

Epigenetic Regulation Mediated by Sphingolipids in Cancer





Abstract
1. Introduction
2. Epigenetic Modifications
3. Epigenetics in Cancer
4. The Sphingolipid Pathway
5. The Sphingolipid Pathway and Epigenetics
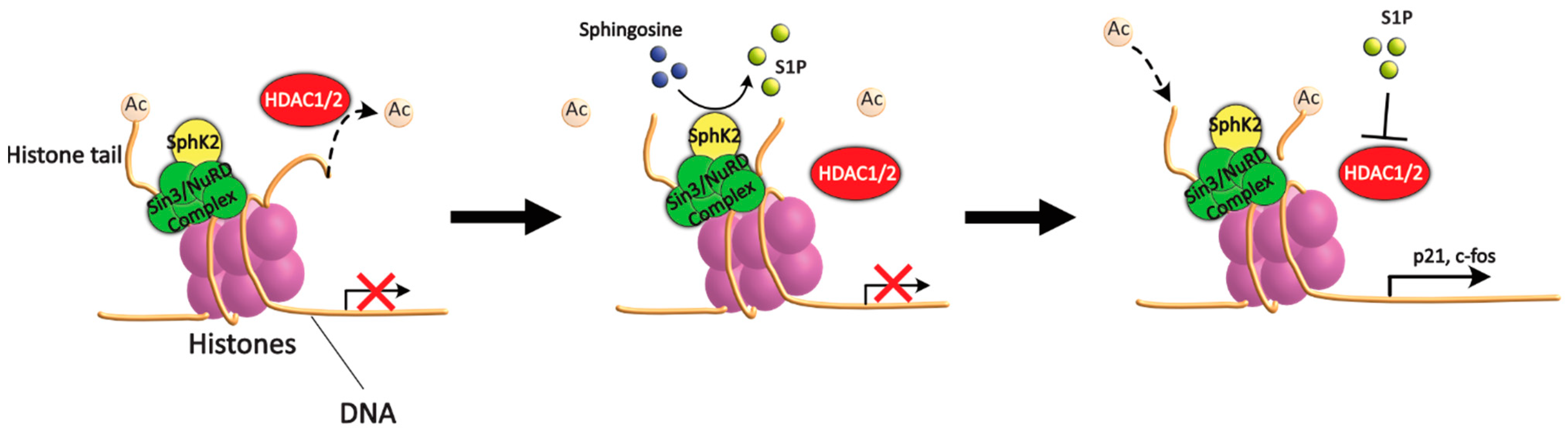
5.1. Sphingolipids and HDAC
5.2. Δ2-HDE as a Novel Epigenetic Regulator
5.3. Ceramide and Epigenetics
6. The Tumour Microenvironment and Epigenetic Balance
6.1. Hypoxia and the Sphingolipid Pathway
6.2. Acidosis and the Sphingolipid Pathway
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Guo, M.; Peng, Y.; Gao, A.; Du, C.; Herman, J.G. Epigenetic heterogeneity in cancer. Biomark. Res. 2019, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho, D.D.; Sharma, S.; You, J.S.; Su, S.F.; Taberlay, P.C.; Kelly, T.K.; Yang, X.; Liang, G.; Jones, P.A. DNA methylation screening identifies driver epigenetic events of cancer cell survival. Cancer Cell 2012, 21, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Alegria-Torres, J.A.; Baccarelli, A.; Bollati, V. Epigenetics and lifestyle. Epigenomics 2011, 3, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Bendridi, N.; Selmi, A.; Balcerczyk, A.; Pirola, L. Ketone Bodies as Metabolites and Signalling Molecules at the Crossroad between Inflammation and Epigenetic Control of Cardiometabolic Disorders. Int. J. Mol. Sci. 2022, 23, 14564. [Google Scholar] [CrossRef]
- Druesne, N.; Pagniez, A.; Mayeur, C.; Thomas, M.; Cherbuy, C.; Duee, P.H.; Martel, P.; Chaumontet, C. Diallyl disulfide (DADS) increases histone acetylation and p21(waf1/cip1) expression in human colon tumor cell lines. Carcinogenesis 2004, 25, 1227–1236. [Google Scholar] [CrossRef]
- Johnson, I.T.; Belshaw, N.J. Environment, diet and CpG island methylation: Epigenetic signals in gastrointestinal neoplasia. Food Chem. Toxicol. 2008, 46, 1346–1359. [Google Scholar] [CrossRef]
- Nagahashi, M.; Ramachandran, S.; Kim, E.Y.; Allegood, J.C.; Rashid, O.M.; Yamada, A.; Zhao, R.; Milstien, S.; Zhou, H.; Spiegel, S.; et al. Sphingosine-1-phosphate produced by sphingosine kinase 1 promotes breast cancer progression by stimulating angiogenesis and lymphangiogenesis. Cancer Res. 2012, 72, 726–735. [Google Scholar] [CrossRef]
- Wang, P.; Yuan, Y.; Lin, W.; Zhong, H.; Xu, K.; Qi, X. Roles of sphingosine-1-phosphate signaling in cancer. Cancer Cell Int. 2019, 19, 295. [Google Scholar] [CrossRef]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Buck-Koehntop, B.A.; Defossez, P.A. On how mammalian transcription factors recognize methylated DNA. Epigenetics 2013, 8, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Spruijt, C.G.; Vermeulen, M. DNA methylation: Old dog, new tricks? Nat. Struct. Mol. Biol. 2014, 21, 949–954. [Google Scholar] [CrossRef] [PubMed]
- McGinty, R.K.; Tan, S. Nucleosome structure and function. Chem. Rev. 2015, 115, 2255–2273. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Kuo, M.H.; Allis, C.D. Roles of histone acetyltransferases and deacetylases in gene regulation. BioEssays News Rev. Mol. Cell. Dev. Biol. 1998, 20, 615–626. [Google Scholar] [CrossRef]
