Res. 2012, 159, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Pandey, S.K.; Kumar, A.; Kujur, P.K.; Singh, R.P.; Singh, S.M. Antitumor and chemosensitizing action of 3-bromopyruvate: Implication of deregulated metabolism. Chem. Biol. Interact. 2017, 270, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, L.M.; Forshell, T.Z.; Rimpi, S.; Kreutzer, C.; Pretsch, W.; Bornkamm, G.W.; Nilsson, J.A. Mouse genetics suggests cell-context dependency for Myc-regulated metabolic enzymes during tumorigenesis. PLoS Genet. 2012, 8, e1002573. [Google Scholar] [CrossRef]
- Curtis, N.J.; Mooney, L.; Hopcroft, L.; Michopoulos, F.; Whalley, N.; Zhong, H.; Murray, C.; Logie, A.; Revill, M.; Byth, K.F.; et al. Pre-clinical pharmacology of AZD3965, a selective inhibitor of MCT1, DLBCL, NHL and Burkitt’s lymphoma anti-tumor activity. Oncotarget 2017, 8, 69219–69236. [Google Scholar] [CrossRef]
- Beloueche-Babari, M.; Wantuch, S.; Casals Galobart, T.; Koniordou, M.; Parkes, H.G.; Arunan, V.; Chung, Y.L.; Eykyn, T.R.; Smith, P.D.; Leach, M.O. MCT1 Inhibitor AZD3965 Increases Mitochondrial Metabolism, Facilitating Combination Therapy and Noninvasive Magnetic Resonance Spectroscopy. Cancer Res. 2017, 77, 5913–5924. [Google Scholar] [CrossRef]
- Schultz, I.R.; Shangraw, R.E. Effect of short-term drinking water exposure to dichloroacetate on its pharmacokinetics and oral bioavailability in human volunteers: A stable isotope study. Toxicol. Sci. 2006, 92, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Schwartz, R.; Diamond, M.P.; Raju, R.P. A Combination Treatment Strategy for Hemorrhagic Shock in a Rat Model Modulates Autophagy. Front. Med. 2019, 6, 281. [Google Scholar] [CrossRef]
- Kumar, A.; Kant, S.; Singh, S.M. Novel molecular mechanisms of antitumor action of dichloroacetate against T cell lymphoma: Implication of altered glucose metabolism, pH homeostasis and cell survival regulation. Chem. Biol. Interact. 2012, 199, 29–37. [Google Scholar] [CrossRef]
- Grassian, A.R.; Parker, S.J.; Davidson, S.M.; Divakaruni, A.S.; Green, C.R.; Zhang, X.; Slocum, K.L.; Pu, M.; Lin, F.; Vickers, C.; et al. IDH1 mutations alter citric acid cycle metabolism and increase dependence on oxidative mitochondrial metabolism. Cancer Res. 2014, 74, 3317–3331. [Google Scholar] [CrossRef] [PubMed]
- Tateishi, K.; Wakimoto, H.; Iafrate, A.J.; Tanaka, S.; Loebel, F.; Lelic, N.; Wiederschain, D.; Bedel, O.; Deng, G.; Zhang, B.; et al. Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell 2015, 28, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, K.; Suzuki, H.I.; Fujimura, T.; Ono, A.; Kaga, N.; Isobe, Y.; Sasaki, M.; Taka, H.; Miyazono, K.; Komatsu, N. A clinically attainable dose of L-asparaginase targets glutamine addiction in lymphoid cell lines. Cancer Sci. 2015, 106, 1534–1543. [Google Scholar] [CrossRef] [PubMed]
- Dengler, M.A.; Weilbacher, A.; Gutekunst, M.; Staiger, A.M.; Vöhringer, M.C.; Horn, H.; Ott, G.; Aulitzky, W.E.; van der Kuip, H. Discrepant NOXA (PMAIP1) transcript and NOXA protein levels: A potential Achilles’ heel in mantle cell lymphoma. Cell Death Dis. 2014, 5, e1013. [Google Scholar] [CrossRef] [PubMed]
