
Fabry Disease and Central Nervous System Involvement: From Big to Small, from Brain to Synapse



{kind=link}
Abstract
1. Introduction
2. The Framework of LSDs and Fabry Disease
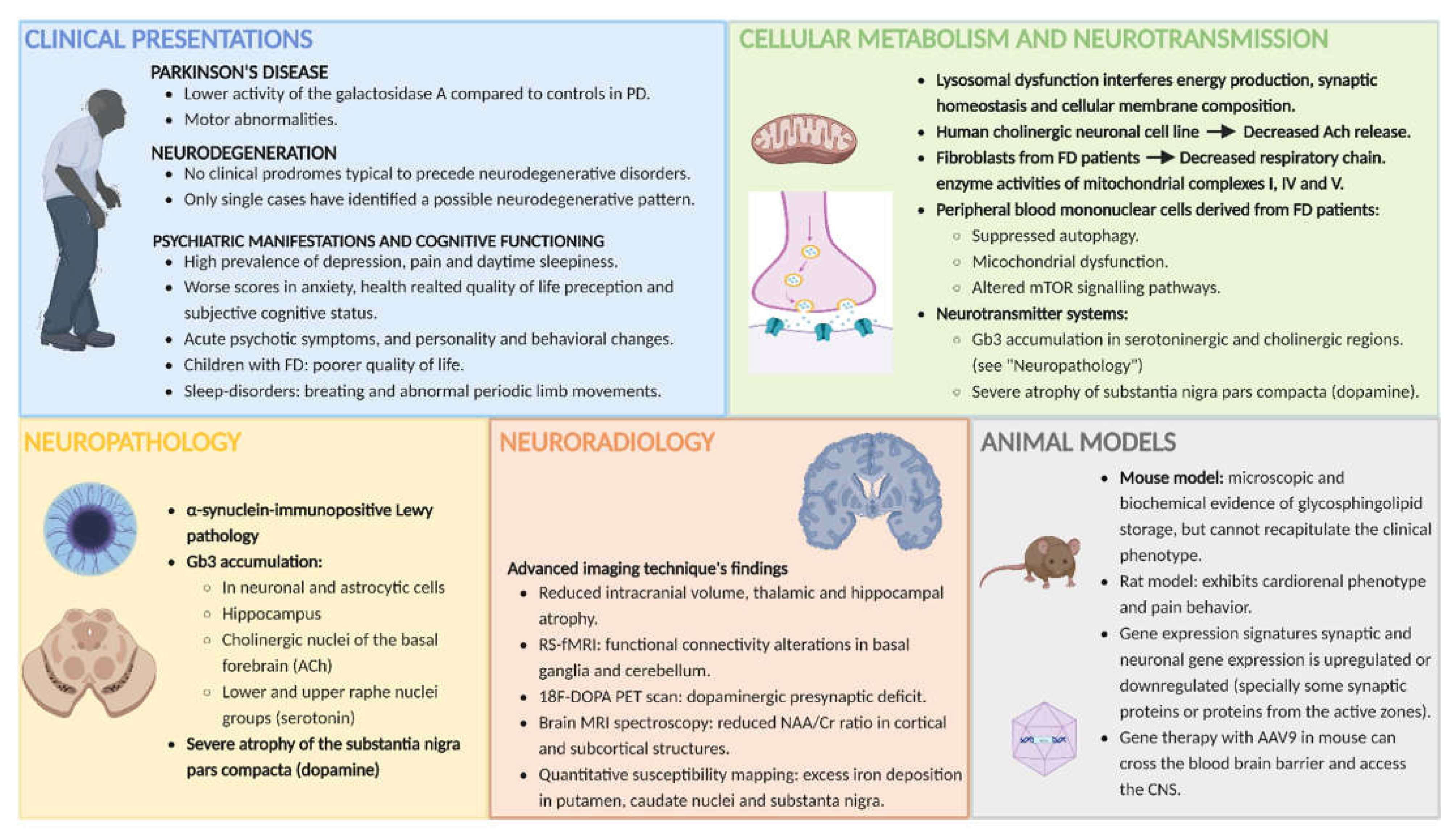
3. Clinical Manifestations Related to CNS Involvement in FD
3.1. The Pain in Fabry Disease
3.2. Parkinson’s Disease (PD)
3.3. Neurodegeneration
3.4. Psychiatric Manifestations and Cognitive Functioning
4. MRI Abnormalities (Other Than Cerebrovascular Disease) in FD
4.1. Reduced Intracranial Volume and Thalamic and Hippocampal Atrophy
4.2. Motor Cortex and Cerebellar and Nigrostriatal Pathway Involvement
4.3. Neurodegeneration, Neuronal Dysfunction and Hypometabolic Brain Regions
4.4. MRI Changes in Children and Adolescents with FD
5. Neuropathology
6. Animal Models
6.1. Mouse and Rat Models and the Recapitulation of FD Phenotype
6.2. Alterations in the Autophagy–Lysosome Pathway (ALP)
6.3. PD and Brain Protein Aggregation
6.4. Gene Expression Related to Neuronal and Synaptic Dysfunction
6.5. New Treatment Options with Impact on the CNS
7. Induced Pluripotent Stem Cell Models in FD
8. Energy Metabolism and Neurotransmission in FD
8.1. Energy Metabolism and Neurotransmission in the CNS, Lysosomal Disorders and Glycosphingolipodoses
8.2. Mitochondrial Involvement, Impaired Autophagic Function and Altered Neurotransmitter Release in FD
8.3. Different Neurotransmitter Systems Altered in FD, and Their Possible Role in Clinical Manifestations
8.3.1. Acetylcholine (ACh)
8.3.2. Dopamine (DA)
8.3.3. Dopamine and Acetylcholine
8.3.4. Serotonin (5-HT)
9. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carnicer-Cáceres, C.; Arranz-Amo, J.; Cea-Arestin, C.; Camprodon-Gomez, M.; Moreno-Martinez, D.; Lucas-Del-Pozo, S.; Moltó-Abad, M.; Tigri-Santiña, A.; Agraz-Pamplona, I.; Rodriguez-Palomares, J.; et al. Biomarkers in Fabry Disease. Implications for Clinical Diagnosis and Follow-up. J. Clin. Med. 2021, 10, 1664. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Clarke, J.T.R.; Giugliani, R.; Elliott, P.; Linhart, A.; Beck, M.; Sunder-Plassmann, G. Natural course of Fabry disease: Changing pattern of causes of death in FOS—Fabry Outcome Survey. J. Med. Genet. 2009, 46, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Arends, M.; Wanner, C.; Hughes, D.; Mehta, A.; Oder, D.; Watkinson, O.T.; Elliott, P.M.; Linthorst, G.E.; Wijburg, F.A.; Biegstraaten, M.; et al. Characterization of Classical and Nonclassical Fabry Disease: A Multicenter Study. J. Am. Soc. Nephrol. 2017, 28, 1631–1641. [Google Scholar] [CrossRef]
