

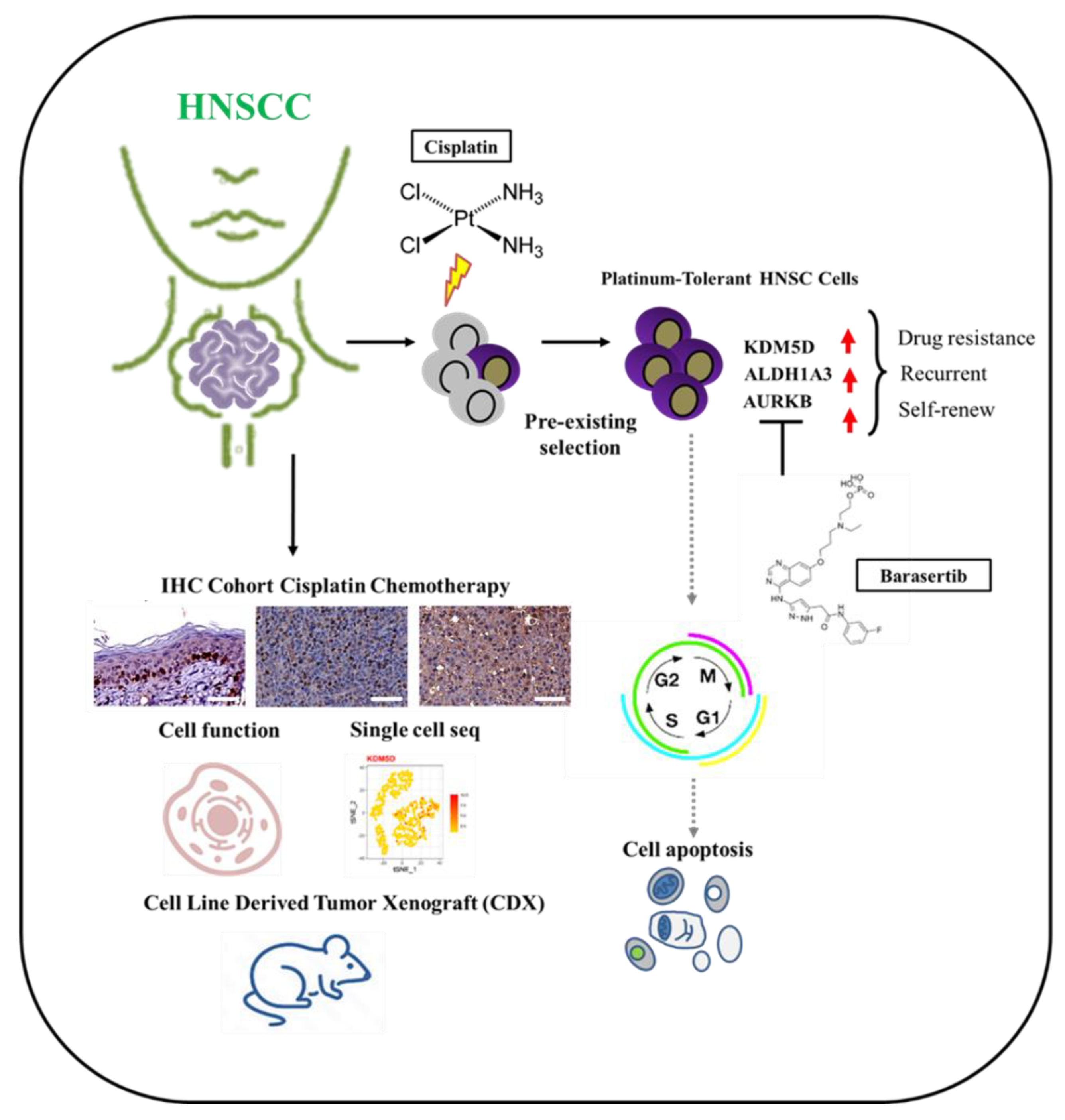
KDM5D Histone Demethylase Identifies Platinum-Tolerant Head and Neck Cancer Cells Vulnerable to Mitotic Catastrophe

 ,
, 
Abstract
1. Introduction
2. Results
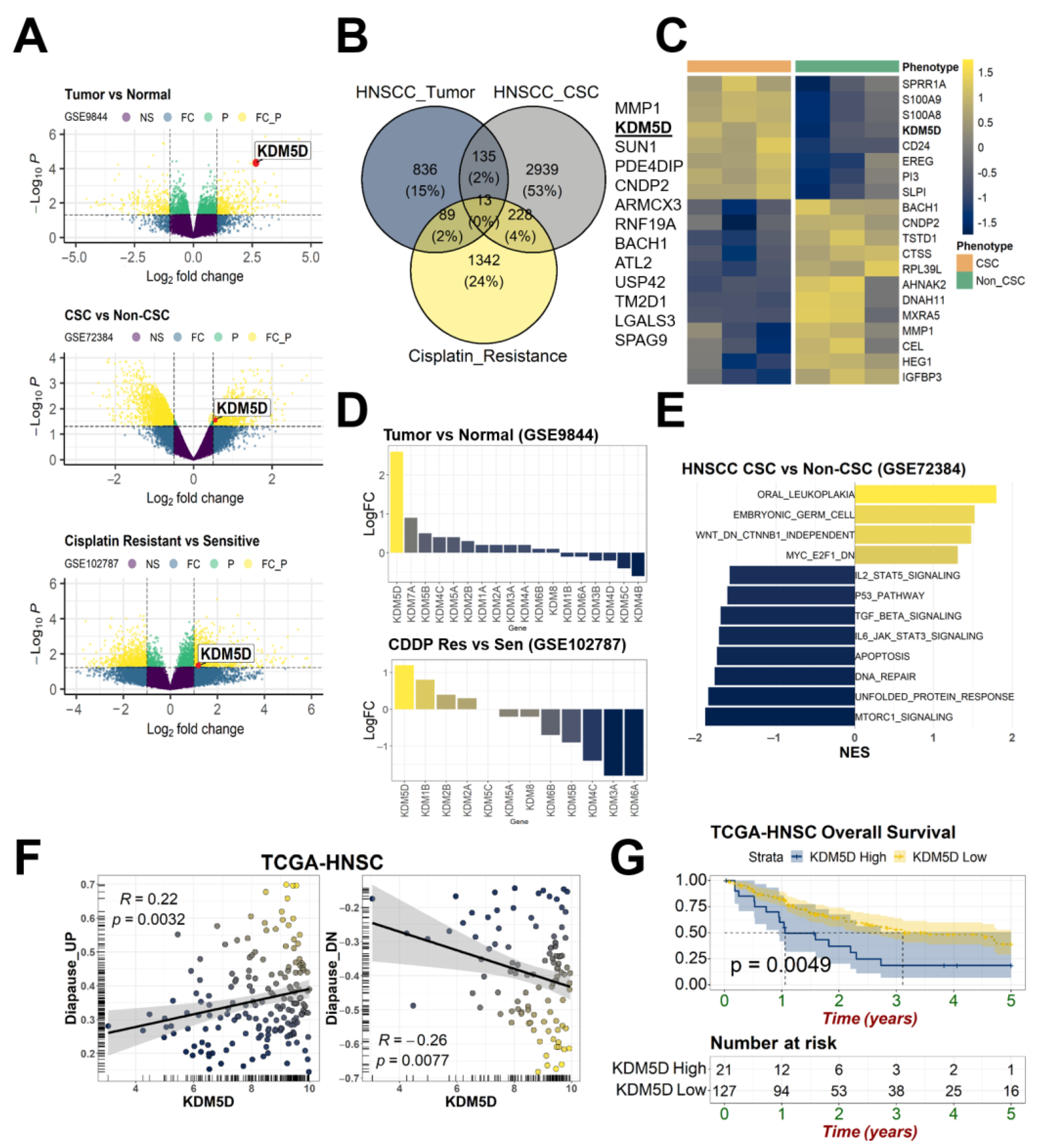
2.1. KDM5D Underlies the Relationships between Treatment Tolerance, Diapause State, and Cancer Stemness
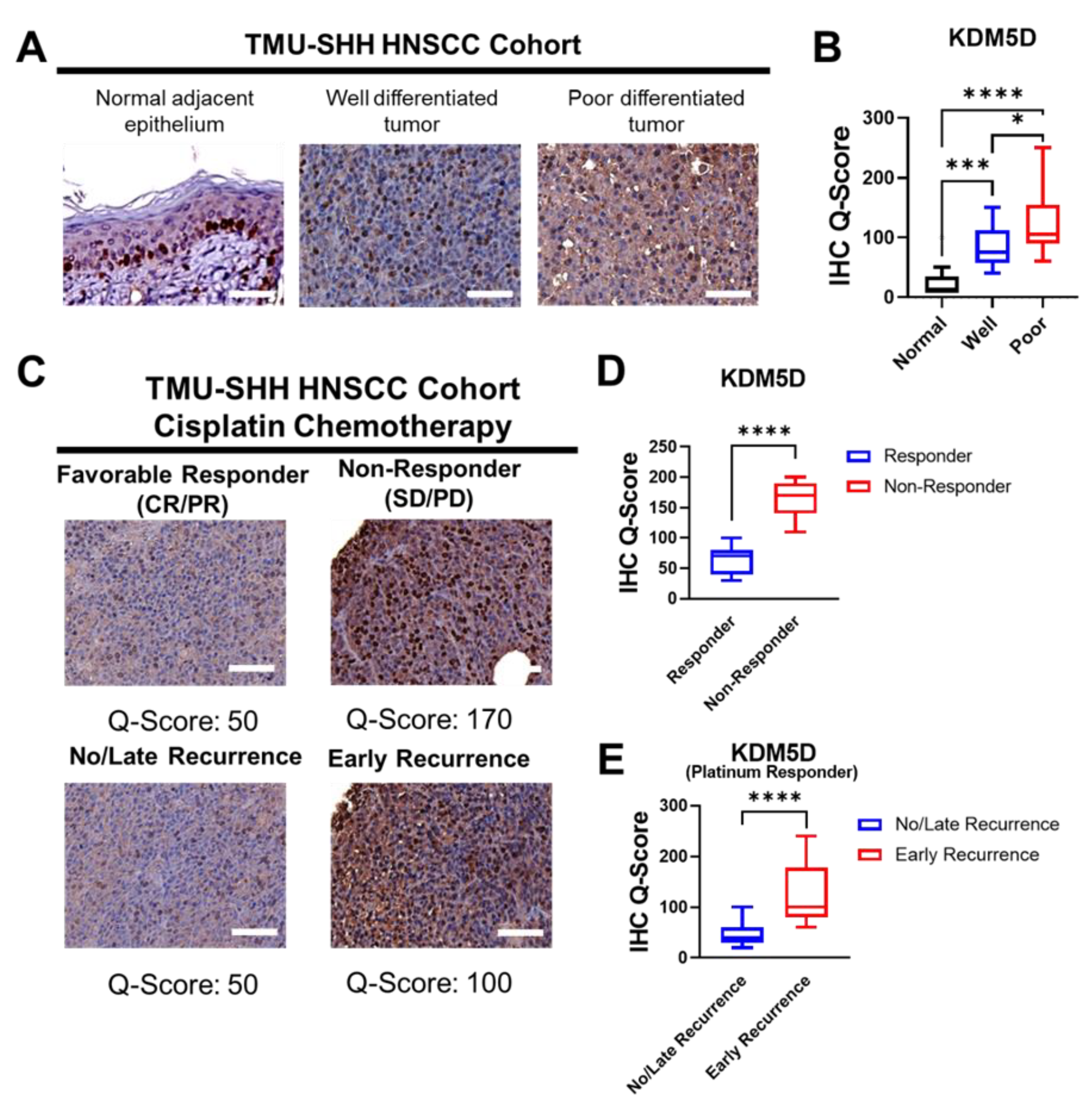
2.2. KDM5D Is Associated with Poor Clinical Outcomes in Patients with HNSCC
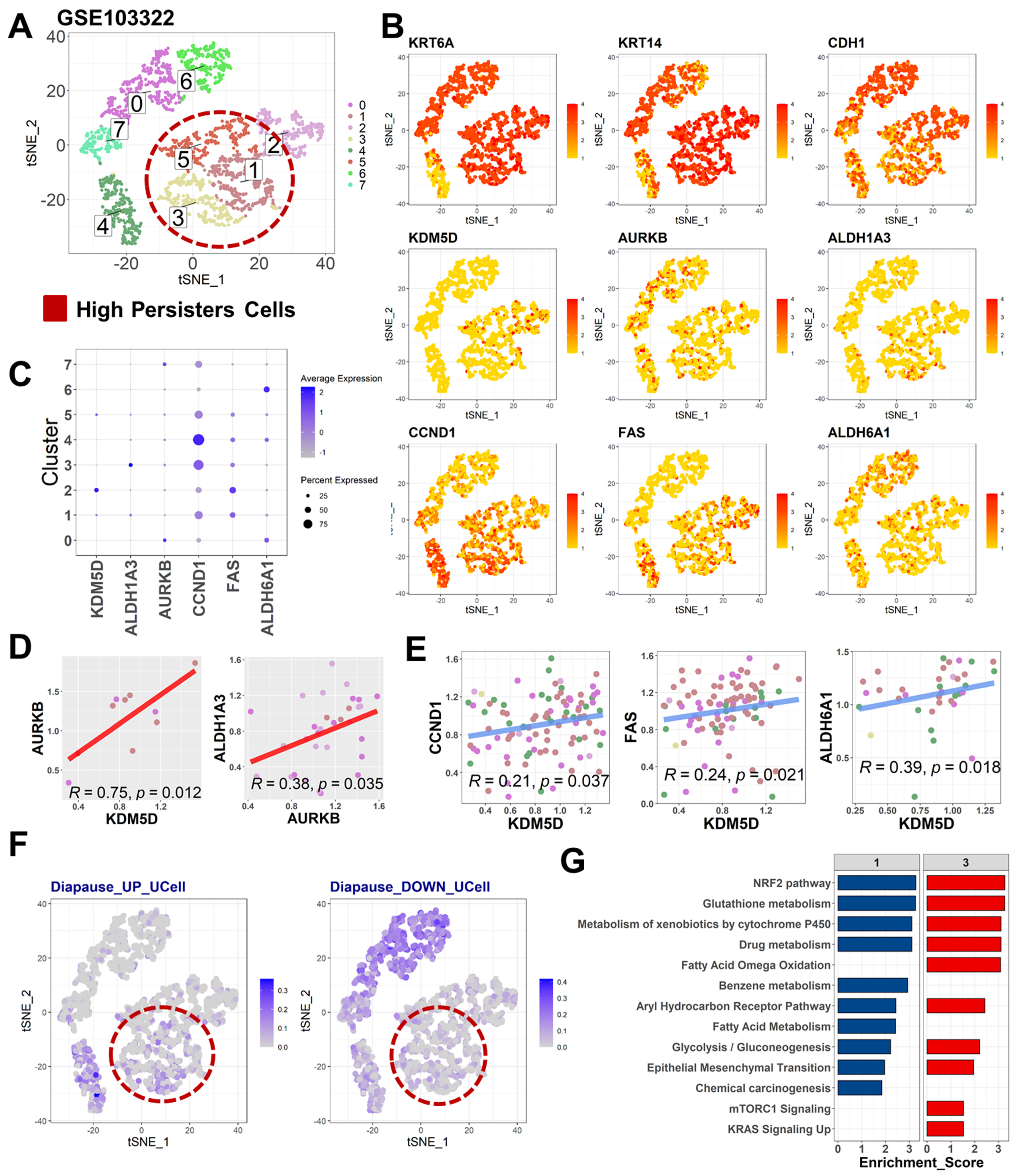
2.3. Diapause State in Persister Cells Is Enriched by KDM5D/AURKB Coexpression
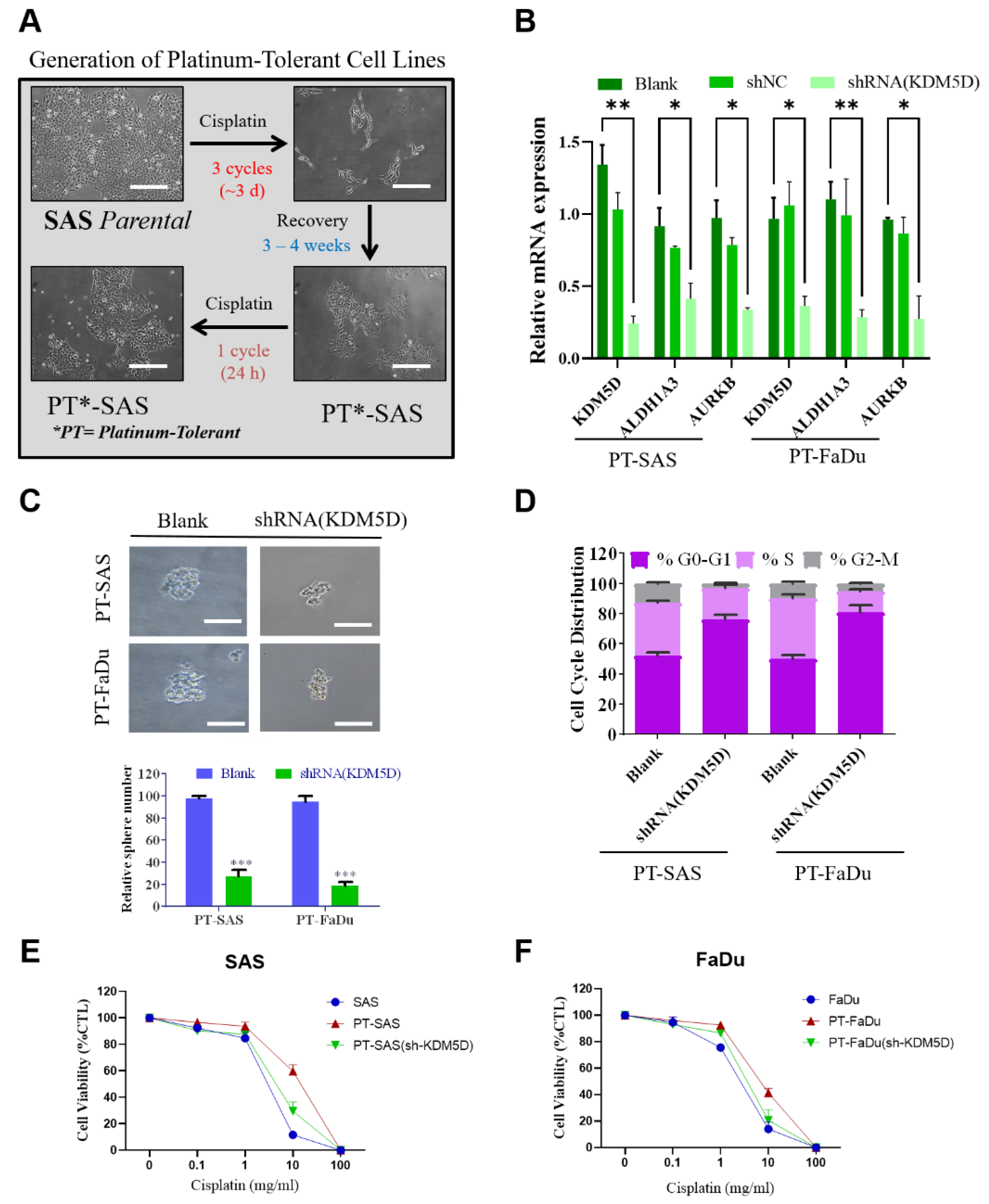
2.4. KDM5D Promoted Persister Cell Development by Modulating AURKB Expression
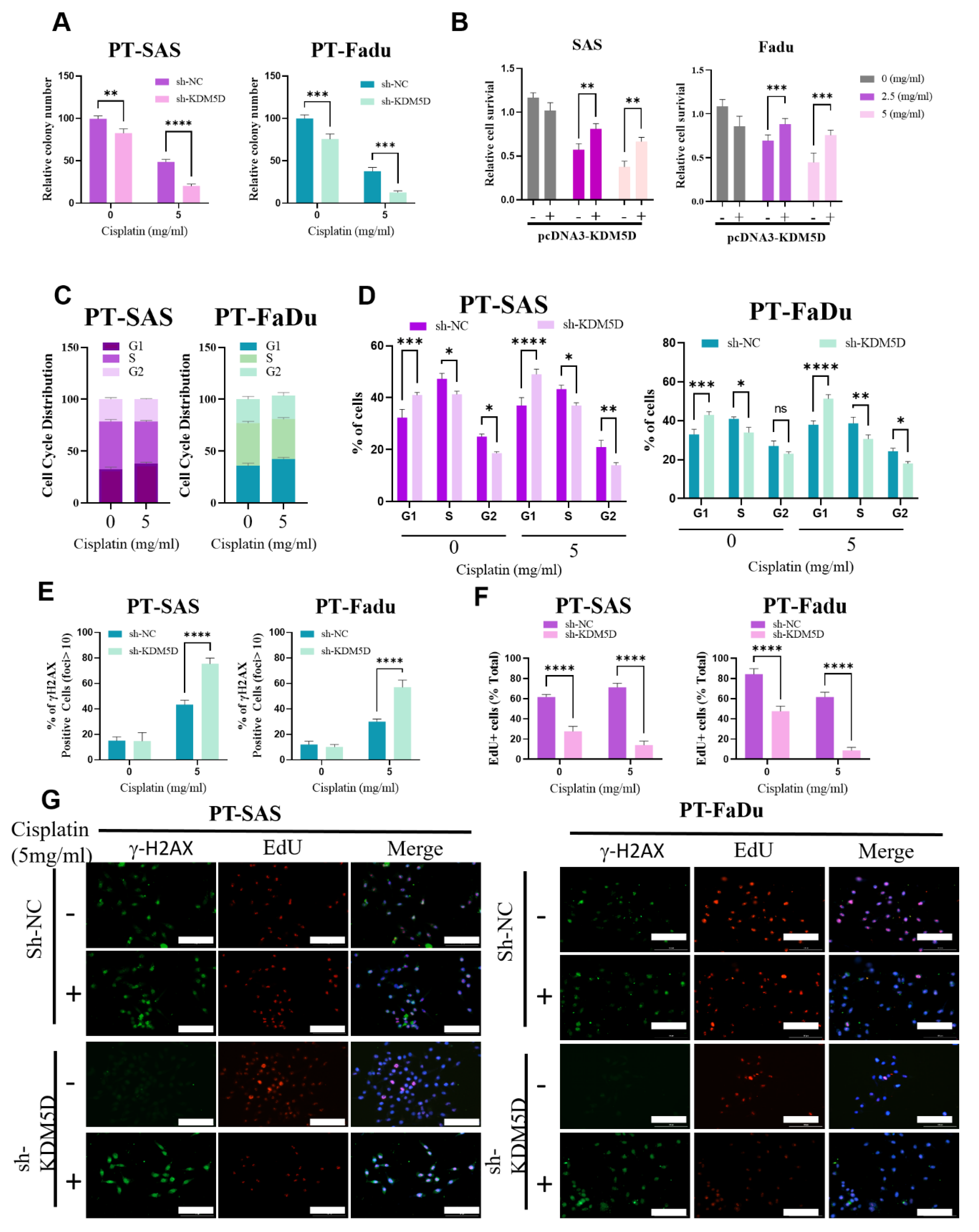
2.5. KDM5D Protects DNA Damage following Platinum Treatment in Persister Cells
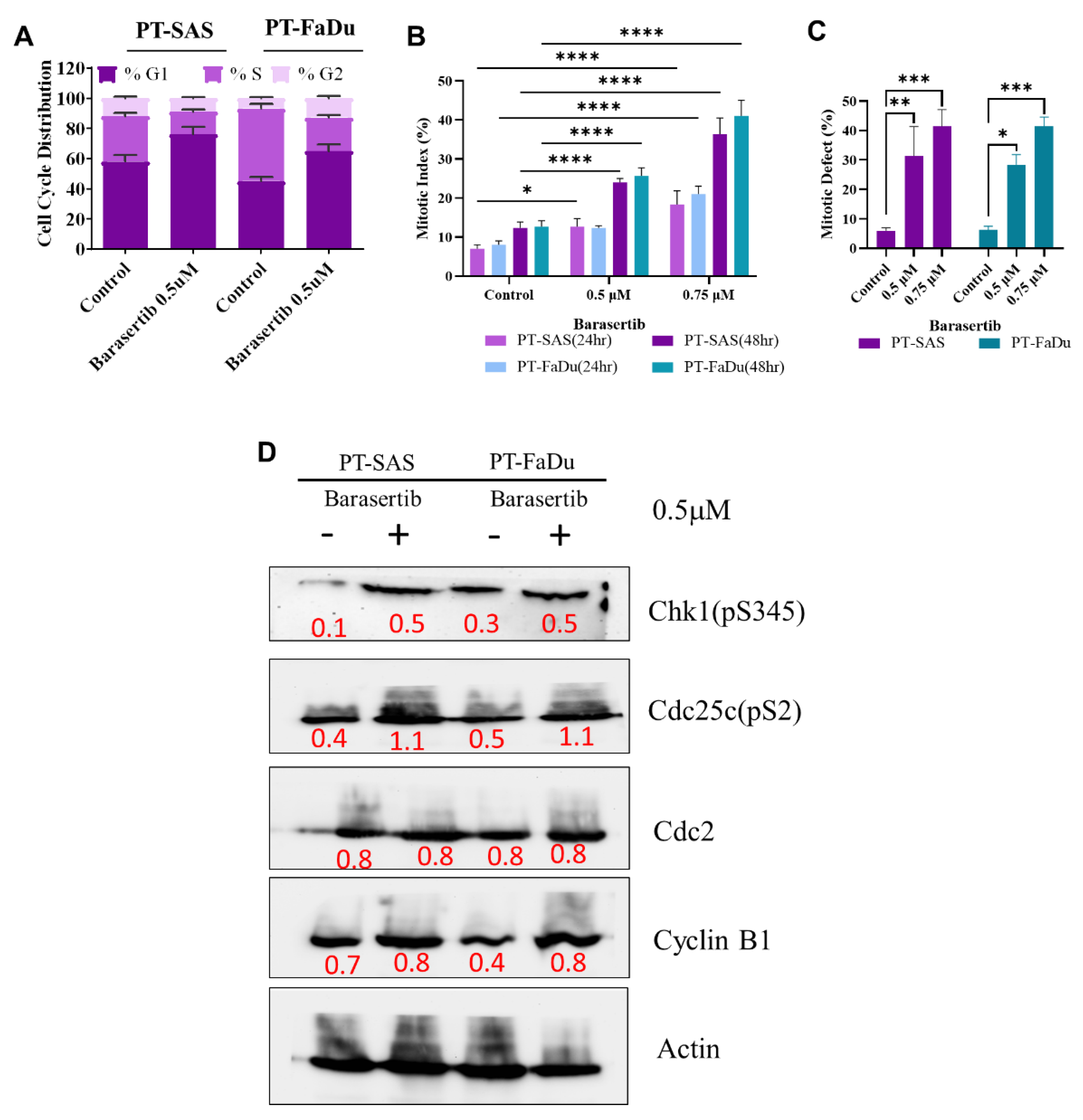
2.6. AURKB-Induced Mitotic Catastrophe Is Disrupted in Persister Cells
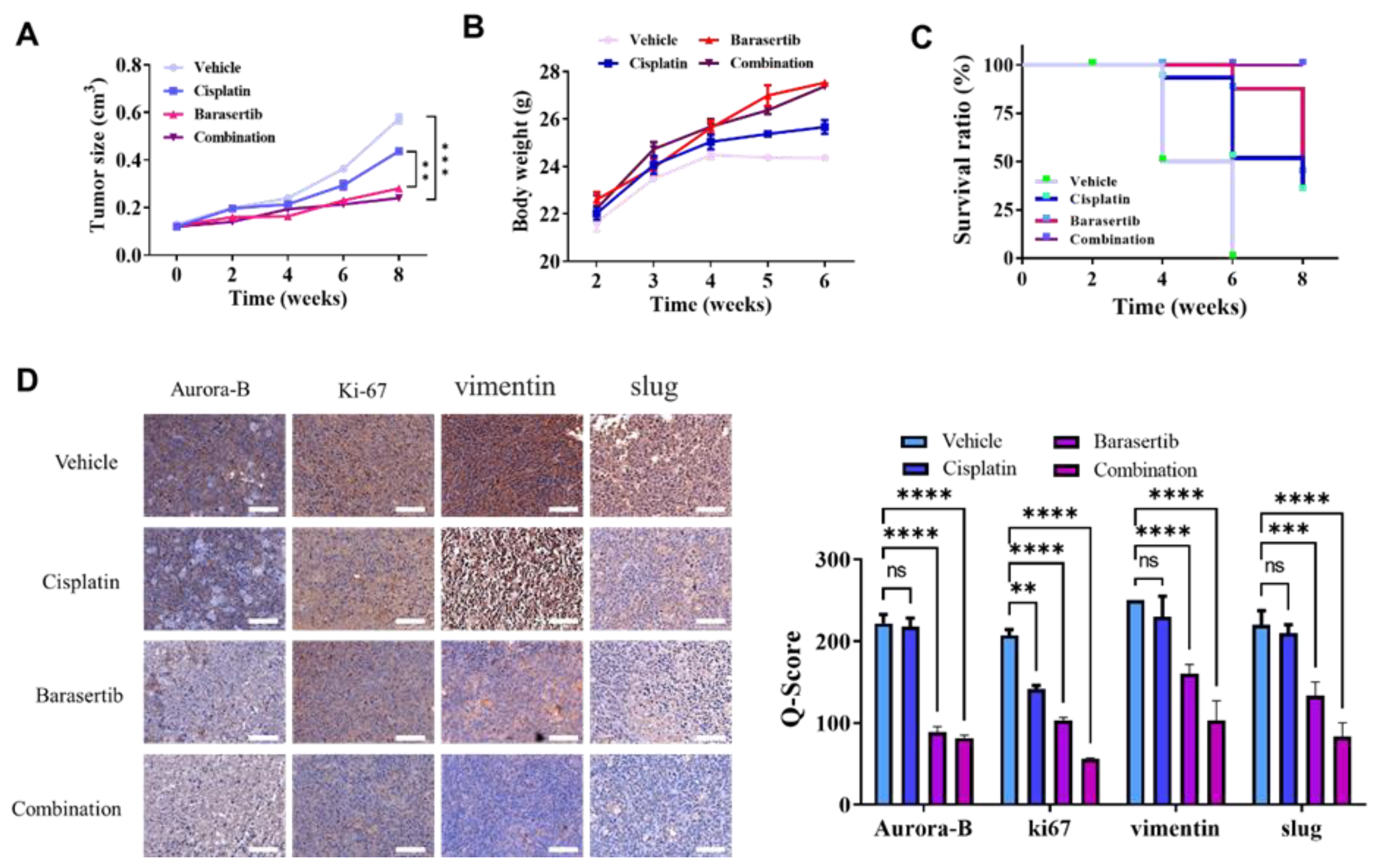
2.7. Cotreatment of Cisplatin and Barasertib Prolonged Tumor Suppression Potential
3. Discussion
4. Materials and Methods
4.1. Microarray and Bulk Tumor RNA Sequencing Data Acquisition
4.2. Single-Cell Profiling of HNSCC
4.3. Identification of Differentially Expressed Genes
4.4. Pathway Enrichment Analysis
4.5. Immunohistochemistry Staining
4.6. Cell Line Culture
4.7. shRNA-Mediated KDM5D Knockdown
4.8. Establishment of Platinum-Tolerant Persister Cells
4.9. Tumorsphere Formation
4.10. Immunofluorescence Staining
4.11. Cell Cycle Distribution Analysis
4.12. Mitotic Index and Defect Measurement
4.13. Sodium Dodecyl Sulfate–Polyacrylamide Gel Electrophoresis and Western Blot Analysis
4.14. Tumor Xenograft Animal Study
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Prim. 2020, 6, 92. [Google Scholar] [CrossRef]
- Vermorken, J.B.; Mesia, R.; Rivera, F.; Remenar, E.; Kawecki, A.; Rottey, S.; Erfan, J.; Zabolotnyy, D.; Kienzer, H.-R.; Cupissol, D.; et al. Platinum-Based Chemotherapy plus Cetuximab in Head and Neck Cancer. New Engl. J. Med. 2008, 359, 1116–1127. [Google Scholar] [CrossRef]