- Yang, X.J.; Seto, E. The Rpd3/Hda1 family of lysine deacetylases: From bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 2008, 9, 206–218. [Google Scholar] [CrossRef]
- Taunton, J.; Hassig, C.A.; Schreiber, S.L. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science 1996, 272, 408–411. [Google Scholar] [CrossRef]
- Yang, X.J.; Gregoire, S. Class II histone deacetylases: From sequence to function, regulation, and clinical implication. Mol. Cell Biol. 2005, 25, 2873–2884. [Google Scholar] [CrossRef]
- Seidel, C.; Schnekenburger, M.; Dicato, M.; Diederich, M. Histone deacetylase 6 in health and disease. Epigenomics 2015, 7, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Cueto, M.A.; Asselbergs, F.; Atadja, P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J. Biol. Chem. 2002, 277, 25748–25755. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Hu, Q.; Kaufman, A.; D’Ercole, A.J.; Ye, P. Developmental expression of histone deacetylase 11 in the murine brain. J. Neurosci. Res. 2008, 86, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J.; Seto, E. Collaborative spirit of histone deacetylases in regulating chromatin structure and gene expression. Curr. Opin. Genet. Dev. 2003, 13, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, Z.; Jia, J.; Du, T.; Zhang, N.; Tang, Y.; Fang, Y.; Fang, D. Overview of Histone Modification. Adv. Exp. Med. Biol. 2021, 1283, 1–16. [Google Scholar] [CrossRef]
- Zha, J.J.; Tang, Y.; Wang, Y.L. Role of mono-ADP-ribosylation histone modification (Review). Exp. Ther. Med. 2021, 21, 577. [Google Scholar] [CrossRef]
- Ryu, H.Y.; Hochstrasser, M. Histone sumoylation and chromatin dynamics. Nucleic Acids. Res. 2021, 49, 6043–6052. [Google Scholar] [CrossRef]
- Mattiroli, F.; Penengo, L. Histone Ubiquitination: An Integrative Signaling Platform in Genome Stability. Trend Genet. 2021, 37, 566–581. [Google Scholar] [CrossRef]
- Yao, Q.; Chen, Y.; Zhou, X. The roles of microRNAs in epigenetic regulation. Curr. Opin. Chem. Biol. 2019, 51, 11–17. [Google Scholar] [CrossRef]
- Orom, U.A.; Nielsen, F.C.; Lund, A.H. MicroRNA-10a binds the 5’UTR of ribosomal protein mRNAs and enhances their translation. Mol. Cell 2008, 30, 460–471. [Google Scholar] [CrossRef]
- Esteller, M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev. Genet. 2007, 8, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Ravenel, J.D.; Broman, K.W.; Perlman, E.J.; Niemitz, E.L.; Jayawardena, T.M.; Bell, D.W.; Haber, D.A.; Uejima, H.; Feinberg, A.P. Loss of imprinting of insulin-like growth factor-II (IGF2) gene in distinguishing specific biologic subtypes of Wilms tumor. J. Natl. Cancer Inst. 2001, 93, 1698–1703. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, A.; Feinberg, A.P. Loss of imprinting of IGF2: A common epigenetic modifier of intestinal tumor risk. Cancer Res. 2005, 65, 11236–11240. [Google Scholar] [CrossRef] [PubMed]
- Avnet, S.; Sciacca, L.; Salerno, M.; Gancitano, G.; Cassarino, M.F.; Longhi, A.; Zakikhani, M.; Carboni, J.M.; Gottardis, M.; Giunti, A.; et al. Insulin receptor isoform A and insulin-like growth factor II as additional treatment targets in human osteosarcoma. Cancer Res. 2009, 69, 2443–2452. [Google Scholar] [CrossRef] [PubMed]
