
High Glucose Promotes Inflammation and Weakens Placental Defenses against E. coli and S. agalactiae Infection: Protective Role of Insulin and Metformin

 , , , ,
, , , ,  , and
, and 
Abstract

1. Introduction
2. Results
2.1. A Hyperglycemic Model in Human Term Placenta
2.2. Insulin and Metformin Diminish Placental Pro-Inflammatory Cytokines Secretion under a Hyperglycemic Condition
2.3. Hyperglycemia Diminishes Placental Beta Defensins Synthesis. Metformin and Insulin Do Not Restore HBDs
2.4. Innate Defense against Bacterial Infections in Pre-Exposed Hyperglycemic Placenta
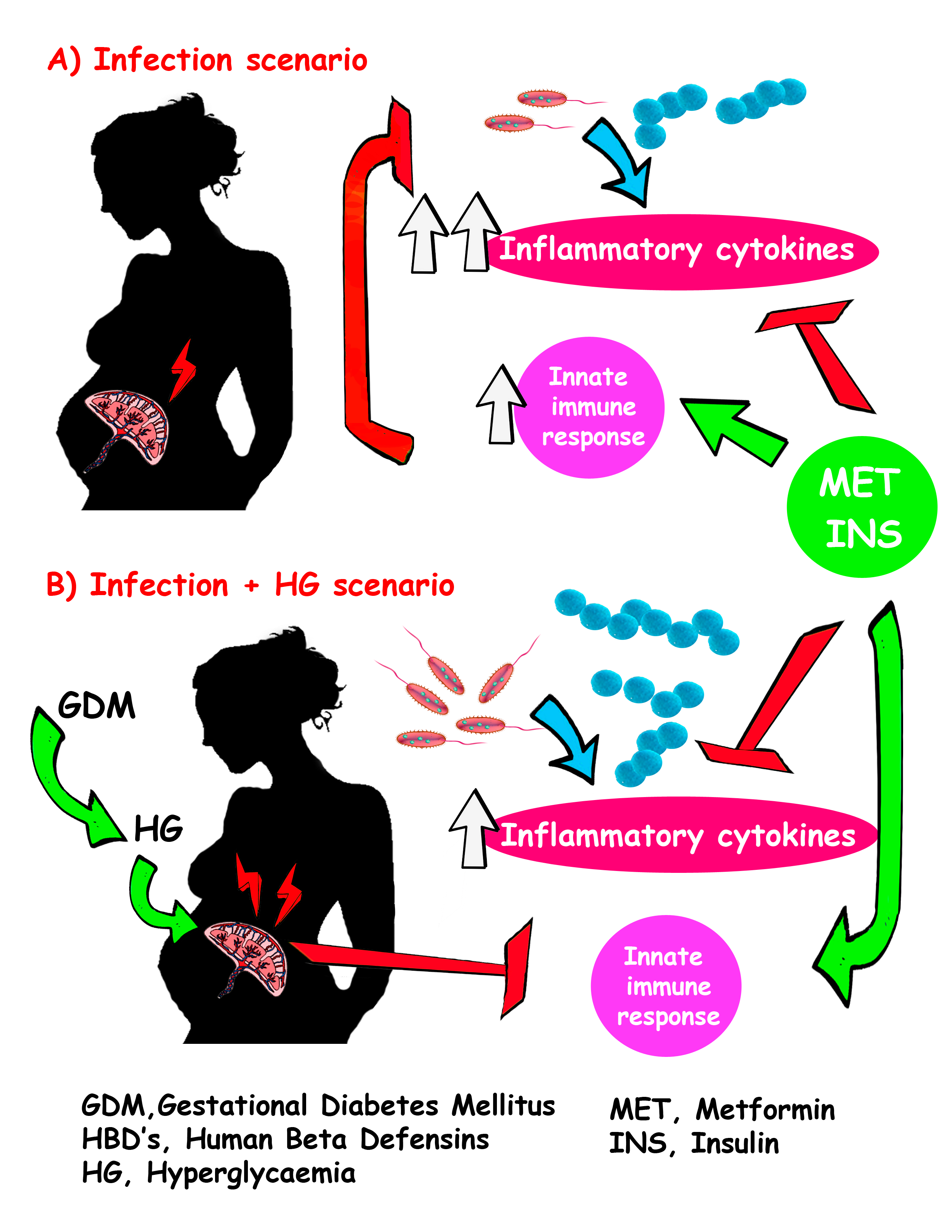
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Sample Collection
4.3. Reagents
4.4. Cotyledon Explant Culture and Experimental Procedures
4.5. Quantification of Cytokines and Adipokines by ELISA
4.6. Tissue Protein Extraction and Human Beta Defensins Quantification by ELISA
4.7. Placental Infection with Escherichia coli or Streptococcus agalactiae
4.8. Staining Techniques
4.9. XTT Cell Viability Assay
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pantham, P.; Aye, I.L.M.H.; Powell, T.L. Inflammation in Maternal Obesity and Gestational Diabetes Mellitus. Placenta 2015, 36, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Olmos-ortiz, A.; Flores-espinosa, P.; Díaz, L.; Velázquez, P.; Ramírez-isarraraz, C.; Zaga-clavellina, V. Immunoendocrine Dysregulation during Gestational Diabetes Mellitus: The Central Role of the Placenta. Int. J. Mol. Sci. 2021, 22, 8087. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhou, Y.; Gui, J.; Li, A.Z.; Su, X.L.; Feng, L. Assessment of the Number and Function of Macrophages in the Placenta of Gestational Diabetes Mellitus Patients. J. Huazhong Univ. Sci. Technol.–Med. Sci. 2013, 33, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Mrizak, I.; Grissa, O.; Henault, B.; Fekih, M.; Bouslema, A.; Boumaiza, I.; Zaouali, M.; Tabka, Z.; Khan, N.A. Placental Infiltration of Inflammatory Markers in Gestational Diabetic Women. Gen. Physiol. Biophys. 2014, 33, 169–176. [Google Scholar] [CrossRef]
- Radaelli, T.; Varastehpour, A.; Catalano, P.; Hauguel-De Mouzon, S. Gestational Diabetes Induces Placental Genes for Chronic Stress and Inflammatory Pathways. Diabetes 2003, 52, 2951–2958. [Google Scholar] [CrossRef]