- Burtness, B.; Harrington, K.J.; Greil, R.; Soulières, D.; Tahara, M.; de Castro, G., Jr. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): A randomised, open-label, phase 3 study. Lancet 2019, 394, 1915–1928. [Google Scholar] [CrossRef]
- Chang, J.H.; Wu, C.C.; Yuan, K.S.; Wu, A.T.H.; Wu, S.Y. Locoregionally recurrent head and neck squamous cell carcinoma: Incidence, survival, prognostic factors, and treatment outcomes. Oncotarget 2017, 8, 55600–55612. [Google Scholar] [CrossRef]
- Wakasugi, T.; Nguyen, T.N.; Takeuchi, S.; Ohkubo, J.-I.; Suzuki, H. Pattern of Recurrence After Platinum-Containing Definitive Therapy and Efficacy of Salvage Treatment for Recurrence in Patients with Squamous Cell Carcinoma of the Head and Neck. Front. Oncol. 2022, 12, 876193. [Google Scholar] [CrossRef]
- Leonce, C.; Saintigny, P.; Ortiz-Cuaran, S. Cell-Intrinsic Mechanisms of Drug Tolerance to Systemic Therapies in Cancer. Mol. Cancer Res. 2022, 20, 11–29. [Google Scholar] [CrossRef]
- De Conti, G.; Dias, M.H.; Bernards, R. Fighting Drug Resistance through the Targeting of Drug-Tolerant Persister Cells. Cancers 2021, 13, 1118. [Google Scholar] [CrossRef]
- Qin, S.; Jiang, J.; Lu, Y.; Nice, E.C.; Huang, C.; Zhang, J.; He, W. Emerging role of tumor cell plasticity in modifying therapeutic response. Signal Transduct. Target. Ther. 2020, 5, 228. [Google Scholar] [CrossRef]
- Rehman, S.K.; Haynes, J.; Collignon, E.; Brown, K.R.; Wang, Y.; Nixon, A.M.L.; Bruce, J.P.; Wintersinger, J.A.; Singh Mer, A.; Lo, E.B.L.; et al. Colorectal Cancer Cells Enter a Diapause-like DTP State to Survive Chemotherapy. Cell 2021, 184, 226–242.e21. [Google Scholar] [CrossRef] [PubMed]
- Hussein, A.M.; Balachandar, N.; Mathieu, J.; Ruohola-Baker, H. Molecular Regulators of Embryonic Diapause and Cancer Diapause-like State. Cells 2022, 11, 2929. [Google Scholar] [CrossRef] [PubMed]