- Bastos, D.C.; Paupert, J.; Maillard, C.; Seguin, F.; Carvalho, M.A.; Agostini, M.; Coletta, R.D.; Noël, A.; Graner, E. Effects of fatty acid synthase inhibitors on lymphatic vessels: An in vitro and in vivo study in a melanoma model. Lab. Investig. 2017, 97, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Molyneux, E.; Merrick, B.; Khanim, F.L.; Banda, K.; Dunn, J.A.; Iqbal, G.; Bunce, C.M.; Drayson, M.T. Bezafibrate and medroxyprogesterone acetate in resistant and relapsed endemic Burkitt lymphoma in Malawi; an open-label, single-arm, phase 2 study (ISRCTN34303497). Br. J. Haematol. 2014, 164, 888–890. [Google Scholar] [CrossRef]
- Huang, J.; Das, S.K.; Jha, P.; Al Zoughbi, W.; Schauer, S.; Claudel, T.; Sexl, V.; Vesely, P.; Birner-Gruenberger, R.; Kratky, D.; et al. The PPARα agonist fenofibrate suppresses B-cell lymphoma in mice by modulating lipid metabolism. BioChim. Biophys. Acta 2013, 1831, 1555–1565. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, S.; Evens, A.M.; Prachand, S.; Schumacker, P.T.; Gordon, L.I. Paradoxical regulation of hypoxia inducible factor-1α (HIF-1α) by histone deacetylase inhibitor in diffuse large B-cell lymphoma. PLoS ONE 2013, 8, e81333. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Yu, D.; Liu, J.; Yang, K.; Wu, G.; Liu, H. Antitumor activity of SAHA, a novel histone deacetylase inhibitor, against murine B cell lymphoma A20 cells in vitro and in vivo. Tumour Biol. 2015, 36, 5051–5061. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.Y.; Wang, H.; Teriete, P.; Yap, J.L.; Chen, L.; Lanning, M.E.; Hu, A.; Lambert, L.J.; Holien, T.; Sundan, A.; et al. Perturbation of the c-Myc-Max protein-protein interaction via synthetic α-helix mimetics. J. Med. Chem. 2015, 58, 3002–3024. [Google Scholar] [CrossRef]
- Huang, M.J.; Cheng, Y.C.; Liu, C.R.; Lin, S.; Liu, H.E. A small-molecule c-Myc inhibitor, 10058-F4, induces cell-cycle arrest, apoptosis, and myeloid differentiation of human acute myeloid leukemia. Exp. Hematol. 2006, 34, 1480–1489. [Google Scholar] [CrossRef]
- Whitfield, J.R.; Beaulieu, M.E.; Soucek, L. Strategies to Inhibit Myc and Their Clinical Applicability. Front. Cell Dev. Biol. 2017, 5, 10. [Google Scholar] [CrossRef]
- Broecker-Preuss, M.; Becher-Boveleth, N.; Bockisch, A.; Dührsen, U.; Müller, S. Regulation of glucose uptake in lymphoma cell lines by c-MYC- and PI3K-dependent signaling pathways and impact of glycolytic pathways on cell viability. J. Transl. Med. 2017, 15, 158. [Google Scholar] [CrossRef]
- Erdmann, T.; Klener, P.; Lynch, J.T.; Grau, M.; Vočková, P.; Molinsky, J.; Tuskova, D.; Hudson, K.; Polanska, U.M.; Grondine, M.; et al. Sensitivity to PI3K and AKT inhibitors is mediated by divergent molecular mechanisms in subtypes of DLBCL. Blood 2017, 130, 310–322. [Google Scholar] [CrossRef]
- Mediani, L.; Gibellini, F.; Bertacchini, J.; Frasson, C.; Bosco, R.; Accordi, B.; Basso, G.; Bonora, M.; Calabrò, M.L.; Mattiolo, A.; et al. Reversal of the glycolytic phenotype of primary effusion lymphoma cells by combined targeting of cellular metabolism and PI3K/Akt/ mTOR signaling. Oncotarget 2016, 7, 5521–5537. [Google Scholar] [CrossRef]