- Han, C.S.; Herrin, M.A.; Pitruzzello, M.C.; Mulla, M.J.; Werner, E.F.; Pettker, C.M.; Flannery, C.A.; Abrahams, V.M. Glucose and Metformin Modulate Human First Trimester Trophoblast Function: A Model and Potential Therapy for Diabetes-Associated Uteroplacental Insufficiency. Am. J. Reprod. Immunol. 2015, 73, 362–371. [Google Scholar] [CrossRef]
- Heim, K.R.; Mulla, M.J.; Potter, J.A.; Han, C.S.; Guller, S.; Abrahams, V.M. Excess Glucose Induce Trophoblast Inflammation and Limit Cell Migration through HMGB1 Activation of Toll-Like Receptor 4. Am. J. Reprod. Immunol. 2018, 80, e13044. [Google Scholar] [CrossRef]
- Zhang, X.; Liao, Q.; Wang, F.; Li, D. Association of Gestational Diabetes Mellitus and Abnormal Vaginal Flora with Adverse Pregnancy Outcomes. Medicine 2018, 97, e11891. [Google Scholar] [CrossRef]
- Yao, H.; Xu, D.; Zhu, Z.; Wang, G. Gestational Diabetes Mellitus Increases the Detection Rate and the Number of Oral Bacteria in Pregnant Women. Medicine 2019, 98, e14903. [Google Scholar] [CrossRef]
- Rafat, D.; Singh, S.; Nawab, T.; Khan, F.; Khan, A.U.; Khalid, S. Association of Vaginal Dysbiosis and Gestational Diabetes Mellitus with Adverse Perinatal Outcomes. Int. J. Gynecol. Obstet. 2021, 158, 70–78. [Google Scholar] [CrossRef]
- Edwards, J.M.; Watson, N.; Focht, C.; Wynn, C.; Todd, C.A.; Walter, E.B.; Phillips Heine, R.; Swamy, G.K. Group B Streptococcus (GBS) Colonization and Disease among Pregnant Women: A Historical Cohort Study. Infect. Dis. Obs. Gynecol. 2019, 2019, 1–6. [Google Scholar] [CrossRef]
- Nguyen, L.M.; Omage, J.I.; Noble, K.; McNew, K.L.; Moore, D.J.; Aronoff, D.M.; Doster, R.S. Group B Streptococcal Infection of the Genitourinary Tract in Pregnant and Non-Pregnant Patients with Diabetes Mellitus: An Immunocompromised Host or Something More? Am. J. Reprod. Immunol. 2021, 86, e13501. [Google Scholar] [CrossRef] [PubMed]
- Russell, N.J.; Seale, A.C.; O’Driscoll, M.; O’Sullivan, C.; Bianchi-Jassir, F.; Gonzalez-Guarin, J.; Lawn, J.E.; Baker, C.J.; Bartlett, L.; Cutland, C.; et al. Maternal Colonization with Group B Streptococcus and Serotype Distribution Worldwide: Systematic Review and Meta-Analyses. Clin. Infect. Dis. 2017, 65, S100–S111. [Google Scholar] [CrossRef] [PubMed]