- Mikubo, M.; Inoue, Y.; Liu, G.; Tsao, M.-S. Mechanism of Drug Tolerant Persister Cancer Cells: The Landscape and Clinical Implication for Therapy. J. Thorac. Oncol. 2021, 16, 1798–1809. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Marsolier, J.; Prompsy, P.; Durand, A.; Lyne, A.-M.; Landragin, C.; Trouchet, A.; Bento, S.T.; Eisele, A.; Foulon, S.; Baudre, L.; et al. H3K27me3 conditions chemotolerance in triple-negative breast cancer. Nat. Genet. 2022, 54, 459–468. [Google Scholar] [CrossRef]
- Van Gils, N.; Verhagen, H.J.M.P.; Broux, M.; Martiáñez, T.; Denkers, F.; Vermue, E.; Rutten, A.; Csikós, T.; Demeyer, S.; Çil, M.; et al. Targeting histone methylation to reprogram the transcriptional state that drives survival of drug-tolerant myeloid leukemia persisters. iScience 2022, 25, 105013. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, J.; Ma, Y.; Wu, C.; Cui, W.; Wang, L. Histone methyltransferase and drug resistance in cancers. J. Exp. Clin. Cancer Res. 2020, 39, 173. [Google Scholar] [CrossRef]
- Højfeldt, J.W.; Agger, K.; Helin, K. Histone lysine demethylases as targets for anticancer therapy. Nat. Rev. Drug Discov. 2013, 12, 917–930. [Google Scholar] [CrossRef]
- Gaillard, S.; Charasson, V.; Ribeyre, C.; Salifou, K.; Pillaire, M.-J.; Hoffmann, J.-S.; Constantinou, A.; Trouche, D.; Vandromme, M. KDM5A and KDM5B histone-demethylases contribute to HU-induced replication stress response and tolerance. Biology Open 2021, 10, bio057729. [Google Scholar] [CrossRef]
- Vinogradova, M.; Gehling, V.S.; Gustafson, A.; Arora, S.; Tindell, C.A.; Wilson, C.; Williamson, K.E.; Guler, G.D.; Gangurde, P.; Manieri, W.; et al. An inhibitor of KDM5 demethylases reduces survival of drug-tolerant cancer cells. Nat. Chem. Biol. 2016, 12, 531–538. [Google Scholar] [CrossRef]