- Hopkin, R.J.; Bissler, J.; Banikazemi, M.; Clarke, L.; Eng, C.M.; Germain, D.P.; Lemay, R.; Tylki-Szymanska, A.; Wilcox, W.R. Characterization of Fabry Disease in 352 Pediatric Patients in the Fabry Registry. Pediatr. Res. 2008, 64, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Dobrovolny, R.; Dvorakova, L.; Ledvinova, J.; Magage, S.; Bultas, J.; Lubanda, J.C.; Elleder, M.; Karetova, D.; Pavlikova, M.; Hrebicek, M. Relationship between X-inactivation and clinical involvement in Fabry heterozygotes. Eleven novel mutations in the α-galactosidase A gene in the Czech and Slovak population. J. Mol. Med. 2005, 83, 647–654. [Google Scholar] [CrossRef]
- Platt, F.M.; D’Azzo, A.; Davidson, B.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Prim. 2018, 4, 27. [Google Scholar] [CrossRef]
- Pará, C.; Bose, P.; Pshezhetsky, A.V. Neuropathophysiology of Lysosomal Storage Diseases: Synaptic Dysfunction as a Starting Point for Disease Progression. J. Clin. Med. 2020, 9, 616. [Google Scholar] [CrossRef]
- Löhle, M.; Hughes, D.; Milligan, A.; Richfield, L.; Reichmann, H.; Mehta, A.; Schapira, A.H. Clinical prodromes of neurodegeneration in Anderson-Fabry disease. Neurology 2015, 84, 1454–1464. [Google Scholar] [CrossRef]
- Miller, J.J.; Kanack, A.J.; Dahms, N.M. Progress in the understanding and treatment of Fabry disease. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129437. [Google Scholar] [CrossRef]
- Møller, A.T.; Jensen, T.S. Neurological manifestations in Fabry’s disease. Nat. Clin. Pract. Neurol. 2007, 3, 95–106. [Google Scholar] [CrossRef]
- Moore, D.F.; Schiffmann, R. Fabry Disease: Neurological Manifestations; Oxford PharmaGenesis: Oxford, UK, 2006. [Google Scholar]
- Mehta, A.; Ginsberg, L. Natural history of the cerebrovascular complications of Fabry disease. Acta Paediatr. Suppl. 2005, 94, 24–27. [Google Scholar] [CrossRef]
- Burlina, A.P. Neurological manifestations and psychological aspects of Fabry disease. Clin. Ther. 2010, 32 (Suppl. 3), S88–S89. [Google Scholar] [CrossRef]
- Watson, J.C.; Sandroni, P. Central Neuropathic Pain Syndromes. Mayo Clin. Proc. 2016, 91, 372–385. [Google Scholar] [CrossRef]
- Burand, A.J.; Stucky, C.L. Fabry disease pain: Patient and preclinical parallels. Pain 2021, 162, 1305–1321. [Google Scholar] [CrossRef]
- Ramaswami, U.; Whybra, C.; Parini, R.; Pintos-Morell, G.; Mehta, A.; Sunder-Plassmann, G.; Widmer, U.; Beck, M.; On Behalf of the fos European Investigators. Clinical manifestations of Fabry disease in children: Data from the Fabry Outcome Survey. Acta Paediatr. Int. J. Paediatr. 2006, 95, 86–92. [Google Scholar] [CrossRef]
- Forstenpointner, J.; Sendel, M.; Moeller, P.; Reimer, M.; Canaan-Kühl, S.; Gaedeke, J.; Rehm, S.; Hüllemann, P.; Gierthmühlen, J.; Baron, R. Bridging the Gap between Vessels and Nerves in Fabry Disease. Front. Neurosci. 2020, 14, 448. [Google Scholar] [CrossRef] [PubMed]
