
Analysis of the Electron Density of a Water Molecule Encapsulated by Two Cholic Acid Residues

 ,
,  ,
, 
Abstract
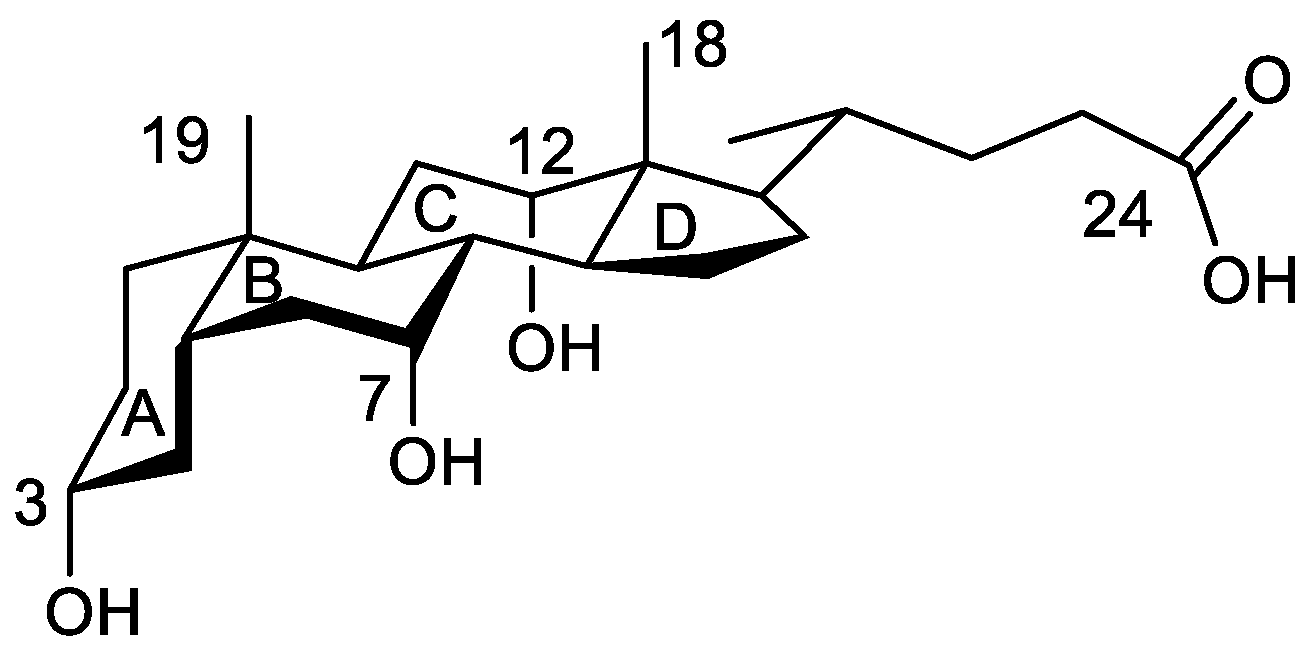
1. Introduction
2. Results and Discussion
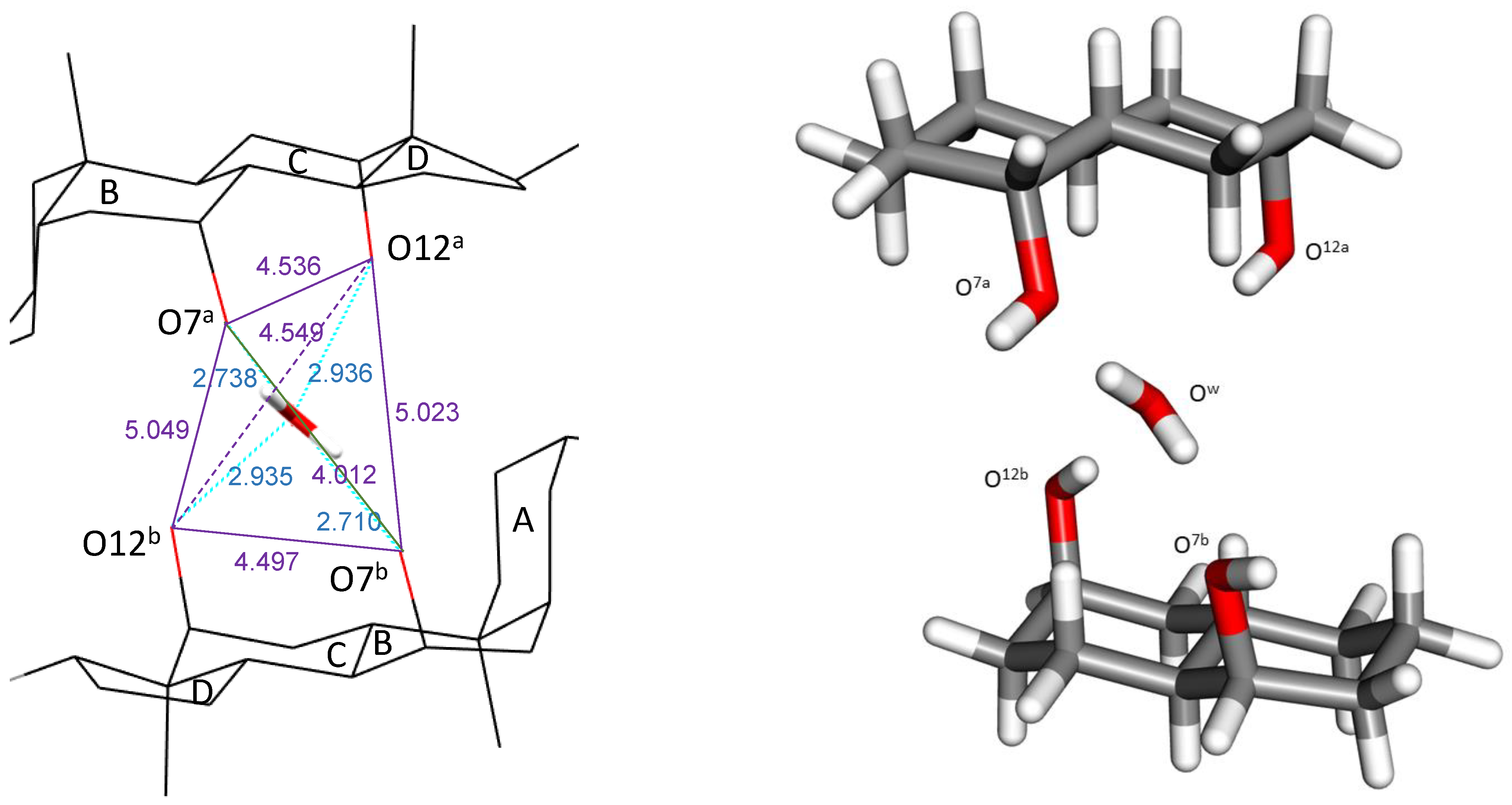
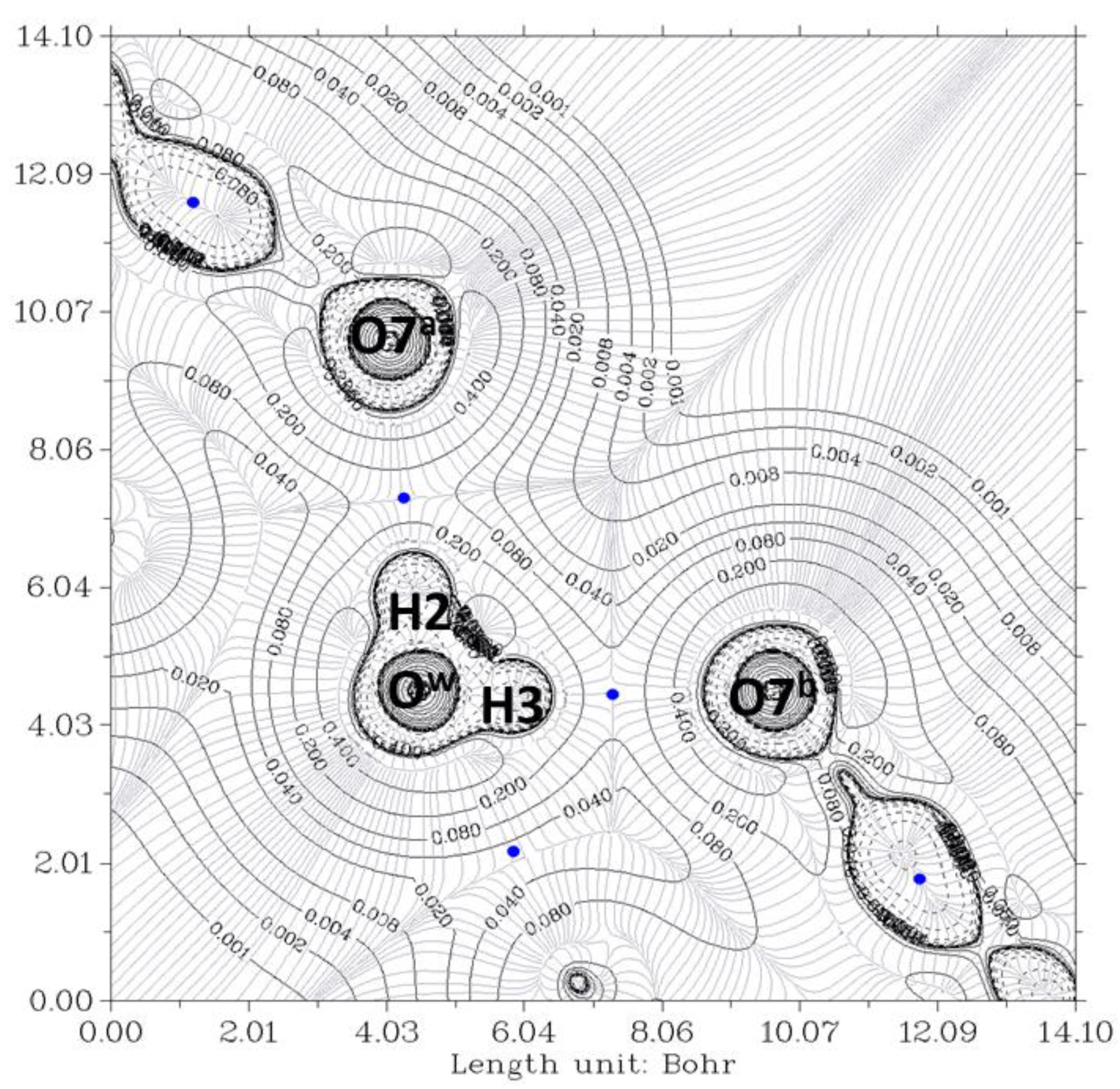
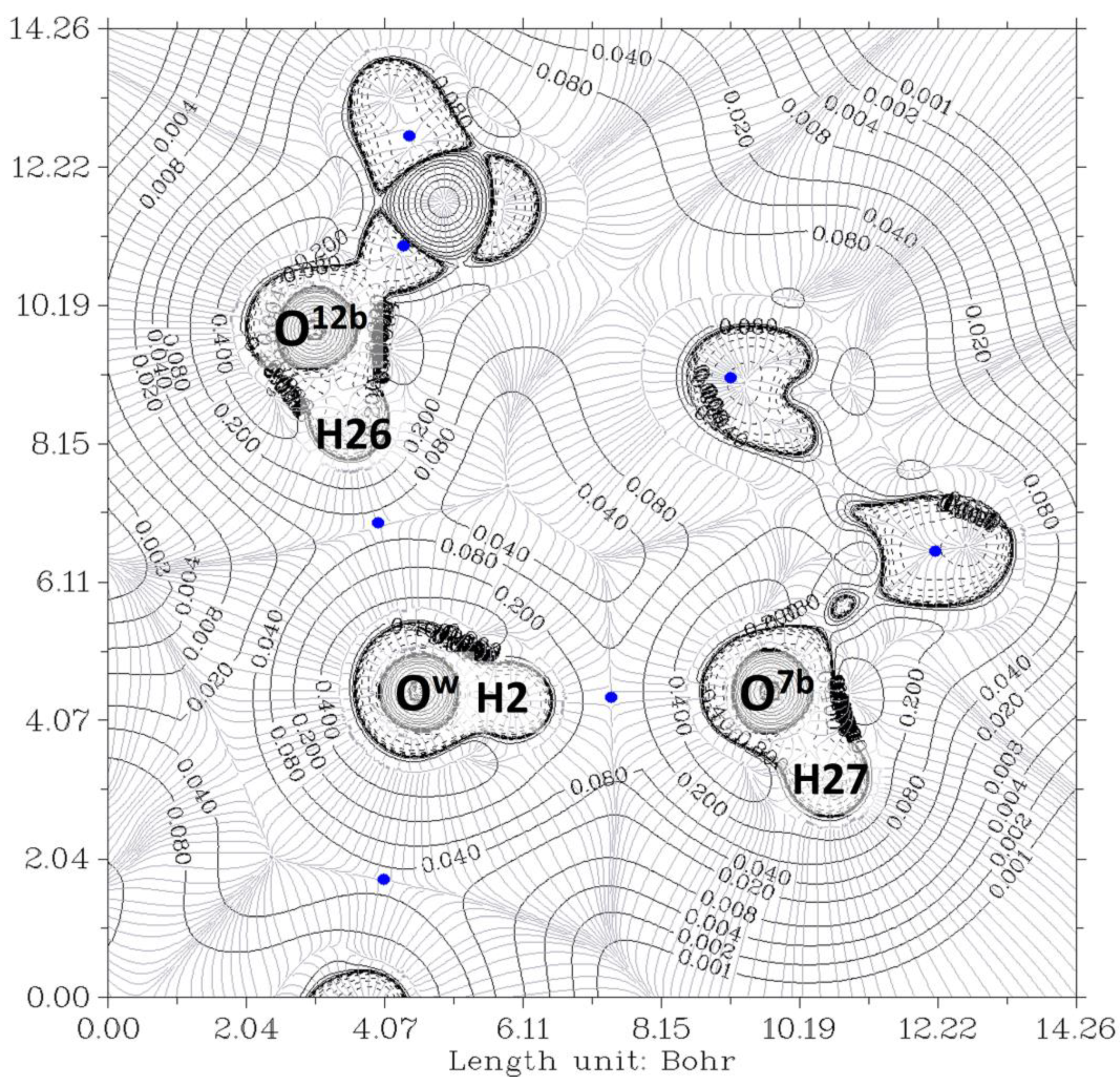
2.1. Complex O12a-H…Ow-H…O7a//O12b-H…Ow-H…O7b
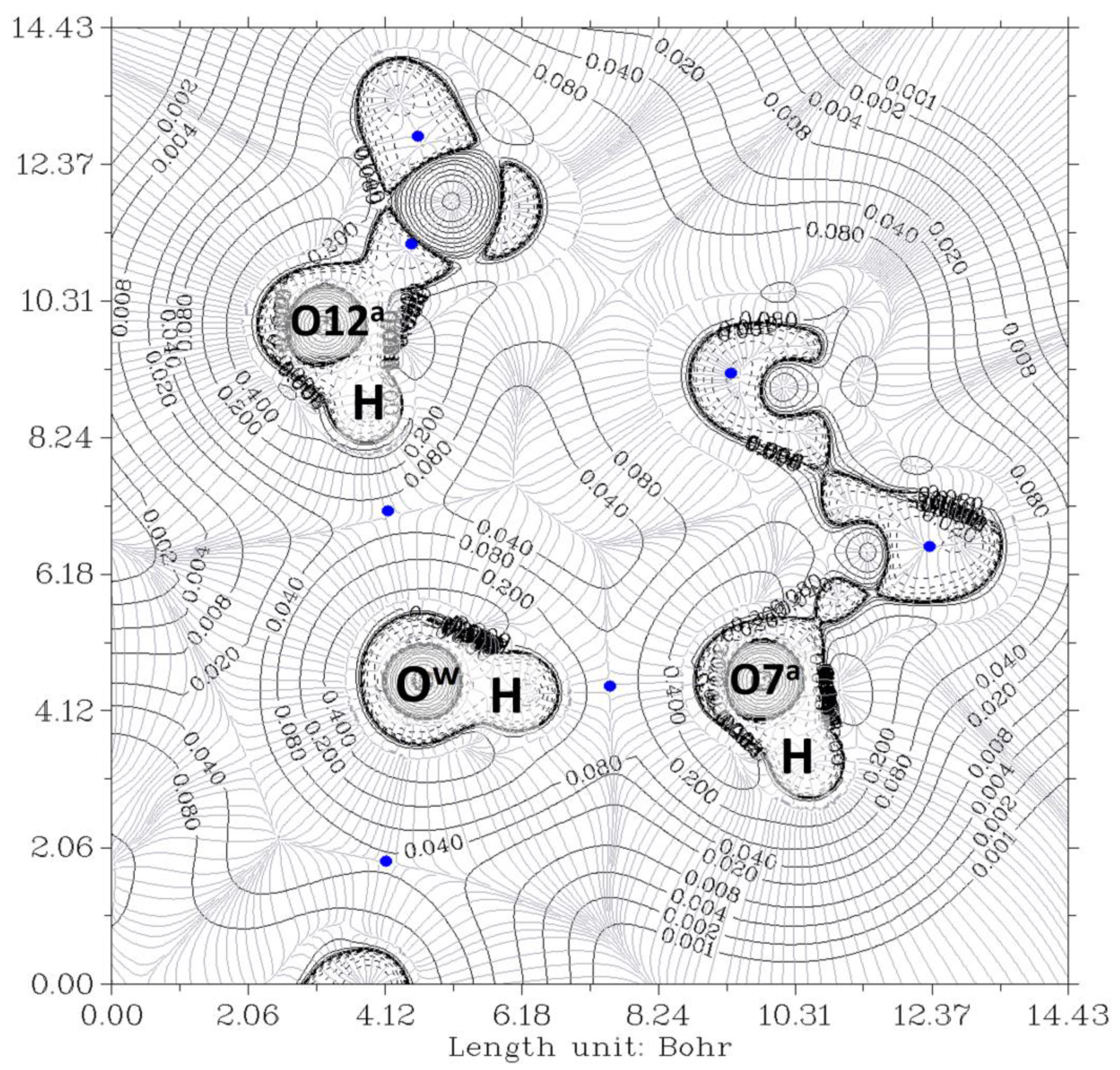
2.2. Complexes O12a-H…Ow-H…O7a, O12b-H…Ow-H…O7b and O12a-H/H…O7a//O12b-H/H…O7b
2.3. Energy of Hydrogen Bonds
3. Materials and Methods
Crystal Structure and Computational Details
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Menger, F.M. Supramolecular chemistry and self-assembly. Proc. Natl. Acad. Sci. USA 2002, 99, 4818–4822. [Google Scholar] [CrossRef] [PubMed]
- Lehn, J.-M. Toward complex matter: Supramolecular chemistry and self-organization. Proc. Natl. Acad. Sci. USA 2002, 99, 4763–4768. [Google Scholar] [CrossRef]
- Hofmann, A.F.; Hagey, L.R.; Krasowski, M.D. Bile salts of vertebrates: Structural variation and possible evolutionary significance. J. Lipid Res. 2010, 51, 226–246. [Google Scholar] [CrossRef]
- Soto, V.H.; Jover, A.; Meijide, F.; Vázquez Tato, J.; Galantini, L.; Pavel, N.V. Supramolecular structures generated by a p-tert-butylphenyl-amide derivative of cholic acid. From vesicles to molecular tubes. Adv. Mater. 2007, 19, 1752–1756. [Google Scholar] [CrossRef]
- Miragaya, J.; Jover, A.; Fraga, F.; Meijide, F.; Tato, J.V. Enantioresolution and Chameleonic Mimicry of 2-Butanol with an Adamantylacetyl Derivative of Cholic Acid. Cryst. Growth Des. 2010, 10, 1124–1129. [Google Scholar] [CrossRef]
- Manghisi, N.; Leggio, C.; Jover, A.; Meijide, F.; Pavel, N.V.; Tellini, V.H.S.; Tato, J.V.; Agostino, R.G.; Galantini, L. Catanionic Tubules with Tunable Charge. Angew. Chem. Int. Ed. 2010, 49, 6604–6607. [Google Scholar] [CrossRef]
- Galantini, L.; di Gregorio, M.C.; Gubitosi, M.; Travaglini, L.; Vázquez Tato, J.; Jover, A.; Meijbe, F.; Tellini, V.H.S.; Pavel, N.V. Bile salts and derivatives: Rigid un-conventional amphiphiles as dispersants, carriers and superstructure building blocks. Curr. Opin. Colloid Interface Sci. 2015, 20, 170–182. [Google Scholar] [CrossRef]