- Dorna, D.; Paluszczak, J. The Emerging Significance of Histone Lysine Demethylases as Prognostic Markers and Therapeutic Targets in Head and Neck Cancers. Cells 2022, 11, 1023. [Google Scholar] [CrossRef] [PubMed]
- Plch, J.; Hrabeta, J.; Eckschlager, T. KDM5 demethylases and their role in cancer cell chemoresistance. Int. J. Cancer 2019, 144, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Chauvistré, H.; Shannan, B.; Daignault-Mill, S.M.; Ju, R.J.; Picard, D.; Egetemaier, S.; Váraljai, R.; Gibhardt, C.S.; Sechi, A.; Kaschani, F.; et al. Persister state-directed transitioning and vulnerability in melanoma. Nat. Commun. 2022, 13, 3055. [Google Scholar] [CrossRef] [PubMed]
- Spangle, J.M.; Dreijerink, K.M.; Groner, A.C.; Cheng, H.; Ohlson, C.E.; Reyes, J.; Lin, C.Y.; Bradner, J.; Zhao, J.J.; Roberts, T.M.; et al. PI3K/AKT Signaling Regulates H3K4 Methylation in Breast Cancer. Cell Rep. 2016, 15, 2692–2704. [Google Scholar] [CrossRef] [PubMed]
- Broad, A.J.; DeLuca, K.F.; DeLuca, J.G. Aurora B kinase is recruited to multiple discrete kinetochore and centromere regions in human cells. J. Cell Biol. 2020, 219, e201905144. [Google Scholar] [CrossRef] [PubMed]
- Su, C.-C.; Chen, N.-C.; Chyau, C.-C.; Tseng, H.-C.; Chou, F.-P. Induction of Mitotic Catastrophe via Inhibition of Aurora B by Ionizing Radiation with Additive of Mulberry Water Extract in Human Bladder Cancer Cells. Integr. Cancer Ther. 2018, 18, 1534735418808586. [Google Scholar] [CrossRef] [PubMed]
- Borah, N.A.; Reddy, M.M. Aurora Kinase B Inhibition: A Potential Therapeutic Strategy for Cancer. Molecules 2021, 26, 1981. [Google Scholar] [CrossRef]
- Arseneault, M.; Monlong, J.; Vasudev, N.S.; Laskar, R.S.; Safisamghabadi, M.; Harnden, P.; Egevad, L.; Nourbehesht, N.; Panichnantakul, P.; Holcatova, I.; et al. Loss of chromosome Y leads to down regulation of KDM5D and KDM6C epigenetic modifiers in clear cell renal cell carcinoma. Sci. Rep. 2017, 7, 44876. [Google Scholar] [CrossRef]