- Robak, L.A.; Jansen, I.E.; Van Rooij, J.; Uitterlinden, A.G.; Kraaij, R.; Jankovic, J.; Heutink, P.; Shulman, J.M. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 2017, 140, 3191–3203. [Google Scholar] [CrossRef] [PubMed]
- Blumenreich, S.; Jenkins, B.J.; Barav, O.B.; Milenkovic, I.; Futerman, A.H. The Lysosome and Nonmotor Symptoms: Linking Parkinson′s Disease and Lysosomal Storage Disorders. Mov. Disord. 2020, 35, 2150–2155. [Google Scholar] [CrossRef] [PubMed]
- Moors, T.; Paciotti, S.; Chiasserini, D.; Calabresi, P.; Parnetti, L.; Beccari, T.; van de Berg, W.D.J. Lysosomal Dysfunction and α-Synuclein Aggregation in Parkinson’s Disease: Diagnostic Links. Mov. Disord. 2016, 31, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Gago, M.F.; Azevedo, O.; Guimarães, A.; Vide, A.T.; Lamas, N.J.; Gil Oliveira, T.; Gaspar, P.; Bicho, E.; Miltenberger-Miltenyi, G.; Ferreira, J.; et al. Parkinson’s Disease and Fabry Disease: Clinical, Biochemical and Neuroimaging Analysis of Three Pedigrees. J. Park. Dis. 2020, 10, 141–152. [Google Scholar] [CrossRef]
- Alcalay, R.; Wolf, P.; Levy, O.; Kang, U.; Waters, C.; Fahn, S.; Ford, B.; Kuo, S.; Vanegas, N.; Shah, H.; et al. Alpha galactosidase A activity in Parkinson’s disease. Neurobiol. Dis. 2018, 112, 85–90. [Google Scholar] [CrossRef]
- Del Tredici, K.; Ludolph, A.C.; Feldengut, S.; Jacob, C.; Reichmann, H.; Bohl, J.R.; Braak, H. Fabry Disease with Concomitant Lewy Body Disease. J. Neuropathol. Exp. Neurol. 2020, 79, 378–392. [Google Scholar] [CrossRef]
- Bolsover, F.E.; Murphy, E.; Cipolotti, L.; Werring, D.J.; Lachmann, R.H. Cognitive dysfunction and depression in Fabry disease: A systematic review. J. Inherit. Metab. Dis. 2014, 37, 177–187. [Google Scholar] [CrossRef]
- Loeb, J.; Feldt-Rasmussen, U.; Madsen, C.V.; Vogel, A. Cognitive Impairments and Subjective Cognitive Complaints in Fabry Disease: A Nationwide Study and Review of the Literature. In JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2018; Volume 41. [Google Scholar] [CrossRef]
- Cole, A.L.; Lee, P.J.; Hughes, D.A.; Deegan, P.B.; Waldek, S.; Lachmann, R.H. Depression in adults with Fabry disease: A common and under-diagnosed problem. J. Inherit. Metab. Dis. 2007, 30, 943–951. [Google Scholar] [CrossRef]
- Varela, P.; Carvalho, G.; Martin, R.P.; Pesquero, J.B. Fabry disease: GLA deletion alters a canonical splice site in a family with neuropsychiatric manifestations. Metab. Brain Dis. 2021, 36, 265–272. [Google Scholar] [CrossRef]
- Crosbie, T.W.; Packman, W.; Packman, S. Psychological aspects of patients with Fabry disease. J. Inherit. Metab. Dis. 2009, 32, 745–753. [Google Scholar] [CrossRef]