- Yahiaoui, A.; Meadows, S.A.; Sorensen, R.A.; Cui, Z.H.; Keegan, K.S.; Brockett, R.; Chen, G.; Quéva, C.; Li, L.; Tannheimer, S.L. PI3Kδ inhibitor idelalisib in combination with BTK inhibitor ONO/GS-4059 in diffuse large B cell lymphoma with acquired resistance to PI3Kδ and BTK inhibitors. PLoS ONE 2017, 12, e0171221. [Google Scholar] [CrossRef]
- Ferreira, A.C.; Robaina, M.C.; Rezende, L.M.; Severino, P.; Klumb, C.E. Histone deacetylase inhibitor prevents cell growth in Burkitt’s lymphoma by regulating PI3K/Akt pathways and leads to upregulation of miR-143, miR-145, and miR-101. Ann. Hematol. 2014, 93, 983–993. [Google Scholar] [PubMed]
- Sekihara, K.; Saitoh, K.; Han, L.; Ciurea, S.; Yamamoto, S.; Kikkawa, M.; Kazuno, S.; Taka, H.; Kaga, N.; Arai, H.; et al. Targeting mantle cell lymphoma metabolism and survival through simultaneous blockade of mTOR and nuclear transporter exportin-1. Oncotarget 2017, 8, 34552–34564. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, A.P.; Bhende, P.M.; Sin, S.H.; Roy, D.; Dittmer, D.P.; Damania, B. Dual inhibition of PI3K and mTOR inhibits autocrine and paracrine proliferative loops in PI3K/Akt/mTOR-addicted lymphomas. Blood 2010, 115, 4455–4463. [Google Scholar] [CrossRef] [PubMed]
- Tarantelli, C.; Gaudio, E.; Arribas, A.J.; Kwee, I.; Hillmann, P.; Rinaldi, A.; Cascione, L.; Spriano, F.; Bernasconi, E.; Guidetti, F.; et al. PQR309 Is a Novel Dual PI3K/mTOR Inhibitor with Preclinical Antitumor Activity in Lymphomas as a Single Agent and in Combination Therapy. Clin. Cancer Res. 2018, 24, 120–129. [Google Scholar] [CrossRef]
- Choudhary, G.S.; Al-Harbi, S.; Mazumder, S.; Hill, B.T.; Smith, M.R.; Bodo, J.; Hsi, E.D.; Almasan, A. MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell Death Dis. 2015, 6, e1593. [Google Scholar] [CrossRef]
- Campàs, C.; Santidrián, A.F.; Domingo, A.; Gil, J. Acadesine induces apoptosis in B cells from mantle cell lymphoma and splenic marginal zone lymphoma. Leukemia 2005, 19, 292–294. [Google Scholar] [CrossRef]
- Campàs, C.; Lopez, J.M.; Santidrián, A.F.; Barragán, M.; Bellosillo, B.; Colomer, D.; Gil, J. Acadesine activates AMPK and induces apoptosis in B-cell chronic lymphocytic leukemia cells but not in T lymphocytes. Blood 2003, 101, 3674–3680. [Google Scholar] [CrossRef]
- Santidrián, A.F.; González-Gironès, D.M.; Iglesias-Serret, D.; Coll-Mulet, L.; Cosialls, A.M.; de Frias, M.; Campàs, C.; González-Barca, E.; Alonso, E.; Labi, V.; et al. AICAR induces apoptosis independently of AMPK and p53 through up-regulation of the BH3-only proteins BIM and NOXA in chronic lymphocytic leukemia cells. Blood 2010, 116, 3023–3032. [Google Scholar] [CrossRef]
- Sengupta, T.K.; Leclerc, G.M.; Hsieh-Kinser, T.T.; Leclerc, G.J.; Singh, I.; Barredo, J.C. Cytotoxic effect of 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside (AICAR) on childhood acute lymphoblastic leukemia (ALL) cells: Implication for targeted therapy. Mol. Cancer 2007, 6, 46. [Google Scholar] [CrossRef]