- Shen, X.; Hu, K.; Cheng, G.; Xu, L.; Chen, Z.; Du, P.; Zhuang, Z. KDM5D inhibit epithelial-mesenchymal transition of gastric cancer through demethylation in the promoter of Cul4A in male. J. Cell. Biochem. 2019, 120, 12247–12258. [Google Scholar] [CrossRef]
- Komura, K.; Yoshikawa, Y.; Shimamura, T.; Chakraborty, G.; Gerke, T.A.; Hinohara, K.; Chadalavada, K.; Jeong, S.H.; Armenia, J.; Du, S.-Y.; et al. ATR inhibition controls aggressive prostate tumors deficient in Y-linked histone demethylase KDM5D. J. Clin. Investig. 2018, 128, 2979–2995. [Google Scholar] [CrossRef]
- Therasse, P.; Arbuck, S.G.; Eisenhauer, E.A.; Wanders, J.; Kaplan, R.S.; Rubinstein, L.; Verweij, J.; Van Glabbeke, M.; van Oosterom, A.T.; Christian, M.C.; et al. New Guidelines to Evaluate the Response to Treatment in Solid Tumors. JNCI J. Natl. Cancer Inst. 2000, 92, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Sano, D.; Fujisawa, T.; Tokuhisa, M.; Shimizu, M.; Sakagami, T.; Hatano, T.; Nishimura, G.; Ichikawa, Y.; Iwai, H.; Oridate, N. Real-world Treatment Outcomes of the EXTREME Regimen as First-line Therapy for Recurrent/Metastatic Squamous Cell Carcinoma of the Head and Neck: A Multi-center Retrospective Cohort Study in Japan. Anticancer. Res. 2019, 39, 6819. [Google Scholar] [CrossRef]
- Kawakami, R.; Mashima, T.; Kawata, N.; Kumagai, K.; Migita, T.; Sano, T.; Mizunuma, N.; Yamaguchi, K.; Seimiya, H. ALDH1A3-mTOR axis as a therapeutic target for anticancer drug-tolerant persister cells in gastric cancer. Cancer Sci. 2020, 111, 962–973. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Dericks, L.; Froment, L.; Boesch, R.; Schmid, R.A.; Karoubi, G. Cisplatin-resistant cells in malignant pleural mesothelioma cell lines show ALDHhighCD44+ phenotype and sphere-forming capacity. BMC Cancer 2014, 14, 304. [Google Scholar] [CrossRef] [PubMed]