- Beksac, A.T.; Orgul, G.; Tanacan, A.; Uckan, H.; Sancak, B.; Portakal, O.; Beksac, M.S. Uropathogens and Gestational Outcomes of Urinary Tract Infections in Pregnancies That Necessitate Hospitalization. Curr. Urol. 2019, 13, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Dautt-Leyva, J.G.; Canizalez-Román, A.; Acosta Alfaro, L.F.; Gonzalez-Ibarra, F.; Murillo-Llanes, J. Maternal and Perinatal Complications in Pregnant Women with Urinary Tract Infection Caused by Escherichia Coli. J. Obstet. Gynaecol. Res. 2018, 44, 1384–1390. [Google Scholar] [CrossRef] [PubMed]
- Thakur, M.; Lata, S.; Pal, A.; Sharma, H.; Dhiman, B. Relationship between Histologic Chorioamnionitis and Genital Tract Cultures in Pre Term Labour. J. Obs. Gynaecol. 2021, 41, 721–725. [Google Scholar] [CrossRef]
- Romero, R.; Gomez-Lopez, N.; Winters, A.D.; Jung, E.; Shaman, M.; Bieda, J.; Panaitescu, B.; Pacora, P.; Erez, O.; Greenberg, J.M.; et al. Evidence That Intra-Amniotic Infections Are Often the Result of an Ascending Invasion—A Molecular Microbiological Study. J. Perinat. Med. 2019, 47, 915–931. [Google Scholar] [CrossRef]
- Kalinderi, K.; Delkos, D.; Kalinderis, M.; Athanasiadis, A.; Kalogiannidis, I. Urinary Tract Infection during Pregnancy: Current Concepts on a Common Multifaceted Problem. J. Obstet. Gynaecol. 2018, 38, 448–453. [Google Scholar] [CrossRef] [PubMed]
- ACOG Practice Bulletin No. 190: Gestational Diabetes Mellitus. Obstet. Gynecol. 2018, 131, e49–e64. [CrossRef]
- Management of Diabetes in Pregnancy: Standards of Medical Care in Diabetes-2020. Diabetes Care 2020, 43, S165–S172. [CrossRef]
- Schäfer-Graf, U.M.; Gembruch, U.; Kainer, F.; Groten, T.; Hummel, S.; Hösli, I.; Grieshop, M.; Kaltheuner, M.; Bührer, C.; Kautzky-Willer, A.; et al. Gestational Diabetes Mellitus (GDM)—Diagnosis, Treatment and Follow-UpGuideline of the DDG and DGGG (S3 Level, AWMF Registry Number 057/008, February 2018). Geburtshilfe Frauenheilkd 2018, 78, 1219–1231. [Google Scholar] [PubMed]
- Diabetes in Pregnancy: Management from Preconception to the Postnatal Period | Guidance and Guidelines | NICE. ISBN: 978-1-4731-0993-3. Published: 25 February 2015. Available online: https://www.nice.org.uk/guidance/ng3/resources/diabetes-in-pregnancy-management-from-preconception-to-the-postnatal-period-pdf-51038446021 (accessed on 10 December 2022).