- Segal, P.; Kohn, Y.; Pollak, Y.; Altarescu, G.; Galili-Weisstub, E.; Raas-Rothschild, A. Psychiatric and cognitive profile in Anderson-Fabry patients: A preliminary study. J. Inherit. Metab. Dis. 2010, 33, 429–436. [Google Scholar] [CrossRef]
- Ali, N.; Caceres, A.; Hall, E.; Laney, D. Attention Deficits and ADHD Symptoms in Adults with Fabry Disease—A Pilot Investigation. J. Clin. Med. 2021, 10, 3367. [Google Scholar] [CrossRef]
- Körver, S.; Geurtsen, G.J.; Hollak, C.E.M.; Van Schaik, I.N.; Longo, M.G.F.; Lima, M.R.; Dijkgraaf, M.G.W.; Langeveld, M. Cognitive functioning and depressive symptoms in Fabry disease: A follow-up study. J. Inherit. Metab. Dis. 2020, 43, 1070–1081. [Google Scholar] [CrossRef]
- Naylor, P.E.; Hudson, K.; Aoki, C.D.; Cordova, M.J.; Packman, W.; Bugescu, N. The Psychosocial Impact of Fabry Disease on Pediatric Patients. J. Pediatr. Genet. 2016, 5, 141–149. [Google Scholar] [CrossRef]
- Talbot, A.; Hammerschlag, G.; Goldin, J.; Nicholls, K. Sleep disturbance, obstructive sleep apnoea and abnormal periodic leg movements: Very common problems in fabry disease. In JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2017; Volume 31. [Google Scholar]
- Cocozza, S.; Russo, C.; Pontillo, G.; Pisani, A.; Brunetti, A. Neuroimaging in Fabry disease: Current knowledge and future directions. Insights Imaging 2018, 9, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Cocozza, S.; Russo, C.; Pisani, A.; Olivo, G.; Riccio, E.; Cervo, A.; Pontillo, G.; Feriozzi, S.; Veroux, M.; Battaglia, Y.; et al. Redefining the Pulvinar Sign in Fabry Disease. Am. J. Neuroradiol. 2017, 38, 2264–2269. [Google Scholar] [CrossRef] [PubMed]
- Ulivi, L.; Kanber, B.; Prados, F.; Davagnanam, I.; Merwick, A.; Chan, E.; Williams, F.; Hughes, D.; Murphy, E.; Lachmann, R.H.; et al. White matter integrity correlates with cognition and disease severity in Fabry disease. Brain 2021, 143, 3331–3342. [Google Scholar] [CrossRef] [PubMed]
- Buechner, S.; Moretti, M.; Burlina, A.P.; Cei, G.; Manara, R.; Ricci, R.; Mignani, R.; Parini, R.; Di Vito, R.; Giordano, G.P.; et al. Central nervous system involvement in Anderson-Fabry disease: A clinical and MRI retrospective study. J. Neurol. Neurosurg. Psychiatry 2008, 79, 1249–1254. [Google Scholar] [CrossRef] [PubMed]
- Pontillo, G.; Cocozza, S.; Brunetti, A.; Morra, V.B.; Riccio, E.; Russo, C.; Saccà, F.; Tedeschi, E.; Pisani, A.; Mario Quarantelli on behalf of the AFFINITY study group. Reduced Intracranial Volume in Fabry Disease: Evidence of Abnormal Neurodevelopment? Front. Neurol. 2018, 9, 672. [Google Scholar] [CrossRef]