- Kurth, I.; Hein, L.; Mäbert, K.; Peitzsch, C.; Koi, L.; Cojoc, M.; Kunz-Schughart, L.; Baumann, M.; Dubrovska, A. Cancer stem cell related markers of radioresistance in head and neck squamous cell carcinoma. Oncotarget 2015, 6, 34494–34509. [Google Scholar] [CrossRef] [PubMed]
- Gully, C.P.; Velazquez-Torres, G.; Shin, J.-H.; Fuentes-Mattei, E.; Wang, E.; Carlock, C.; Chen, J.; Rothenberg, D.; Adams, H.P.; Choi, H.H.; et al. Aurora B kinase phosphorylates and instigates degradation of p53. Proc. Natl. Acad. Sci. USA 2012, 109, E1513–E1522. [Google Scholar] [CrossRef]
- Durinikova, E.; Kozovska, Z.; Poturnajova, M.; Plava, J.; Cierna, Z.; Babelova, A.; Bohovic, R.; Schmidtova, S.; Tomas, M.; Kucerova, L.; et al. ALDH1A3 upregulation and spontaneous metastasis formation is associated with acquired chemoresistance in colorectal cancer cells. BMC Cancer 2018, 18, 848. [Google Scholar] [CrossRef] [PubMed]
- Owiti, N.A.; Nagel, Z.D.; Engelward, B.P. Fluorescence Sheds Light on DNA Damage, DNA Repair, and Mutations. Trends Cancer 2021, 7, 240–248. [Google Scholar] [CrossRef]
- Kitamura, N.; Sento, S.; Yoshizawa, Y.; Sasabe, E.; Kudo, Y.; Yamamoto, T. Current Trends and Future Prospects of Molecular Targeted Therapy in Head and Neck Squamous Cell Carcinoma. Int. J. Mol. Sci. 2020, 22, 240. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, G.; Condello, S.; Huang, H.; Cardenas, H.; Tanner, E.J.; Wei, J.; Ji, Y.; Li, J.; Tan, Y.; et al. Frizzled-7 Identifies Platinum-Tolerant Ovarian Cancer Cells Susceptible to Ferroptosis. Cancer Res. 2021, 81, 384–399. [Google Scholar] [CrossRef]
- You, J.H.; Lee, J.; Roh, J.-L. PGRMC1-dependent lipophagy promotes ferroptosis in paclitaxel-tolerant persister cancer cells. J. Exp. Clin. Cancer Res. 2021, 40, 350. [Google Scholar] [CrossRef] [PubMed]