- Tarry-Adkins, J.L.; Aiken, C.E.; Ozanne, S.E. Comparative Impact of Pharmacological Treatments for Gestational Diabetes on Neonatal Anthropometry Independent of Maternal Glycaemic Control: A Systematic Review and Meta-Analysis. PLoS Med. 2020, 17, e1003126. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.Q.; Xu, G.X.; Teng, X.Y.; Xu, J.W.; Tang, L.F.; Feng, C.; Rao, J.P.; Jin, M.; Wang, L.Q. Glycemic Control and Neonatal Outcomes in Women with Gestational Diabetes Mellitus Treated Using Glyburide, Metformin, or Insulin: A Pairwise and Network Meta-Analysis. BMC Endocr. Disord. 2021, 21, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Yang, H. Metformin–a Potentially Effective Drug for Gestational Diabetes Mellitus: A Systematic Review and Meta-Analysis. J. Matern.-Fetal Neonatal. Med. 2017, 30, 1874–1881. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, M.G.; Klein, D.; Bolder, U.; Einspanier, R. Insulin Attenuates the Systemic Inflammatory Response in Endotoxemic Rats. Endocrinology 2004, 145, 4084–4093. [Google Scholar] [CrossRef]
- Sun, Q.; Li, J.; Gao, F. New Insights into Insulin: The Anti-Inflammatory Effect and Its Clinical Relevance. World J. Diabetes 2014, 5, 89–96. [Google Scholar] [CrossRef]
- Leffler, M.; Hrach, T.; Stuerzl, M.; Horch, R.E.; Herndon, D.N.; Jeschke, M.G. Insulin Attenuates Apoptosis and Exerts Anti-Inflammatory Effects in Endotoxemic Human Macrophages. J. Surg. Res. 2007, 143, 398–406. [Google Scholar] [CrossRef]
- Brix-Christensen, V.; Andersen, S.K.; Andersen, R.; Mengel, A.; Dyhr, T.; Andersen, N.T.; Larsson, A.; Schmitz, O.; Ørskov, H.; Tønnesen, E. Acute Hyperinsulinemia Restrains Endotoxin-Induced Systemic Inflammatory Response: An Experimental Study in a Porcine Model. Anesthesiology 2004, 100, 861–870. [Google Scholar] [CrossRef]
- Jing, Y.; Wu, F.; Li, D.; Yang, L.; Li, Q.; Li, R. Metformin Improves Obesity-Associated Inflammation by Altering Macrophages Polarization. Mol. Cell. Endocrinol. 2018, 461, 256–264. [Google Scholar] [CrossRef]
- de Araújo, A.A.; Pereira, A.D.S.B.F.; de Medeiros, C.A.C.X.; Brito, G.A.D.C.; Leitão, R.F.D.C.; Araújo, L.D.S.; Guedes, P.M.M.; Hiyari, S.; Pirih, F.Q.; de Araújo, R.F. Effects of Metformin on Inflammation, Oxidative Stress, and Bone Loss in a Rat Model of Periodontitis. PLoS ONE 2017, 12, e0183506. [Google Scholar] [CrossRef]
- Peixoto, L.G.; Teixeira, R.R.; Vilela, D.D.; Barbosa, L.N.; Caixeta, D.C.; Deconte, S.R.; de Assis de Araújo, F.; Sabino-Silva, R.; Espindola, F.S. Metformin Attenuates the TLR4 Inflammatory Pathway in Skeletal Muscle of Diabetic Rats. Acta Diabetol. 2017, 54, 943–951. [Google Scholar] [CrossRef] [PubMed]