- Cocozza, S.; Pisani, A.; Olivo, G.; Saccà, F.; Ugga, L.; Riccio, E.; Migliaccio, S.; Morra, V.B.; Brunetti, A.; Quarantelli, M.; et al. Alterations of functional connectivity of the motor cortex in Fabry disease. Neurology 2017, 88, 1822–1829. [Google Scholar] [CrossRef]
- Albrecht, J.; Dellani, P.R.; Muller, M.J.; Schermuly, I.; Beck, M.; Stoeter, P.; Gerhard, A.; Fellgiebel, A. Voxel based analyses of diffusion tensor imaging in Fabry disease. J. Neurol. Neurosurg. Psychiatry 2007, 78, 964–969. [Google Scholar] [CrossRef]
- Russo, C.; Pontillo, G.; Pisani, A.; Saccà, F.; Riccio, E.; Macera, A.; Rusconi, G.; Stanzione, A.; Borrelli, P.; Morra, V.B.; et al. Striatonigral involvement in Fabry Disease: A quantitative and volumetric Magnetic Resonance Imaging study. Park. Relat. Disord. 2018, 57, 27–32. [Google Scholar] [CrossRef]
- Tedeschi, G.; Bonavita, S.; Banerjee, T.; Virta, A.; Schiffmann, R. Diffuse central neuronal involvement in Fabry disease: A proton MRS imaging study. Neurology 1999, 52, 1663. [Google Scholar] [CrossRef]
- Cocozza, S.; Schiavi, S.; Pontillo, G.; Battocchio, M.; Riccio, E.; Caccavallo, S.; Russo, C.; Di Risi, T.; Pisani, A.; Daducci, A.; et al. Microstructural damage of the cortico-striatal and thalamo-cortical fibers in Fabry disease: A diffusion MRI tractometry study. Neuroradiology 2020, 62, 1459–1466. [Google Scholar] [CrossRef]
- Ravanfar, P.; Loi, S.M.; Syeda, W.T.; Van Rheenen, T.E.; Bush, A.I.; Desmond, P.; Cropley, V.L.; Lane, D.J.R.; Opazo, C.M.; Moffat, B.A.; et al. Systematic Review: Quantitative Susceptibility Mapping (QSM) of Brain Iron Profile in Neurodegenerative Diseases. Front. Neurosci. 2021, 15, 618435. [Google Scholar] [CrossRef]
- Korsholm, K.; Feldt-Rasmussen, U.; Granqvist, H.; Højgaard, L.; Bollinger, B.; Rasmussen, A.K.; Law, I. Positron Emission Tomography and Magnetic Resonance Imaging of the Brain in Fabry Disease: A Nationwide, Long-Time, Prospective Follow-Up. PLoS ONE 2015, 10, e0143940. [Google Scholar] [CrossRef]
- Marchesoni, C.; Cisneros, E.; Pfister, P.; Yáñez, P.; Rollan, C.; Romero, C.; Kisinovsky, I.; Rattagan, L.; Cejas, L.L.; Pardal, A.; et al. Brain MRI findings in children and adolescents with Fabry disease. J. Neurol. Sci. 2018, 395, 131–134. [Google Scholar] [CrossRef]
- Fellgiebel, A.; Gartenschläger, M.; Wildberger, K.; Scheurich, A.; Desnick, R.J.; Sims, K. Enzyme Replacement Therapy Stabilized White Matter Lesion Progression in Fabry Disease. Cerebrovasc. Dis. 2014, 38, 448–456. [Google Scholar] [CrossRef]
- Stefaniak, J.D.; Parkes, L.M.; Parry-Jones, A.R.; Potter, G.M.; Vail, A.; Jovanovic, A.; Smith, C.J. Enzyme replacement therapy and white matter hyperintensity progression in Fabry disease. Neurology 2018, 91, e1413–e1422. [Google Scholar] [CrossRef]