- You, J.H.; Lee, J.; Roh, J.L. Mitochondrial pyruvate carrier 1 regulates ferroptosis in drug-tolerant persister head and neck cancer cells via epithelial-mesenchymal transition. Cancer Lett. 2021, 507, 40–54. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Brady, S.W.; Ma, X.; Shen, S.; Zhang, Y.; Li, Y.; Szlachta, K.; Dong, L.; Liu, Y.; Yang, F.; et al. Therapy-induced mutations drive the genomic landscape of relapsed acute lymphoblastic leukemia. Blood 2020, 135, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Dhimolea, E.; de Matos Simoes, R.; Kansara, D.; Al’Khafaji, A.; Bouyssou, J.; Weng, X.; Sharma, S.; Raja, J.; Awate, P.; Shirasaki, R.; et al. An Embryonic Diapause-like Adaptation with Suppressed Myc Activity Enables Tumor Treatment Persistence. Cancer Cell 2021, 39, 240–256.e11. [Google Scholar] [CrossRef]
- Brier, A.-S.B.; Loft, A.; Madsen, J.G.S.; Rosengren, T.; Nielsen, R.; Schmidt, S.F.; Liu, Z.; Yan, Q.; Gronemeyer, H.; Mandrup, S. The KDM5 family is required for activation of pro-proliferative cell cycle genes during adipocyte differentiation. Nucleic Acids Res. 2017, 45, 1743–1759. [Google Scholar] [CrossRef] [PubMed]
- Porcelli, L.; Guida, G.; Quatrale, A.E.; Cocco, T.; Sidella, L.; Maida, I.; Iacobazzi, R.M.; Ferretta, A.; Stolfa, D.A.; Strippoli, S.; et al. Aurora kinase B inhibition reduces the proliferation of metastatic melanoma cells and enhances the response to chemotherapy. J. Transl. Med. 2015, 13, 26. [Google Scholar] [CrossRef] [PubMed]
- Bavetsias, V.; Linardopoulos, S. Aurora Kinase Inhibitors: Current Status and Outlook. Front. Oncol. 2015, 5, 278. [Google Scholar] [CrossRef]
- Ye, H.; Yu, T.; Temam, S.; Ziober, B.L.; Wang, J.; Schwartz, J.L.; Mao, L.; Wong, D.T.; Zhou, X. Transcriptomic dissection of tongue squamous cell carcinoma. BMC Genom. 2008, 9, 69. [Google Scholar] [CrossRef]
- McDermott, S.C.; Rodriguez-Ramirez, C.; McDermott, S.P.; Wicha, M.S.; Nör, J.E. FGFR signaling regulates resistance of head and neck cancer stem cells to cisplatin. Oncotarget 2018, 9, 25148–25165. [Google Scholar] [CrossRef]