- de Souza Teixeira, A.A.; Souza, C.O.; Biondo, L.A.; Sanches Silveira, L.; Lima, E.A.; Batatinha, H.A.; Araujo, A.P.; Alves, M.J.; Hirabara, S.M.; Curi, R.; et al. Short-Term Treatment with Metformin Reduces Hepatic Lipid Accumulation but Induces Liver Inflammation in Obese Mice. Inflammopharmacology 2018, 26, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Bulatova, N.; Kasabri, V.; Qotineh, A.; AL-Athami, T.; Yousef, A.M.; AbuRuz, S.; Momani, M.; Zayed, A. Effect of Metformin Combined with Lifestyle Modification versus Lifestyle Modification Alone on Proinflammatory-Oxidative Status in Drug-Naïve Pre-Diabetic and Diabetic Patients: A Randomized Controlled Study. Diabetes Metab. Syndr. Clin. Res. Rev. 2018, 12, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Lin, S.; Chen, Y.; Li, X.; Ma, S.; Fu, Y.; Wei, C.; Wang, C.; Xu, W. The Effect of Metformin on the Expression of GPR109A, NF-ΚB and IL-1β in Peripheral Blood Leukocytes from Patients with Type 2 Diabetes Mellitus. Ann. Clin. Lab. Sci. 2017, 47, 556–562. [Google Scholar] [PubMed]
- Han, J.; Li, Y.; Liu, X.; Zhou, T.; Sun, H.; Edwards, P.; Gao, H.; Yu, F.S.; Qiao, X. Metformin Suppresses Retinal Angiogenesis and Inflammation in Vitro and in Vivo. PLoS ONE 2019, 13, e0193031. [Google Scholar] [CrossRef]
- Murtha, M.J.; Eichler, T.; Bender, K.; Metheny, J.; Li, B.; Schwaderer, A.L.; Mosquera, C.; James, C.; Schwartz, L.; Becknell, B.; et al. Insulin Receptor Signaling Regulates Renal Collecting Duct and Intercalated Cell Antibacterial Defenses. J. Clin. Investig. 2018, 128, 5634–5646. [Google Scholar] [CrossRef] [PubMed]
- Barnea, M.; Madar, Z.; Froy, O. Glucose and Insulin Are Needed for Optimal Defensin Expression in Human Cell Lines. Biochem. Biophys. Res. Commun. 2008, 367, 452–456. [Google Scholar] [CrossRef]
- Malik, F.; Mehdi, S.F.; Ali, H.; Patel, P.; Basharat, A.; Kumar, A.; Ashok, F.; Stein, J.; Brima, W.; Malhotra, P.; et al. Is Metformin Poised for a Second Career as an Antimicrobial? Diabetes/Metab. Res. Rev. 2018, 34, e2975. [Google Scholar] [CrossRef]
- Majhi, R.K.; Mohanty, S.; Kamolvit, W.; White, J.K.; Scheffschick, A.; Brauner, H.; Brauner, A. Metformin Strengthens Uroepithelial Immunity against E. Coli Infection. Sci. Rep. 2021, 11, 19263. [Google Scholar] [CrossRef]
- INTERGROWTH-21st The International Fetal and Newborn Growth Consortium for the 21st Century. Standards and Tools. Available online: https://intergrowth21.tghn.org/standards-tools/ (accessed on 25 October 2022).