- Soto, V.H.; Alvarez, M.; Meijide, F.; Trillo, J.V.; Antelo, A.; Jover, A.; Galantini, L.; Tato, J.V. Ice-like encapsulated water by two cholic acid moieties. Steroids 2012, 77, 1228–1232. [Google Scholar] [CrossRef] [PubMed]
- Meijide, F.; de Frutos, S.; Soto, V.H.; Jover, A.; Seijas, J.A.; Vázquez-Tato, M.P.; Fraga, F.; Tato, J.V. A Standard Structure for Bile Acids and Derivatives. Crystals 2018, 8, 86. [Google Scholar] [CrossRef]
- Giglio, E. Structural aspect of inclusion compounds formed by organic host lattices. In Inclusion Compounds of Deoxycholic Acid; Atwood, J.L., Davies, J.E.D., MacNicol, D.D., Eds.; Academic Press: London, UK, 1984; pp. 207–229. [Google Scholar]
- Miyata, M.; Sada, K. Deoxycholic acid and related hosts. In Comprehensive Supra-Molecular Chemistry 6; MacNicol, D.D.T.F., Bishop, R., Eds.; Elsevier: Oxford, UK, 1996; pp. 147–176. [Google Scholar]
- Campanelli, A.R.; De Sanctis, S.C.; Giglio, E.; Pavel, N.V.; Quagliata, C. From crystal to micelle: A new approach to the micellar structure. J. Incl. Phenom. Macrocycl. Chem. 1989, 7, 391–400. [Google Scholar] [CrossRef]
- Sada, K.; Sugahara, M.; Kato, K.; Miyata, M. Controlled Expansion of a Molecular Cavity in a Steroid Host Compound. J. Am. Chem. Soc. 2001, 123, 4386–4392. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, M.; Sada, K.; Miyata, M. A robust structural motif in inclusion crystals of nor-bile acids. Chem. Commun. 1999, 3, 293–294. [Google Scholar] [CrossRef]
- Sugahara, M.; Hirose, J.; Sada, K.; Miyata, M. Inclusion Abilities of Bile Acids with Different Side Chain Length. Mol. Cryst. Liq. Cryst. 2001, 356, 155–162. [Google Scholar] [CrossRef]
- Hishikawa, Y.; Aoki, Y.; Sada, K.; Miyata, M. Selective Inclusion Phenomena in Lithocholamide Crystal Lattices; Design of Bilayered Assemblies through Ladder-type Hydrogen Bonding Network. Chem. Lett. 1998, 27, 1289–1290. [Google Scholar] [CrossRef]