- Gama-Sosa, M.A.; Slagel, V.A.; Trewyn, R.W.; Oxenhandler, R.; Kuo, K.C.; Gehrke, C.W.; Ehrlich, M. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res. 1983, 11, 6883–6894. [Google Scholar] [CrossRef]
- Lee, S.T.; Wiemels, J.L. Genome-wide CpG island methylation and intergenic demethylation propensities vary among different tumor sites. Nucleic Acids Res. 2016, 44, 1105–1117. [Google Scholar] [CrossRef]
- Wolff, E.M.; Byun, H.M.; Han, H.F.; Sharma, S.; Nichols, P.W.; Siegmund, K.D.; Yang, A.S.; Jones, P.A.; Liang, G. Hypomethylation of a LINE-1 promoter activates an alternate transcript of the MET oncogene in bladders with cancer. PLoS Genet. 2010, 6, e1000917. [Google Scholar] [CrossRef]
- Wu, S.P.; Cooper, B.T.; Bu, F.; Bowman, C.J.; Killian, J.K.; Serrano, J.; Wang, S.; Jackson, T.M.; Gorovets, D.; Shukla, N.; et al. DNA Methylation-Based Classifier for Accurate Molecular Diagnosis of Bone Sarcomas. JCO Precis. Oncol. 2017, 2017. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Z.; Yang, X.; Lu, W.; Chen, Y.; Lin, Y.; Wang, J.; Lin, S.; Yun, J.P. H3K27 acetylation activated-COL6A1 promotes osteosarcoma lung metastasis by repressing STAT1 and activating pulmonary cancer-associated fibroblasts. Theranostics 2021, 11, 1473–1492. [Google Scholar] [CrossRef]
- Alhmoud, J.F.; Woolley, J.F.; Al Moustafa, A.E.; Malki, M.I. DNA Damage/Repair Management in Cancers. Cancers 2020, 12, 1050. [Google Scholar] [CrossRef] [PubMed]
- van Attikum, H.; Gasser, S.M. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol. 2009, 19, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Thomson, J.M.; Hemann, M.T.; Hernando-Monge, E.; Mu, D.; Goodson, S.; Powers, S.; Cordon-Cardo, C.; Lowe, S.W.; Hannon, G.J.; et al. A microRNA polycistron as a potential human oncogene. Nature 2005, 435, 828–833. [Google Scholar] [CrossRef]
- Hajibabaie, F.; Abedpoor, N.; Assareh, N.; Tabatabaiefar, M.A.; Shariati, L.; Zarrabi, A. The Importance of SNPs at miRNA Binding Sites as Biomarkers of Gastric and Colorectal Cancers: A Systematic Review. J. Pers. Med. 2022, 12, 456. [Google Scholar] [CrossRef]
- Futerman, A.H.; Riezman, H. The ins and outs of sphingolipid synthesis. Trends Cell Biol. 2005, 15, 312–318. [Google Scholar] [CrossRef]
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An overview of sphingolipid metabolism: From synthesis to breakdown. Adv. Exp. Med. Biol. 2010, 688, 1–23. [Google Scholar] [CrossRef]
- Haimovitz-Friedman, A.; Kolesnick, R.N.; Fuks, Z. Ceramide signaling in apoptosis. Br. Med. Bull. 1997, 53, 539–553. [Google Scholar] [CrossRef]
- Garcia-Gonzalez, V.; Diaz-Villanueva, J.F.; Galindo-Hernandez, O.; Martinez-Navarro, I.; Hurtado-Ureta, G.; Perez-Arias, A.A. Ceramide Metabolism Balance, a Multifaceted Factor in Critical Steps of Breast Cancer Development. Int. J. Mol. Sci. 2018, 19, 2527. [Google Scholar] [CrossRef]
- Carpinteiro, A.; Becker, K.A.; Japtok, L.; Hessler, G.; Keitsch, S.; Pozgajova, M.; Schmid, K.W.; Adams, C.; Muller, S.; Kleuser, B.; et al. Regulation of hematogenous tumor metastasis by acid sphingomyelinase. EMBO Mol. Med. 2015, 7, 714–734. [Google Scholar] [CrossRef]