- Rosilio, C.; Lounnas, N.; Nebout, M.; Imbert, V.; Hagenbeek, T.; Spits, H.; Asnafi, V.; Pontier-Bres, R.; Reverso, J.; Michiels, J.F.; et al. The metabolic perturbators metformin, phenformin and AICAR interfere with the growth and survival of murine PTEN-deficient T cell lymphomas and human T-ALL/T-LL cancer cells. Cancer Lett. 2013, 336, 114–126. [Google Scholar] [CrossRef]
- Offman, M.N.; Krol, M.; Patel, N.; Krishnan, S.; Liu, J.; Saha, V.; Bates, P.A. Rational engineering of L-asparaginase reveals importance of dual activity for cancer cell toxicity. Blood 2011, 117, 1614–1621. [Google Scholar] [CrossRef] [PubMed]
- Abd-Aziz, N.; Stanbridge, E.J.; Shafee, N. Bortezomib attenuates HIF-1- but not HIF-2-mediated transcriptional activation. Oncol. Lett. 2015, 10, 2192–2196. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, R.; Ravi, D.; Evens, A.M.; Parekkadan, B. Scaffold-mediated switching of lymphoma metabolism in culture. Cancer Metab. 2022, 10, 15. [Google Scholar] [CrossRef]
- Welcker, M.; Orian, A.; Jin, J.; Grim, J.E.; Harper, J.W.; Eisenman, R.N.; Clurman, B.E. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc. Natl. Acad. Sci. USA 2004, 101, 9085–9090. [Google Scholar] [CrossRef]
- Bahram, F.; von der Lehr, N.; Cetinkaya, C.; Larsson, L.G. c-Myc hot spot mutations in lymphomas result in inefficient ubiquitination and decreased proteasome-mediated turnover. Blood 2000, 95, 2104–2110. [Google Scholar] [CrossRef]
- Popov, N.; Wanzel, M.; Madiredjo, M.; Zhang, D.; Beijersbergen, R.; Bernards, R.; Moll, R.; Elledge, S.J.; Eilers, M. The ubiquitin-specific protease USP28 is required for MYC stability. Nat. Cell Biol. 2007, 9, 765–774. [Google Scholar] [CrossRef]
- Xie, H.; Tang, C.H.; Song, J.H.; Mancuso, A.; Del Valle, J.R.; Cao, J.; Xiang, Y.; Dang, C.V.; Lan, R.; Sanchez, D.J.; et al. IRE1α RNase-dependent lipid homeostasis promotes survival in Myc-transformed cancers. J. Clin. Investig. 2018, 128, 1300–1316. [Google Scholar] [CrossRef] [PubMed]
- Dreyling, M.; Santoro, A.; Mollica, L.; Leppä, S.; Follows, G.A.; Lenz, G.; Kim, W.S.; Nagler, A.; Panayiotidis, P.; Demeter, J.; et al. Phosphatidylinositol 3-Kinase Inhibition by Copanlisib in Relapsed or Refractory Indolent Lymphoma. J. Clin. Oncol. 2017, 35, 3898–3905. [Google Scholar] [CrossRef]
- Horwitz, S.M.; Koch, R.; Porcu, P.; Oki, Y.; Moskowitz, A.; Perez, M.; Myskowski, P.; Officer, A.; Jaffe, J.D.; Morrow, S.N.; et al. Activity of the PI3K-δ,γ inhibitor duvelisib in a phase 1 trial and preclinical models of T-cell lymphoma. Blood 2018, 131, 888–898. [Google Scholar] [CrossRef]
- Gu, L.; Gao, J.; Li, Q.; Zhu, Y.P.; Jia, C.S.; Fu, R.Y.; Chen, Y.; Liao, Q.K.; Ma, Z. Rapamycin reverses NPM-ALK-induced glucocorticoid resistance in lymphoid tumor cells by inhibiting mTOR signaling pathway, enhancing G1 cell cycle arrest and apoptosis. Leukemia 2008, 22, 2091–2096. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Xie, L.; Zuo, C.; Ma, Z.; Zhang, Y.; Zhu, Y.; Gao, J. Targeting mTOR/p70S6K/glycolysis signaling pathway restores glucocorticoid sensitivity to 4E-BP1 null Burkitt Lymphoma. BMC Cancer 2015, 15, 529. [Google Scholar] [CrossRef] [PubMed]
- Drew, B.G.; Kingwell, B.A. Acadesine, an adenosine-regulating agent with the potential for widespread indications. Expert Opin. Pharmacother. 2008, 9, 2137–2144. [Google Scholar] [CrossRef]