- Miyata, M.; Tohnai, N.; Hisaki, I. Supramolecular Chirality in Crystalline Assemblies of Bile Acids and Their Derivatives; Three-Axial, Tilt, Helical, and Bundle Chirality. Molecules 2007, 12, 1973–2000. [Google Scholar] [CrossRef]
- Fletcher, N.H. The Chemical Physics of Ice. The Chemical Physics of Ice; Cambridge University Press: Cambridge, UK, 1970. [Google Scholar]
- Bergmann, U.; Di Cicco, A.; Wernet, P.; Principi, E.; Glatzel, P.; Nilsson, A. Nearest-neighbor oxygen distances in liquid water and ice observed by x-ray Raman based extended x-ray absorption fine structure. J. Chem. Phys. 2007, 127, 174504. [Google Scholar] [CrossRef]
- Steiner, T. The Hydrogen Bond in the Solid State. Angew Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- Atwood, J.L.; Barbour, L.J.; Ness, T.J.; Raston, C.L.; Raston, P.L. A Well-Resolved Ice-like (H2O)8 Cluster in an Organic Supramolecular Complex. J. Am. Chem. Soc. 2001, 123, 7192–7193. [Google Scholar] [CrossRef]
- Barbour, L.J.; Orr, G.W.; Atwood, J.L. An intermolecular (H2O)10 cluster in a solid-state supramolecular complex. Nature 1998, 393, 671–673. [Google Scholar] [CrossRef]
- Barbour, L.; Orr, G.; Atwood, J. CCDC 145404: Experimental Crystal Structure Determination. Chem. Commun. 2000, 859. [Google Scholar] [CrossRef]
- Hoelzl, C.; Horinek, D. Pressure increases the ice-like order of water at hydrophobic interfaces. Phys. Chem. Chem. Phys. 2018, 20, 21257–21261. [Google Scholar] [CrossRef]
- Saha, B.K.; Nangia, A. First example of an ice-like water hexamer boat tape structure in a supramolecular organic host. Chem. Commun. 2006, 12, 1825–1827. [Google Scholar] [CrossRef]
- Smolin, N.; Daggett, V. Formation of Ice-like Water Structure on the Surface of an Antifreeze Protein. J. Phys. Chem. B 2008, 112, 6193–6202. [Google Scholar] [CrossRef] [PubMed]
- Mahatabuddin, S.; Fukami, D.; Arai, T.; Nishimiya, Y.; Shimizu, R.; Shibazaki, C.; Kondo, H.; Adachi, M.; Tsuda, S. Polypentagonal ice-like water networks emerge solely in an activity-improved variant of ice-binding protein. Proc. Natl. Acad. Sci. USA 2018, 115, 5456–5461. [Google Scholar] [CrossRef] [PubMed]