- Gomez-Munoz, A.; Gangoiti, P.; Granado, M.H.; Arana, L.; Ouro, A. Ceramide-1-phosphate in cell survival and inflammatory signaling. Adv. Exp. Med. Biol. 2010, 688, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Coant, N.; Sakamoto, W.; Mao, C.; Hannun, Y.A. Ceramidases, roles in sphingolipid metabolism and in health and disease. Adv. Biol. Regul. 2017, 63, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Cao, Y. The sphingomyelin synthase family: Proteins, diseases, and inhibitors. Biol. Chem. 2017, 398, 1319–1325. [Google Scholar] [CrossRef]
- Taniguchi, M.; Okazaki, T. The role of sphingomyelin and sphingomyelin synthases in cell death, proliferation and migration-from cell and animal models to human disorders. Biochim. Biophys. Acta 2014, 1841, 692–703. [Google Scholar] [CrossRef]
- Igarashi, N.; Okada, T.; Hayashi, S.; Fujita, T.; Jahangeer, S.; Nakamura, S. Sphingosine kinase 2 is a nuclear protein and inhibits DNA synthesis. J. Biol. Chem. 2003, 278, 46832–46839. [Google Scholar] [CrossRef] [PubMed]
- Pitson, S.M.; Moretti, P.A.; Zebol, J.R.; Lynn, H.E.; Xia, P.; Vadas, M.A.; Wattenberg, B.W. Activation of sphingosine kinase 1 by ERK1/2-mediated phosphorylation. EMBO J. 2003, 22, 5491–5500. [Google Scholar] [CrossRef]
- Ding, G.; Sonoda, H.; Yu, H.; Kajimoto, T.; Goparaju, S.K.; Jahangeer, S.; Okada, T.; Nakamura, S.I. Protein kinase D-mediated phosphorylation and nuclear export of sphingosine kinase 2. J. Biol. Chem. 2007, 282, 27493–27502. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Liu, Y.; Zou, F. Sensitization of human colon cancer cells to sodium butyrate-induced apoptosis by modulation of sphingosine kinase 2 and protein kinase D. Exp. Cell Res. 2012, 318, 43–52. [Google Scholar] [CrossRef]
- Weigert, A.; Schiffmann, S.; Sekar, D.; Ley, S.; Menrad, H.; Werno, C.; Grosch, S.; Geisslinger, G.; Brune, B. Sphingosine kinase 2 deficient tumor xenografts show impaired growth and fail to polarize macrophages towards an anti-inflammatory phenotype. Int. J. Cancer 2009, 125, 2114–2121. [Google Scholar] [CrossRef]
- Liu, W.; Ning, J.; Li, C.; Hu, J.; Meng, Q.; Lu, H.; Cai, L. Overexpression of Sphk2 is associated with gefitinib resistance in non-small cell lung cancer. Tumour Biol. 2016, 37, 6331–6336. [Google Scholar] [CrossRef]
- Rosen, H.; Stevens, R.C.; Hanson, M.; Roberts, E.; Oldstone, M.B. Sphingosine-1-phosphate and its receptors: Structure, signaling, and influence. Annu. Rev. Biochem. 2013, 82, 637–662. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, J.H.; Song, W.K.; Kim, J.H.; Chun, J.S. Sphingosine 1-phosphate activates Erk-1/-2 by transactivating epidermal growth factor receptor in rat-2 cells. IUBMB Life 2000, 50, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Bonnaud, S.; Niaudet, C.; Legoux, F.; Corre, I.; Delpon, G.; Saulquin, X.; Fuks, Z.; Gaugler, M.H.; Kolesnick, R.; Paris, F. Sphingosine-1-phosphate activates the AKT pathway to protect small intestines from radiation-induced endothelial apoptosis. Cancer Res. 2010, 70, 9905–9915. [Google Scholar] [CrossRef]