- Van Den Neste, E.; Van den Berghe, G.; Bontemps, F. AICA-riboside (acadesine), an activator of AMP-activated protein kinase with potential for application in hematologic malignancies. Expert Opin. Investig. Drugs 2010, 19, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Vakana, E.; Platanias, L.C. AMPK in BCR-ABL expressing leukemias. Regulatory effects and therapeutic implications. Oncotarget 2011, 2, 1322–1328. [Google Scholar] [CrossRef]
- Liu, X.; Chhipa, R.R.; Pooya, S.; Wortman, M.; Yachyshin, S.; Chow, L.M.; Kumar, A.; Zhou, X.; Sun, Y.; Quinn, B.; et al. Discrete mechanisms of mTOR and cell cycle regulation by AMPK agonists independent of AMPK. Proc. Natl. Acad. Sci. USA 2014, 111, E435–E444. [Google Scholar] [CrossRef] [PubMed]
- Ly, P.; Kim, S.B.; Kaisani, A.A.; Marian, G.; Wright, W.E.; Shay, J.W. Aneuploid human colonic epithelial cells are sensitive to AICAR-induced growth inhibition through EGFR degradation. Oncogene 2013, 32, 3139–3146. [Google Scholar] [CrossRef]
- Jose, C.; Bellance, N.; Chatelain, E.H.; Benard, G.; Nouette-Gaulain, K.; Rossignol, R. Antiproliferative activity of levobupivacaine and aminoimidazole carboxamide ribonucleotide on human cancer cells of variable bioenergetic profile. Mitochondrion 2012, 12, 100–109. [Google Scholar] [CrossRef]
- Rattan, R.; Giri, S.; Singh, A.K.; Singh, I. 5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside inhibits cancer cell proliferation in vitro and in vivo via AMP-activated protein kinase. J. Biol. Chem. 2005, 280, 39582–39593. [Google Scholar] [CrossRef]
- Van Den Neste, E.; Cazin, B.; Janssens, A.; González-Barca, E.; Terol, M.J.; Levy, V.; Pérez de Oteyza, J.; Zachee, P.; Saunders, A.; de Frias, M.; et al. Acadesine for patients with relapsed/refractory chronic lymphocytic leukemia (CLL): A multicenter phase I/II study. Cancer Chemo. Ther. Pharmacol. 2013, 71, 581–591. [Google Scholar] [CrossRef]
- Wang, H.; Shao, R.; Liu, W.; Tang, H.; Lu, Y. Identification of a prognostic metabolic gene signature in diffuse large B-cell lymphoma. J. Cell Mol. Med. 2021, 25, 7066–7077. [Google Scholar] [CrossRef]
- He, J.; Chen, Z.; Xue, Q.; Sun, P.; Wang, Y.; Zhu, C.; Shi, W. Identification of molecular subtypes and a novel prognostic model of diffuse large B-cell lymphoma based on a metabolism-associated gene signature. J. Transl. Med. 2022, 20, 186. [Google Scholar] [CrossRef]
- Zhang, J.; Medina-Cleghorn, D.; Bernal-Mizrachi, L.; Bracci, P.M.; Hubbard, A.; Conde, L.; Riby, J.; Nomura, D.K.; Skibola, C.F. The potential relevance of the endocannabinoid, 2-arachidonoylglycerol, in diffuse large B-cell lymphoma. Oncoscience 2016, 3, 31–41. [Google Scholar] [CrossRef][Green Version]
- Yang, F.; Du, J.; Zhang, H.; Ruan, G.; Xiang, J.; Wang, L.; Sun, H.; Guan, A.; Shen, G.; Liu, Y.; et al. Serum Metabolomics of Burkitt Lymphoma Mouse Models. PLoS ONE 2017, 12, e0170896. [Google Scholar] [CrossRef] [PubMed]
- Stenson, M.; Pedersen, A.; Hasselblom, S.; Nilsson-Ehle, H.; Karlsson, B.G.; Pinto, R.; Andersson, P.O. Serum nuclear magnetic resonance-based metabolomics and outcome in diffuse large B-cell lymphoma patients-a pilot study. Leuk. Lymphoma 2016, 57, 1814–1822. [Google Scholar] [CrossRef] [PubMed]