- Odendahl, N.L.; Geissler, P.L. Local Ice-like Structure at the Liquid Water Surface. J. Am. Chem. Soc. 2022, 144, 11178–11188. [Google Scholar] [CrossRef] [PubMed]
- Bonn, M.; Bakker, H.J.; Tong, Y.; Backus, E.H.G. No Ice-Like Water at Aqueous Biological Interfaces. Biointerphases 2012, 7, 20. [Google Scholar] [CrossRef]
- He, Z.; Zhou, J.; Lu, X.; Corry, B. Ice-like Water Structure in Carbon Nanotube (8,8) Induces Cationic Hydration Enhancement. J. Phys. Chem. C 2013, 117, 11412–11420. [Google Scholar] [CrossRef]
- Takaiwa, D.; Hatano, I.; Koga, K.; Tanaka, H. Phase diagram of water in carbon nanotubes. Proc. Natl. Acad. Sci. USA 2008, 105, 39–43. [Google Scholar] [CrossRef]
- Pascal, T.A.; Goddard, W.A.; Jung, Y. Entropy and the driving force for the filling of carbon nanotubes with water. Proc. Natl. Acad. Sci. USA 2011, 108, 11794–11798. [Google Scholar] [CrossRef]
- Agrawal, K.V.; Shimizu, S.; Drahushuk, L.W.; Kilcoyne, D.; Strano, M.S. Observation of extreme phase transition temperatures of water confined inside isolated carbon nanotubes. Nat. Nanotechnol. 2017, 12, 267–273. [Google Scholar] [CrossRef]
- Sugiyama, Y.; Futamura, R.; Iiyama, T. Ice-like Structure of Water Confined in Hydrophobic Sub-nanometer Spaces at Room Temperature. Chem. Lett. 2022, 51, 760–764. [Google Scholar] [CrossRef]
- Banerjee, D.; Bhat, S.N.; Bhat, S.V.; Leporini, D. Molecular probe dynamics reveals suppression of ice-like regions in strongly confined supercooled water. PLoS ONE 2012, 7, e44382. [Google Scholar] [CrossRef] [PubMed]
- Weissmann, M.; Blum, L.; Cohan, N.V. On the hydrogen bond in an ice-like structure. Chem. Phys. Lett. 1967, 1, 95–98. [Google Scholar] [CrossRef]
- Sun, D.; Xu, H.-R.; Yang, C.-F.; Wei, Z.-H.; Zhang, N.; Huang, R.-B.; Zheng, L.-S. Encapsulated Diverse Water Aggregates in Two Ag(I)/4,4’-Bipyridine/Dicarboxylate Hosts: 1D Water Tape and Chain. Crystal Growth Des. 2010, 10, 4642–4649. [Google Scholar] [CrossRef]
- Ma, B.-Q.; Sun, H.-L.; Gao, S. Cyclic water pentamer in a tape-like structure. Chem. Commun. 2004, 19, 2220–2221. [Google Scholar] [CrossRef]