- Sato, K.; Kon, J.; Tomura, H.; Osada, M.; Murata, N.; Kuwabara, A.; Watanabe, T.; Ohta, H.; Ui, M.; Okajima, F. Activation of phospholipase C-Ca2+ system by sphingosine 1-phosphate in CHO cells transfected with Edg-3, a putative lipid receptor. FEBS Lett. 1999, 443, 25–30. [Google Scholar] [CrossRef]
- Dusaban, S.S.; Chun, J.; Rosen, H.; Purcell, N.H.; Brown, J.H. Sphingosine 1-phosphate receptor 3 and RhoA signaling mediate inflammatory gene expression in astrocytes. J. Neuroinflamm. 2017, 14, 111. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, H.A.; Pham, D.H.; Zebol, J.R.; Moretti, P.A.; Peterson, A.L.; Leclercq, T.M.; Chan, H.; Powell, J.A.; Pitman, M.R.; Samuel, M.S.; et al. An oncogenic role for sphingosine kinase 2. Oncotarget 2016, 7, 64886–64899. [Google Scholar] [CrossRef] [PubMed]
- Le Stunff, H.; Peterson, C.; Liu, H.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate and lipid phosphohydrolases. Biochim. Biophys. Acta 2002, 1582, 8–17. [Google Scholar] [CrossRef]
- Serra, M.; Saba, J.D. Sphingosine 1-phosphate lyase, a key regulator of sphingosine 1-phosphate signaling and function. Adv. Enzym. Regul. 2010, 50, 349–362. [Google Scholar] [CrossRef]
- Kumar, A.; Byun, H.S.; Bittman, R.; Saba, J.D. The sphingolipid degradation product trans-2-hexadecenal induces cytoskeletal reorganization and apoptosis in a JNK-dependent manner. Cell Signal 2011, 23, 1144–1152. [Google Scholar] [CrossRef]
- Amaegberi, N.V.; Semenkova, G.N.; Kvacheva, Z.B.; Lisovskaya, A.G.; Pinchuk, S.V.; Shadyro, O.I. 2-Hexadecenal inhibits growth of C6 glioma cells. Cell Biochem. Funct. 2019, 37, 281–289. [Google Scholar] [CrossRef]
- Hait, N.C.; Allegood, J.; Maceyka, M.; Strub, G.M.; Harikumar, K.B.; Singh, S.K.; Luo, C.; Marmorstein, R.; Kordula, T.; Milstien, S.; et al. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 2009, 325, 1254–1257. [Google Scholar] [CrossRef] [PubMed]
- Al Bitar, S.; Gali-Muhtasib, H. The Role of the Cyclin Dependent Kinase Inhibitor p21(cip1/waf1) in Targeting Cancer: Molecular Mechanisms and Novel Therapeutics. Cancers 2019, 11, 1475. [Google Scholar] [CrossRef] [PubMed]
- Tulchinsky, E. Fos family members: Regulation, structure and role in oncogenic transformation. Histol. Histopathol. 2000, 15, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Ihlefeld, K.; Claas, R.F.; Koch, A.; Pfeilschifter, J.M.; Meyer Zu Heringdorf, D. Evidence for a link between histone deacetylation and Ca(2)+ homoeostasis in sphingosine-1-phosphate lyase-deficient fibroblasts. Biochem. J. 2012, 447, 457–464. [Google Scholar] [CrossRef]
- Kariya, Y.; Kihara, A.; Ikeda, M.; Kikuchi, F.; Nakamura, S.; Hashimoto, S.; Choi, C.H.; Lee, Y.M.; Igarashi, Y. Products by the sphingosine kinase/sphingosine 1-phosphate (S1P) lyase pathway but not S1P stimulate mitogenesis. Genes Cells 2005, 10, 605–615. [Google Scholar] [CrossRef]
- Ebenezer, D.L.; Ramchandran, R.; Fu, P.; Mangio, L.A.; Suryadevara, V.; Ha, A.W.; Berdyshev, E.; Van Veldhoven, P.P.; Kron, S.J.; Schumacher, F.; et al. Nuclear Sphingosine-1-phosphate Lyase Generated ∆2-hexadecenal is A Regulator of HDAC Activity and Chromatin Remodeling in Lung Epithelial Cells. Cell Biochem. Biophys. 2021, 79, 575–592. [Google Scholar] [CrossRef]