- Le, Y.; Shen, X.; Kang, H.; Wang, Q.; Li, K.; Zheng, J.; Yu, Y. Accelerated, untargeted metabolomics analysis of cutaneous T-cell lymphoma reveals metabolic shifts in plasma and tumor adjacent skins of xenograft mice. J. Mass Spectrom 2018, 53, 739. [Google Scholar] [CrossRef] [PubMed]
- Eberlin, L.S.; Gabay, M.; Fan, A.C.; Gouw, A.M.; Tibshirani, R.J.; Felsher, D.W.; Zare, R.N. Alteration of the lipid profile in lymphomas induced by MYC overexpression. Proc. Natl. Acad. Sci. USA 2014, 111, 10450–10455. [Google Scholar] [CrossRef] [PubMed]
- Leone, R.D.; Powell, J.D. Metabolism of immune cells in cancer. Nat. Rev. Cancer 2020, 20, 516–531. [Google Scholar] [CrossRef]
- Deshpande, R.P.; Sharma, S.; Watabe, K. The Confounders of Cancer Immunotherapy: Roles of Lifestyle, Metabolic Disorders and Sociological Factors. Cancers 2020, 12, 2983. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, R.; Gu, H.; Zhang, E.; Qu, J.; Cao, W.; Huang, X.; Yan, H.; He, J.; Cai, Z. Metabolic reprogramming in macrophage responses. Biomark. Res. 2021, 9, 1. [Google Scholar] [CrossRef]
- Dai, X.; Bu, X.; Gao, Y.; Guo, J.; Hu, J.; Jiang, C.; Zhang, Z.; Xu, K.; Duan, J.; He, S.; et al. Energy status dictates PD-L1 protein abundance and anti-tumor immunity to enable checkpoint blockade. Mol. Cell 2021, 81, 2317–2331.e6. [Google Scholar] [CrossRef]
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Myklebust, J.H.; Varghese, B.; Gill, S.; Jan, M.; Cha, A.C.; Chan, C.K.; Tan, B.T.; et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell 2010, 142, 699–713. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Xie, L.; Yu, L.; Wang, P. Targeting CD47-SIRPα axis for Hodgkin and non-Hodgkin lymphoma immunotherapy. Genes Dis. 2023, in press. [Google Scholar] [CrossRef]



| Agent | Target | Mechanism | Type of Diseases |
|---|---|---|---|
| methotrexate | DHFR | folate to THF conversion | prophylaxis and treatment of CNS lymphoma |
| IM156 | mitochondrial complex I inhibitor | mitochondrial oxidative phosphorylation and NADH oxidation | lymphomas (NCT03272256) |
| IACS-010759 | oxidative phosphorylation inhibitor | mitochondrial oxidative phosphorylation and NADH oxidation | R/R AML (NCT02882321) |
| AZD-3965 | MCT1 | mediate the bidirectional transport of lactatein and out of cells | DLBCL, BL (NCT01791595) |
| CPI-613 | mitochondria | oxidative metabolism | T-cell NHL (NCT04217317), R/R BL or HGBCL (NCT03793140) |
| IM156 | mitochondria | complex I | advanced lymphoma (NCT03272256) |
| IACS-010759 | mitochondria | complex I | R/R AML (NCT02882321) |
| ibrutinib | BTK inhibitor | downstream pro-proliferative kinase of BCR signal | MCL, CLL/SLL, WM, MZL, cGVHD (NCT02169180, NCT04771507, NCT02604511, NCT04212013, NCT05348096) |
| acalabrutinib | MCL, CLL/SLL, MZL (NCT02213926, NCT04505254, NCT04646395) | ||
| rapalogs | mTORC1 inhibitor | rapamycin analogues | MCL |
| temsirolimus | mTORC1 inhibitor | cell cycle arrest in the G1 phase, inhibits tumor angiogenesis by reducing synthesis of VEGF | MCL (NCT01078142, NCT01180049), HL (NCT01902160), R/R NHL (NCT01281917) |