- Popelier, P.L.A. On the full topology of the Laplacian of the electron densityOn the full topology of the Laplacian of the electron density. Coord. Chem Rev. 2000, 197, 169–189. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules. Acc. Chem. Res. 1985, 18, 9–15. [Google Scholar] [CrossRef]
- Grabowski, S.J. Hydrogen Bond—Definitions, Criteria of Existence and Various Types. In Understanding Hydrogen Bonds: Theoretical and Experimental Views; From Book Series: Theoretical and Computational Chemistry Series; Royal Society of Chemistry: London, UK, 2020; Chapter 1; pp. 1–40. [Google Scholar] [CrossRef]
- Popelier, P.L.A. Characterization of a Dihydrogen Bond on the Basis of the Electron Density. J. Phys. Chem. A 1998, 102, 1873–1878. [Google Scholar] [CrossRef]
- Carroll, M.T.; Chang, C.; Bader, R.F. Prediction of the structures of hydrogen-bonded complexes using the laplacian of the charge density. Mol. Phys. 1988, 63, 387–405. [Google Scholar] [CrossRef]
- Koch, U.; Popelier, P.L.A. Characterization of C-H-O Hydrogen Bonds on the Basis of the Charge Density. J. Phys. Chem. 1995, 99, 9747–9754. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Carroll, M.T.; Cheeseman, J.R.; Chang, C. Properties of atoms in molecules: Atomic volumes. J. Am. Chem. Soc. 1987, 109, 7968–7979. [Google Scholar] [CrossRef]
- Alkorta, I.; Rozas, I.; Elguero, J. Bond Length–Electron Density Relationships: From Covalent Bonds to Hydrogen Bond Interac-tions. Struct. Chem. 1998, 9, 243–247. [Google Scholar] [CrossRef]
- Tang, T.-H.; Deretey, E.; Jensen, S.J.K.; Csizmadia, I.G. Hydrogen bonds: Relation between lengths and electron densities at bond critical points. Eur. Phys. J. D 2005, 37, 217–222. [Google Scholar] [CrossRef]
- Mukhopadhyay, A.; Cole, W.T.; Saykally, R.J. The water dimer I: Experimental characterization. Chem. Phys. Lett. 2015, 633, 13–26. [Google Scholar] [CrossRef]
- Mukhopadhyay, A.; Xantheas, S.S.; Saykally, R.J. The water dimer II: Theoretical investigations. Chem. Phys. Lett. 2018, 700, 163–175. [Google Scholar] [CrossRef]