- De Veber, G.A.; Schwarting, G.A.; Kolodny, E.H.; Kowall, N.W. Fabry disease: Immunocytochemical characterization of neuronal involvement. Ann. Neurol. 1992, 31, 409–415. [Google Scholar] [CrossRef]
- Kaye, E.M.; Kolodny, E.H.; Logigian, E.L.; Ullman, M.D. Nervous system involvement in Fabry’s disease: Clinicopathological and biochemical correlation. Ann. Neurol. 1988, 23, 505–509. [Google Scholar] [CrossRef]
- Ohshima, T.; Murray, G.J.; Swaim, W.D.; Longenecker, G.; Quirk, J.M.; Cardarelli, C.O.; Sugimoto, Y.; Pastan, I.; Gottesman, M.M.; Brady, R.O.; et al. α-Galactosidase A deficient mice: A model of Fabry disease. Proc. Natl. Acad. Sci. USA 1997, 94, 2540–2544. [Google Scholar] [CrossRef]
- Rodrigues, L.; Ferraz, M.; Rodrigues, D.; Pais-Vieira, M.; Lima, D.; Brady, R.; Sousa, M.; Sá-Miranda, M. Neurophysiological, behavioral and morphological abnormalities in the Fabry knockout mice. Neurobiol. Dis. 2009, 33, 48–56. [Google Scholar] [CrossRef]
- Hofmann, L.; Karl, F.; Sommer, C.; Üçeyler, N. Affective and cognitive behavior in the alpha-galactosidase A deficient mouse model of Fabry disease. PLoS ONE 2017, 12, e0180601. [Google Scholar] [CrossRef]
- Biferi, M.G.; Cohen-Tannoudji, M.; García-Silva, A.; Souto-Rodríguez, O.; Viéitez-González, I.; San-Millán-Tejado, B.; Fernández-Carrera, A.; Pérez-Márquez, T.; Teijeira-Bautista, S.; Barrera, S.; et al. Systemic Treatment of Fabry Disease Using a Novel AAV9 Vector Expressing α-Galactosidase A. Mol. Ther. Methods Clin. Dev. 2021, 20, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Bangari, D.S.; Ashe, K.M.; Desnick, R.J.; Maloney, C.; Lydon, J.; Piepenhagen, P.; Budman, E.; Leonard, J.P.; Cheng, S.H.; Marshall, J.; et al. α-Galactosidase A Knockout Mice: Progressive organ pathology resembles the type 2 later-onset phenotype of fabry disease. Am. J. Pathol. 2015, 185, 651–665. [Google Scholar] [CrossRef] [PubMed]
- Lakoma, J.; Rimondini, R.; Ferrer-Montiel, A.; Donadio, V.; Liguori, R.; Caprini, M. Increased expression of Trpv1 in peripheral terminals mediates thermal nociception in Fabry disease mouse model. Mol. Pain 2016, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.P.; Tse, T.E.; O’Quinn, D.B.; Percival, S.M.; Jaimes, E.A.; Warnock, D.G.; Shacka, J.J. Autophagy-lysosome pathway associated neuropathology and axonal degeneration in the brains of alpha-galactosidase A-deficient mice. Acta Neuropathol. Commun. 2014, 2, 20. [Google Scholar] [CrossRef]
- Loos, B.; Engelbrecht, A.-M.; Lockshin, R.A.; Klionsky, D.J.; Zakeri, Z. The variability of autophagy and cell death susceptibility: Unanswered questions. Autophagy 2013, 9, 1270–1285. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017, 3, 17013. [Google Scholar] [CrossRef]