| idelalisib | PI3Kδ inhibitor | against PI3Kδ isoforms | iNHL (NCT01282424), HL (NCT01393106), FL (NCT03568929), CLL (NCT03582098) |
| copanlisib | PI3Kδ and PI3Kα inhibitor | against PI3K-α and PI3K-δ isoforms | iNHL, DLBCL, MCL, PTCL (NCT05217914, NCT04433182, NCT04939272, NCT03877055, NCT03052933) |
| duvelisib | PI3Kδ and PI3Kγinhibitor | against PI3Kδ and PI3Kγ isoforms | CLL/SLL, DLBCL, PTCL (NCT02004522, NCT04890236, NCT04803201) |
| umbralisib | PI3Kδ and casein kinase-1 epsilon inhibitor | against PI3Kδisoforms and casein kinase-1 epsilon | MZL, FL, CLL, WM (NCT03919175, NCT03364231, NCT02535286) |
| L-asparaginase | asparagine | inhibit protein biosynthesis in lymphoblasts | ALL (NCT01518517, NCT00506597), NK/T cell lymphoma (NCT00854425) |
| Telaglenastat (CB-839) | glutaminase inhibitor | glutamine to glutamate conversion | NHL (NCT02071888), including MCL, WM, TCL |
| AG-270 | MAT2A | production of S-adenosylmethionine | advanced lymphoma (NCT03361358, NCT03435250) |
| Devimistat (CPI-613) | lipoate analog | mitochondrial oxidative metabolism | R/R T-cell NHL (NCT04217317) |
| Subtype | Control | Metabolites | Type of Samples | Origin | Method | Clinical Relevance | Ref |
|---|---|---|---|---|---|---|---|
| DLBCL | healthy people | 2-AG | serum | patients | LC-MS/MS | pathogenesis or progression | [252] |
| BL | Normal mice | glutamate, glycerol, and choline | serum | mice | NMR and MS | diagnosis and prognosis | [253] |
| Refractory or early relapse DLBCL patients | cured patients | ↑lysine, arginine, cadaverine, 2-HB | serum | patients | NMR | high-risk of failing to immunochemotherapy | [254] |
| CTCL | control samples | ↑GLT ↓adenosine monophosphate ↑CTP ↑prostaglandins, pyrimidine, mevalonate pathway ↓tryptophan ↑PRPP | skin and plasma | mice | UHPLC-QTOF | progress of carcinogenesis, leads to CTCL further development. | [255] |
| MYC-induced lymphomas | normal tissue | ↑glycerophosphoglycerols, cardiolipins and monounsaturated fatty acids | tissue and cells | patients and cell lines | DESI-MSI | MYC regulates cellular metabolism in cancer | [256] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pang, Y.; Lu, T.; Xu-Monette, Z.Y.; Young, K.H. Metabolic Reprogramming and Potential Therapeutic Targets in Lymphoma. Int. J. Mol. Sci. 2023, 24, 5493. https://doi.org/10.3390/ijms24065493
Pang Y, Lu T, Xu-Monette ZY, Young KH. Metabolic Reprogramming and Potential Therapeutic Targets in Lymphoma. International Journal of Molecular Sciences. 2023; 24(6):5493. https://doi.org/10.3390/ijms24065493
Chicago/Turabian StylePang, Yuyang, Tingxun Lu, Zijun Y. Xu-Monette, and Ken H. Young. 2023. "Metabolic Reprogramming and Potential Therapeutic Targets in Lymphoma" International Journal of Molecular Sciences 24, no. 6: 5493. https://doi.org/10.3390/ijms24065493
APA StylePang, Y., Lu, T., Xu-Monette, Z. Y., & Young, K. H. (2023). Metabolic Reprogramming and Potential Therapeutic Targets in Lymphoma. International Journal of Molecular Sciences, 24(6), 5493. https://doi.org/10.3390/ijms24065493