- Dyke, T.R.; Muenter, J.S. Microwave spectrum and structure of hydrogen bonded water dimer. J. Chem. Phys. 1974, 60, 2929–2930. [Google Scholar] [CrossRef]
- Dyke, T.R.; Mack, K.M.; Muenter, J.S. The structure of water dimer from molecular beam electric resonance spectroscopy. J. Chem. Phys. 1977, 66, 498–510. [Google Scholar] [CrossRef]
- Odutola, J.A.; Dyke, T.R. Partially deuterated water dimers: Microwave spectra and structure. J. Chem. Phys. 1980, 72, 5062–5070. [Google Scholar] [CrossRef]
- Lane, J.R. CCSDTQ Optimized Geometry of Water Dimer. J. Chem. Theory Comput. 2013, 9, 316–323. [Google Scholar] [CrossRef]
- Grabowski, S.J. Ab Initio Calculations on Conventional and Unconventional Hydrogen BondsStudy of the Hydrogen Bond Strength. J. Phys. Chem. A 2001, 105, 10739–10746. [Google Scholar] [CrossRef]
- Kumar, P.S.V.; Raghavendra, V.; Subramanian, V. Bader’s Theory of Atoms in Molecules (AIM) and its Applications to Chemical Bonding. J. Chem. Sci. 2016, 128, 1527–1536. [Google Scholar] [CrossRef]
- Isaev, A.N. Ammonia and phosphine complexes with proton donors. Hydrogen bonding from the backside of the N(P) lone pair. Comput. Theor. Chem. 2018, 1142, 28–38. [Google Scholar] [CrossRef]
- Rowland, R.S.; Taylor, R. Intermolecular Nonbonded Contact Distances in Organic Crystal Structures: Comparison with Distances Expected from van der Waals Radii. J. Phys. Chem. 1996, 100, 7384–7391. [Google Scholar] [CrossRef]
- Wendler, K.; Thar, J.; Zahn, S.; Kirchner, B. Estimating the Hydrogen Bond Energy. J. Phys. Chem. A 2010, 114, 9529–9536. [Google Scholar] [CrossRef]
- Jeffrey, G.J. An Introduction to Hydrogen Bonding; Oxford University Press: New York, NY, USA, 1997; p. 12. [Google Scholar]
- Emamian, S.; Lu, T.; Kruse, H.; Emamian, H. Exploring Nature and Predicting Strength of Hydrogen Bonds: A Correlation Analysis Between Atoms-in-Molecules Descriptors, Binding Energies, and Energy Components of Symmetry-Adapted Perturbation Theory. J. Comput. Chem. 2019, 40, 2868–2881. [Google Scholar] [CrossRef]