- Clarke, J.; Kayatekin, C.; Viel, C.; Shihabuddin, L.; Sardi, S. Murine Models of Lysosomal Storage Diseases Exhibit Differences in Brain Protein Aggregation and Neuroinflammation. Biomedicines 2021, 9, 446. [Google Scholar] [CrossRef]
- Kummer, K.; Kalpachidou, T.; Mitrić, M.; Langeslag, M.; Kress, M. Altered Gene Expression in Prefrontal Cortex of a Fabry Disease Mouse Model. Front. Mol. Neurosci. 2018, 11, 201. [Google Scholar] [CrossRef]
- Borger, D.K.; McMahon, B.; Lal, T.R.; Serra-Vinardell, J.; Aflaki, E.; Sidransky, E. Induced pluripotent stem cell models of lysosomal storage disorders. Dis. Model. Mech. 2017, 10, 691–704. [Google Scholar] [CrossRef]
- Itier, J.-M.; Ret, G.; Viale, S.; Sweet, L.; Bangari, D.; Caron, A.; Le-Gall, F.; Bénichou, B.; Leonard, J.; Deleuze, J.-F.; et al. Effective clearance of GL-3 in a human iPSC-derived cardiomyocyte model of Fabry disease. J. Inherit. Metab. Dis. 2014, 37, 1013–1022. [Google Scholar] [CrossRef]
- Birket, M.J.; Raibaud, S.; Lettieri, M.; Adamson, A.D.; Letang, V.; Cervello, P.; Redon, N.; Ret, G.; Viale, S.; Wang, B.; et al. A Human Stem Cell Model of Fabry Disease Implicates LIMP-2 Accumulation in Cardiomyocyte Pathology. Stem Cell Rep. 2019, 13, 380–393. [Google Scholar] [CrossRef]
- Luciani, M.; Gritti, A.; Meneghini, V. Human iPSC-Based Models for the Development of Therapeutics Targeting Neurodegenerative Lysosomal Storage Diseases. Front. Mol. Biosci. 2020, 7, 224. [Google Scholar] [CrossRef]
- Miyajima, T.; Saito, R.; Yanagisawa, H.; Igarashi, M.; Wu, C.; Iwamoto, T.; Eto, Y. Characterization of cellular phenotypes in neurons derived from induced pluripotent stem cells of male patients with Fabry disease. J. Inherit. Metab. Dis. 2023, 46, 143–152. [Google Scholar] [CrossRef]
- McKenna, M.C.; Schuck, P.F.; Ferreira, G.C. Fundamentals of CNS energy metabolism and alterations in lysosomal storage diseases. J. Neurochem. 2019, 148, 590–599. [Google Scholar] [CrossRef]
- Oyarzabal, A.; Marin-Valencia, I. Synaptic energy metabolism and neuronal excitability, in sickness and health. J. Inherit. Metab. Dis. 2019, 42, 220–236. [Google Scholar] [CrossRef]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef]
- Ballabio, A.; Bonifacino, J.S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 101–118. [Google Scholar] [CrossRef]
- Ryckman, A.E.; Brockhausen, I.; Walia, J.S. Metabolism of Glycosphingolipids and Their Role in the Pathophysiology of Lysosomal Storage Disorders. Int. J. Mol. Sci. 2020, 21, 6881. [Google Scholar] [CrossRef]