- Liu, S.; Ren, B.; Gao, H.; Liao, S.; Zhai, Y.-X.; Li, S.; Su, X.-J.; Jin, P.; Stroncek, D.; Xu, Z.; et al. Over-expression of BAG-1 in head and neck squamous cell carcinomas (HNSCC) is associated with cisplatin-resistance. J. Transl. Med. 2017, 15, 189. [Google Scholar] [CrossRef]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624.e24. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Outcome | KDM5D | χ2 | p | |
|---|---|---|---|---|
| High | Low | |||
| Total Cohort (n = 100) | ||||
| No Responses (SD/PD) * | 21 (70.0%) | 9 (30.0%) | 7.83 | 0.004 |
| Favorable Responses (CR/PR) * | 26 (37.1%) | 44 (62.9%) | ||
| Platinum Responder cohort (n = 70) | ||||
| Early Recurrence (<12 months) | 12 (60.0%) | 8 (40.0%) | 6.26 | 0.012 |
| No/Late Recurrence (>12 months) | 14 (28.0%) | 36 (72.0%) | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, T.-M.; Huang, C.-M.; Setiawan, S.A.; Hsieh, M.-S.; Sheen, C.-C.; Yeh, C.-T. KDM5D Histone Demethylase Identifies Platinum-Tolerant Head and Neck Cancer Cells Vulnerable to Mitotic Catastrophe. Int. J. Mol. Sci. 2023, 24, 5310. https://doi.org/10.3390/ijms24065310
Chen T-M, Huang C-M, Setiawan SA, Hsieh M-S, Sheen C-C, Yeh C-T. KDM5D Histone Demethylase Identifies Platinum-Tolerant Head and Neck Cancer Cells Vulnerable to Mitotic Catastrophe. International Journal of Molecular Sciences. 2023; 24(6):5310. https://doi.org/10.3390/ijms24065310
Chicago/Turabian StyleChen, Tsung-Ming, Chih-Ming Huang, Syahru Agung Setiawan, Ming-Shou Hsieh, Chih-Chi Sheen, and Chi-Tai Yeh. 2023. "KDM5D Histone Demethylase Identifies Platinum-Tolerant Head and Neck Cancer Cells Vulnerable to Mitotic Catastrophe" International Journal of Molecular Sciences 24, no. 6: 5310. https://doi.org/10.3390/ijms24065310
APA StyleChen, T.-M., Huang, C.-M., Setiawan, S. A., Hsieh, M.-S., Sheen, C.-C., & Yeh, C.-T. (2023). KDM5D Histone Demethylase Identifies Platinum-Tolerant Head and Neck Cancer Cells Vulnerable to Mitotic Catastrophe. International Journal of Molecular Sciences, 24(6), 5310. https://doi.org/10.3390/ijms24065310