- Rocher-Casterline, B.E.; Ch’Ng, L.C.; Mollner, A.K.; Reisler, H. Communication: Determination of the bond dissociation energy (D0) of the water dimer, (H2O)2, by velocity map imaging. J. Chem. Phys. 2011, 134, 211101. [Google Scholar] [CrossRef]
- Ruscic, B. Active Thermochemical Tables: Water and Water Dimer. J. Phys. Chem. A 2013, 117, 11940–11953. [Google Scholar] [CrossRef]
- Feyereisen, M.W.; Feller, D.; Dixon, D.A. Hydrogen Bond Energy of the Water Dimer. J. Phys. Chem. 1996, 100, 2993–2997. [Google Scholar] [CrossRef]
- Grabowski, S.J. High-Level Ab Initio Calculations of Dihydrogen-Bonded Complexes. J. Phys. Chem. A 2000, 104, 5551–5557. [Google Scholar] [CrossRef]
- Gu, Y.; Kar, T.; Scheiner, S. Fundamental Properties of the CH···O Interaction: Is It a True Hydrogen Bond? J. Am. Chem. Soc. 1999, 121, 9411–9422. [Google Scholar] [CrossRef]
- Shank, A.; Wang, Y.; Kaledin, A.; Braams, B.J.; Bowman, J.M. Accurate ab initio and “hybrid” potential energy surfaces, intramolecular vibrational energies, and classical ir spectrum of the water dimer. J. Chem. Phys. 2009, 130, 144314. [Google Scholar] [CrossRef] [PubMed]
- Moin, S.T.; Hofer, T.S.; Randolf, B.R.; Rode, B.M. Structure and dynamics of methanol in water: A quantum mechanical charge field molecular dynamics study. J. Comput. Chem. 2011, 32, 886–892. [Google Scholar] [CrossRef] [PubMed]
- Boyd, R.; Choi, S.C. Hydrogen bonding between nitriles and hydrogen halides and the topological properties of molecular charge distributions. Chem. Phys. Lett. 1986, 129, 62–65. [Google Scholar] [CrossRef]
- Rozenberg, M. The hydrogen bond—Practice and QTAIM theory. RSC Adv. 2014, 4, 26928–26931. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 RC; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex O12a-H…Ow-H…O7a//O12b-H…Ow-H…O7b | ||||
|---|---|---|---|---|
| Identification HBCP (Figure 5) | 50 | 59 | 17 | 43 |
| Property at HBCP | O12a-H…Ow | Ow-H…O7a | O12b-H…Ow | Ow-H…O7b |
| O-O length/Å crystal | 2.936 | 2.738 | 2.935 | 2.710 |
| O…H length/Å crystal | 2.114 | 1.920 | 2.177 | 1.866 |
| Electron density ρb, au | 0.0154 | 0.0239 | 0.0138 | 0.0270 |