- Ebenezer, D.L.; Fu, P.; Mangio, L.A.; Berdyshev, E.; Schumacher, F.; Kleuser, B.; Van Veldhoven, P.P.; Natarajan, V. Δ-2 Hexadecenal Generated from S1P by Nuclear S1P Lyase Is a Regulator of HDAC1/2 Activity and Histone Acetylation in Lung Epithelial Cells. Biochem. Mol. Biol. 2019, 33, 489.3. [Google Scholar] [CrossRef]
- Mukhopadhyay, A.; Tabanor, K.; Chaguturu, R.; Aldrich, J.V. Targeting inhibitor 2 of protein phosphatase 2A as a therapeutic strategy for prostate cancer treatment. Cancer Biol. Ther. 2013, 14, 962–972. [Google Scholar] [CrossRef]
- Seo, S.B.; McNamara, P.; Heo, S.; Turner, A.; Lane, W.S.; Chakravarti, D. Regulation of histone acetylation and transcription by INHAT, a human cellular complex containing the set oncoprotein. Cell 2001, 104, 119–130. [Google Scholar] [CrossRef]
- He, Q.; Wang, G.; Dasgupta, S.; Dinkins, M.; Zhu, G.; Bieberich, E. Characterization of an apical ceramide-enriched compartment regulating ciliogenesis. Mol. Biol. Cell 2012, 23, 3156–3166. [Google Scholar] [CrossRef]
- Seeley, E.S.; Nachury, M.V. Constructing and deconstructing roles for the primary cilium in tissue architecture and cancer. Methods Cell Biol. 2009, 94, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Basten, S.G.; Willekers, S.; Vermaat, J.S.; Slaats, G.G.; Voest, E.E.; van Diest, P.J.; Giles, R.H. Reduced cilia frequencies in human renal cell carcinomas versus neighboring parenchymal tissue. Cilia 2013, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Wooten-Blanks, L.G.; Song, P.; Senkal, C.E.; Ogretmen, B. Mechanisms of ceramide-mediated repression of the human telomerase reverse transcriptase promoter via deacetylation of Sp3 by histone deacetylase 1. FASEB J. 2007, 21, 3386–3397. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef]
- Pillai, S.R.; Damaghi, M.; Marunaka, Y.; Spugnini, E.P.; Fais, S.; Gillies, R.J. Causes, consequences, and therapy of tumors acidosis. Cancer Metastasis Rev. 2019, 38, 205–222. [Google Scholar] [CrossRef]
- Corbet, C.; Feron, O. Tumour acidosis: From the passenger to the driver’s seat. Nat. Rev. Cancer 2017, 17, 577–593. [Google Scholar] [CrossRef]
- Chano, T.; Avnet, S.; Kusuzaki, K.; Bonuccelli, G.; Sonveaux, P.; Rotili, D.; Mai, A.; Baldini, N. Tumour-specific metabolic adaptation to acidosis is coupled to epigenetic stability in osteosarcoma cells. Am. J. Cancer Res. 2016, 6, 859–875. [Google Scholar]
- Al Tameemi, W.; Dale, T.P.; Al-Jumaily, R.M.K.; Forsyth, N.R. Hypoxia-Modified Cancer Cell Metabolism. Front. Cell Dev. Biol. 2019, 7, 4. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef]
- Semenza, G.L.; Agani, F.; Booth, G.; Forsythe, J.; Iyer, N.; Jiang, B.H.; Leung, S.; Roe, R.; Wiener, C.; Yu, A. Structural and functional analysis of hypoxia-inducible factor 1. Kidney Int. 1997, 51, 553–555. [Google Scholar] [CrossRef] [PubMed]
- Masoud, G.N.; Li, W. HIF-1alpha pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Bruick, R.K.; McKnight, S.L. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 2001, 294, 1337–1340. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, J.F.; Dachs, G.U.; Gleadle, J.M.; Maxwell, P.H.; Pugh, C.W.; Stratford, I.J.; Wood, S.M.; Ratcliffe, P.J. Hypoxia response elements. Oncol. Res. 1997, 9, 327–332. [Google Scholar] [PubMed]