- Kaneski, C.R.; Brady, R.O.; Hanover, J.A.; Schueler, U.H. Development of a model system for neuronal dysfunction in Fabry disease. Mol. Genet. Metab. 2016, 119, 144–150. [Google Scholar] [CrossRef]
- Lücke, T.; Höppner, W.; Schmidt, E.; Illsinger, S.; Das, A.M. Fabry disease: Reduced activities of respiratory chain enzymes with decreased levels of energy-rich phosphates in fibroblasts. Mol. Genet. Metab. 2004, 82, 93–97. [Google Scholar] [CrossRef]
- Ivanova, M.M.; Changsila, E.; Iaonou, C.; Goker-Alpan, O. Impaired autophagic and mitochondrial functions are partially restored by ERT in Gaucher and Fabry diseases. PLoS ONE 2019, 14, e0210617. [Google Scholar] [CrossRef] [PubMed]
- Amalric, M.; Pattij, T.; Sotiropoulos, I.; Silva, J.M.; Sousa, N.; Ztaou, S.; Chiamulera, C.; Wahlberg, L.U.; Emerich, D.F.; Paolone, G. Where Dopaminergic and Cholinergic Systems Interact: A Gateway for Tuning Neurodegenerative Disorders. Front. Behav. Neurosci. 2021, 15, 661973. [Google Scholar] [CrossRef] [PubMed]
- Pourhamzeh, M.; Moravej, F.G.; Arabi, M.; Shahriari, E.; Mehrabi, S.; Ward, R.; Ahadi, R.; Joghataei, M.T. The Roles of Serotonin in Neuropsychiatric Disorders. Cell. Mol. Neurobiol. 2021, 42, 1671–1692. [Google Scholar] [CrossRef] [PubMed]
- MacQueen, G.; Frodl, T. The hippocampus in major depression: Evidence for the convergence of the bench and bedside in psychiatric research? Mol. Psychiatry 2011, 16, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Mahar, I.; Bambico, F.R.; Mechawar, N.; Nobrega, J.N. Stress, serotonin, and hippocampal neurogenesis in relation to depression and antidepressant effects. Neurosci. Biobehav. Rev. 2014, 38, 173–192. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cortés-Saladelafont, E.; Fernández-Martín, J.; Ortolano, S. Fabry Disease and Central Nervous System Involvement: From Big to Small, from Brain to Synapse. Int. J. Mol. Sci. 2023, 24, 5246. https://doi.org/10.3390/ijms24065246
Cortés-Saladelafont E, Fernández-Martín J, Ortolano S. Fabry Disease and Central Nervous System Involvement: From Big to Small, from Brain to Synapse. International Journal of Molecular Sciences. 2023; 24(6):5246. https://doi.org/10.3390/ijms24065246
Chicago/Turabian StyleCortés-Saladelafont, Elisenda, Julián Fernández-Martín, and Saida Ortolano. 2023. "Fabry Disease and Central Nervous System Involvement: From Big to Small, from Brain to Synapse" International Journal of Molecular Sciences 24, no. 6: 5246. https://doi.org/10.3390/ijms24065246
APA StyleCortés-Saladelafont, E., Fernández-Martín, J., & Ortolano, S. (2023). Fabry Disease and Central Nervous System Involvement: From Big to Small, from Brain to Synapse. International Journal of Molecular Sciences, 24(6), 5246. https://doi.org/10.3390/ijms24065246