| ρb calculated according to [47] | 0.0170 | 0.0270 | 0.0146 | 0.0308 |
| Laplacian of the electron density at HBCP, ∇2ρb, au | 0.0667 | 0.106 | 0.0616 | 0.118 |
| HBCP…O length/ Å, r1 | 1.355 | 1.242 | 1.383 | 1.217 |
| HBCP…H length/ Å, r2 | 0.759 | 0.679 | 0.795 | 0.650 |
| r1+ r2 = rO…H length/Å | 2.114 | 1.921 | 2.178 | 1.867 |
| /Å | 0.225 | 0.338 | 0.197 | 0.363 |
| 0.341 | 0.421 | 0.305 | 0.450 | |
| O12a-H…Ow-H…O7a | O12b-H…Ow-H…O7b | |||
|---|---|---|---|---|
| Property at HBCP | CP71 (CP50) O12a-H…Ow | CP74 (CP59) Ow-H…O7a | CP68 (CP17) O12b-H…Ow | CP71 (CP43) Ow-H…O7b |
| Electron density ρb, au | 0.0152 | 0.0239 | 0.0137 | 0.0270 |
| Laplacian of ρb, ∇2ρb, au | 0.0662 | 0.106 | 0.0610 | 0.119 |
| HBCP…O length/Å, r1 | 1.353 | 1.243 | 1.381 | 1.218 |
| HBCP…H length/Å, r2 | 0.761 | 0.679 | 0.797 | 0.650 |
| r1+ r2 = O…H length/Å | 2.115 | 1.922 | 2.177 | 1.867 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vázquez-Tato, M.P.; Seijas, J.A.; Meijide, F.; de Frutos, S.; Vázquez Tato, J. Analysis of the Electron Density of a Water Molecule Encapsulated by Two Cholic Acid Residues. Int. J. Mol. Sci. 2023, 24, 5359. https://doi.org/10.3390/ijms24065359
Vázquez-Tato MP, Seijas JA, Meijide F, de Frutos S, Vázquez Tato J. Analysis of the Electron Density of a Water Molecule Encapsulated by Two Cholic Acid Residues. International Journal of Molecular Sciences. 2023; 24(6):5359. https://doi.org/10.3390/ijms24065359
Chicago/Turabian StyleVázquez-Tato, María Pilar, Julio A. Seijas, Francisco Meijide, Santiago de Frutos, and José Vázquez Tato. 2023. "Analysis of the Electron Density of a Water Molecule Encapsulated by Two Cholic Acid Residues" International Journal of Molecular Sciences 24, no. 6: 5359. https://doi.org/10.3390/ijms24065359
APA StyleVázquez-Tato, M. P., Seijas, J. A., Meijide, F., de Frutos, S., & Vázquez Tato, J. (2023). Analysis of the Electron Density of a Water Molecule Encapsulated by Two Cholic Acid Residues. International Journal of Molecular Sciences, 24(6), 5359. https://doi.org/10.3390/ijms24065359