- Zhang, L.; Song, J.; Xin, X.; Sun, D.; Huang, H.; Chen, Y.; Zhang, T.; Zhang, Y. Hypoxia stimulates the migration and invasion of osteosarcoma via up-regulating the NUSAP1 expression. Open Med. 2021, 16, 1083–1089. [Google Scholar] [CrossRef]
- Collin, L.J.; Maliniak, M.L.; Cronin-Fenton, D.P.; Ahern, T.P.; Christensen, K.B.; Ulrichsen, S.P.; Damkier, P.; Hamilton-Dutoit, S.; Yacoub, R.; Christiansen, P.M.; et al. Hypoxia-inducible factor-1alpha expression and breast cancer recurrence in a Danish population-based case control study. Breast Cancer Res. 2021, 23, 103. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Batie, M.; Frost, J.; Frost, M.; Wilson, J.W.; Schofield, P.; Rocha, S. Hypoxia induces rapid changes to histone methylation and reprograms chromatin. Science 2019, 363, 1222–1226. [Google Scholar] [CrossRef]
- Choudhry, H.; Harris, A.L. Advances in Hypoxia-Inducible Factor Biology. Cell Metab. 2018, 27, 281–298. [Google Scholar] [CrossRef]
- Lee, J.S.; Kim, Y.; Bhin, J.; Shin, H.J.; Nam, H.J.; Lee, S.H.; Yoon, J.B.; Binda, O.; Gozani, O.; Hwang, D.; et al. Hypoxia-induced methylation of a pontin chromatin remodeling factor. Proc. Natl. Acad Sci. USA 2011, 108, 13510–13515. [Google Scholar] [CrossRef]
- Perez-Perri, J.I.; Dengler, V.L.; Audetat, K.A.; Pandey, A.; Bonner, E.A.; Urh, M.; Mendez, J.; Daniels, D.L.; Wappner, P.; Galbraith, M.D.; et al. The TIP60 Complex Is a Conserved Coactivator of HIF1A. Cell Rep. 2016, 16, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liang, X.; Che, G.; Chen, Y.; Luo, L.; Liu, K.; Xie, R.; Zeng, L. Molecular Classification of Genes Associated with Hypoxic Lipid Metabolism in Pancreatic Cancer. Biomolecules 2022, 12, 1533. [Google Scholar] [CrossRef] [PubMed]
- Tobias, F.; Hummon, A.B. Lipidomic comparison of 2D and 3D colon cancer cell culture models. J. Mass Spectrom. 2022, 57, e4880. [Google Scholar] [CrossRef]
- Hafizi, R.; Imeri, F.; Wenger, R.H.; Huwiler, A. S1P Stimulates Erythropoietin Production in Mouse Renal Interstitial Fibroblasts by S1P(1) and S1P(3) Receptor Activation and HIF-2alpha Stabilization. Int. J. Mol. Sci. 2021, 22, 9467. [Google Scholar] [CrossRef] [PubMed]
- Hait, N.C.; Maiti, A.; Xu, P.; Qi, Q.; Kawaguchi, T.; Okano, M.; Takabe, K.; Yan, L.; Luo, C. Regulation of hypoxia-inducible factor functions in the nucleus by sphingosine-1-phosphate. FASEB J. 2020, 34, 4293–4310. [Google Scholar] [CrossRef]
- Di Pompo, G.; Cortini, M.; Baldini, N.; Avnet, S. Acid Microenvironment in Bone Sarcomas. Cancers 2021, 13, 3848. [Google Scholar] [CrossRef]
- Kolosenko, I.; Avnet, S.; Baldini, N.; Viklund, J.; De Milito, A. Therapeutic implications of tumor interstitial acidification. Semin. Cancer Biol. 2017, 43, 119–133. [Google Scholar] [CrossRef]
- Spugnini, E.P.; Sonveaux, P.; Stock, C.; Perez-Sayans, M.; De Milito, A.; Avnet, S.; Garcia, A.G.; Harguindey, S.; Fais, S. Proton channels and exchangers in cancer. Biochim. Biophys. Acta 2015, 1848, 2715–2726. [Google Scholar] [CrossRef]
- Dioum, E.M.; Chen, R.; Alexander, M.S.; Zhang, Q.; Hogg, R.T.; Gerard, R.D.; Garcia, J.A. Regulation of hypoxia-inducible factor 2alpha signaling by the stress-responsive deacetylase sirtuin 1. Science 2009, 324, 1289–1293. [Google Scholar] [CrossRef]
- Lim, J.H.; Lee, Y.M.; Chun, Y.S.; Chen, J.; Kim, J.E.; Park, J.W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef]
- Cortini, M.; Armirotti, A.; Columbaro, M.; Longo, D.L.; Di Pompo, G.; Cannas, E.; Maresca, A.; Errani, C.; Longhi, A.; Righi, A.; et al. Exploring Metabolic Adaptations to the Acidic Microenvironment of Osteosarcoma Cells Unveils Sphingosine 1-Phosphate as a Valuable Therapeutic Target. Cancers 2021, 13, 311. [Google Scholar] [CrossRef] [PubMed]
- Pillai, S.; Mahmud, I.; Mahar, R.; Griffith, C.; Langsen, M.; Nguyen, J.; Wojtkowiak, J.W.; Swietach, P.; Gatenby, R.A.; Bui, M.M.; et al. Lipogenesis mediated by OGR1 regulates metabolic adaptation to acid stress in cancer cells via autophagy. Cell Rep. 2022, 39, 110796. [Google Scholar] [CrossRef] [PubMed]
- Corbet, C.; Pinto, A.; Martherus, R.; Santiago de Jesus, J.P.; Polet, F.; Feron, O. Acidosis Drives the Reprogramming of Fatty Acid Metabolism in Cancer Cells through Changes in Mitochondrial and Histone Acetylation. Cell Metab. 2016, 24, 311–323. [Google Scholar] [CrossRef]
- Gao, Z.; Wang, H.; Xiao, F.J.; Shi, X.F.; Zhang, Y.K.; Xu, Q.Q.; Zhang, X.Y.; Ha, X.Q.; Wang, L.S. SIRT1 mediates Sphk1/S1P-induced proliferation and migration of endothelial cells. Int. J. Biochem. Cell Biol. 2016, 74, 152–160. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
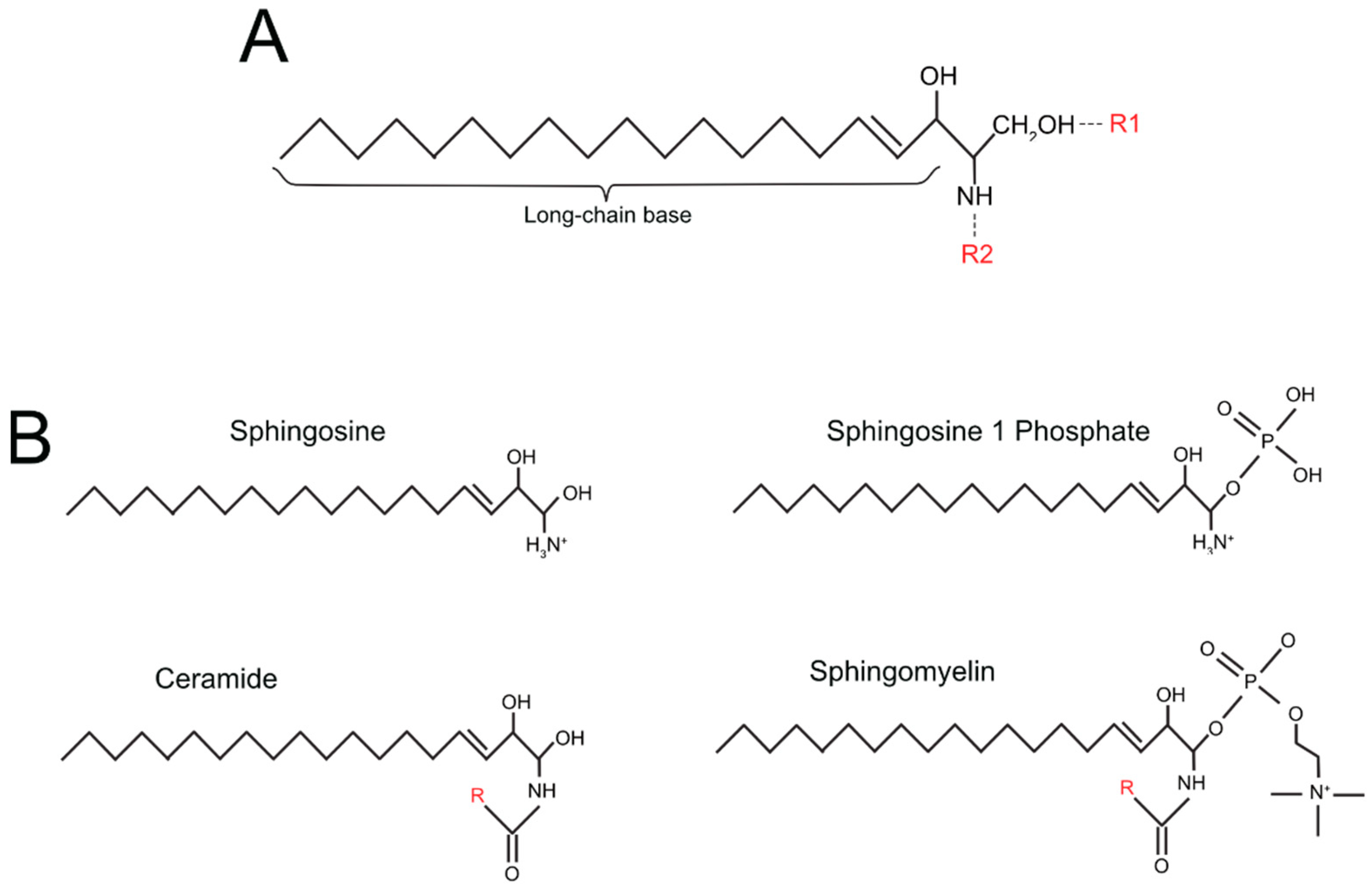
| Simple Sphingolipids | R1 Group (See Figure 1A) | R2 Group (See Figure 1A) |
|---|---|---|
| sphingosine | OH | H |
| sphingosine-1 phosphate (S1P) | PO4 | H |
| ceramide | OH | O + Fatty acid residue |
| ceramide-1-phosphate (C1P) | PO4 | O + Fatty acid residue |
| Complex sphingolipids | ||
| Sphingomyelin | phosphocholine group | O + Fatty acid residue |
| Cerebroside | single sugar residue | O + Fatty acid residue |
| Globoside | di, tri, tetra-saccharide residue | O + Fatty acid residue |
| Sulfatide | single sugar residue + sulphate group | O + Fatty acid residue |
| Ganglioside | oligosaccharide residue + sialic acid | O + Fatty acid residue |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bozzini, N.; Avnet, S.; Baldini, N.; Cortini, M. Epigenetic Regulation Mediated by Sphingolipids in Cancer. Int. J. Mol. Sci. 2023, 24, 5294. https://doi.org/10.3390/ijms24065294
Bozzini N, Avnet S, Baldini N, Cortini M. Epigenetic Regulation Mediated by Sphingolipids in Cancer. International Journal of Molecular Sciences. 2023; 24(6):5294. https://doi.org/10.3390/ijms24065294
Chicago/Turabian StyleBozzini, Nicolò, Sofia Avnet, Nicola Baldini, and Margherita Cortini. 2023. "Epigenetic Regulation Mediated by Sphingolipids in Cancer" International Journal of Molecular Sciences 24, no. 6: 5294. https://doi.org/10.3390/ijms24065294
APA StyleBozzini, N., Avnet, S., Baldini, N., & Cortini, M. (2023). Epigenetic Regulation Mediated by Sphingolipids in Cancer. International Journal of Molecular Sciences, 24(6), 5294. https://doi.org/10.3390/ijms24065294