- Villar, J.; Ismail, L.C.; Victora, C.G.; Ohuma, E.O.; Bertino, E.; Altman, D.G.; Lambert, A.; Papageorghiou, A.T.; Carvalho, M.; Jaffer, Y.A.; et al. International Standards for Newborn Weight, Length, and Head Circumference by Gestational Age and Sex: The Newborn Cross-Sectional Study of the INTERGROWTH-21st Project. Lancet 2014, 384, 857–868. [Google Scholar] [CrossRef]
- Abadpour, S.; Halvorsen, B.; Sahraoui, A.; Korsgren, O.; Aukrust, P.; Scholz, H. Interleukin-22 Reverses Human Islet Dysfunction and Apoptosis Triggered by Hyperglycemia and LIGHT. J. Mol. Endocrinol. 2018, 60, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Huerta-García, E.; Ventura-Gallegos, J.L.; Victoriano, M.E.C.; Montiél-Dávalos, A.; Tinoco-Jaramillo, G.; López-Marure, R. Dehydroepiandrosterone Inhibits the Activation and Dysfunction of Endothelial Cells Induced by High Glucose Concentration. Steroids 2012, 77, 233–240. [Google Scholar] [CrossRef]
- Rice, G.E.; Scholz-Romero, K.; Sweeney, E.; Peiris, H.; Kobayashi, M.; Duncombe, G.; Mitchell, M.D.; Salomon, C. The Effect of Glucose on the Release and Bioactivity of Exosomes from First Trimester Trophoblast Cells. J. Clin. Endocrinol. Metab. 2015, 100, E1280–E1288. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Pu, D.; Sun, Y.; Chen, J.; Luo, C.; Wang, M.; Zhou, J.; Lv, A.; Zhu, S.; Liao, Z.; et al. High Glucose-Induced Defective Thrombospondin-1 Release from Astrocytes via TLR9 Activation Contributes to the Synaptic Protein Loss. Exp. Cell Res. 2018, 363, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Akison, L.K.; Nitert, M.D.; Clifton, V.L.; Moritz, K.M.; Simmons, D.G. Review: Alterations in Placental Glycogen Deposition in Complicated Pregnancies: Current Preclinical and Clinical Evidence. Placenta 2017, 54, 52–58. [Google Scholar] [CrossRef]
- Tsiotra, P.C.; Halvatsiotis, P.; Patsouras, K.; Maratou, E.; Salamalekis, G.; Raptis, S.A.; Dimitriadis, G.; Boutati, E. Circulating Adipokines and MRNA Expression in Adipose Tissue and the Placenta in Women with Gestational Diabetes Mellitus. Peptides 2018, 101, 157–166. [Google Scholar] [CrossRef]
- Miehle, K.; Stepan, H.; Fasshauer, M. Leptin, Adiponectin and Other Adipokines in Gestational Diabetes Mellitus and Pre-Eclampsia. Clin. Endocrinol. 2012, 76, 2–11. [Google Scholar] [CrossRef]
- Hau, C.S.; Kanda, N.; Noda, S.; Tatsuta, A.; Kamata, M.; Shibata, S.; Asano, Y.; Sato, S.; Watanabe, S.; Tada, Y. Visfatin Enhances the Production of Cathelicidin Antimicrobial Peptide, Human β-Defensin-2, Human β-Defensin-3, and S100A7 in Human Keratinocytes and Their Orthologs in Murine Imiquimod-Induced Psoriatic Skin. Am. J. Pathol. 2013, 182, 1705–1717. [Google Scholar] [CrossRef]
- Tang, Y.; Liu, X.; Feng, C.; Zhou, Z.; Liu, S. Nicotinamide Phosphoribosyltransferase (Nampt) of Hybrid Crucian Carp Protects Intestinal Barrier and Enhances Host Immune Defense against Bacterial Infection. Dev. Comp. Immunol. 2022, 128, 104314. [Google Scholar] [CrossRef]
- Hara, C.D.C.P.; França, E.L.; Fagundes, D.L.G.; de Queiroz, A.A.; Rudge, M.V.C.; Honorio-França, A.C.; Calderon, I.D.M.P. Characterization of Natural Killer Cells and Cytokines in Maternal Placenta and Fetus of Diabetic Mothers. J. Immunol. Res. 2016, 2016, 1–8. [Google Scholar] [CrossRef]
- Carrasco-Wong, I.; Moller, A.; Giachini, F.R.; Lima, V.V.; Toledo, F.; Stojanova, J.; Sobrevia, L.; San Martín, S. Placental Structure in Gestational Diabetes Mellitus. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165535. [Google Scholar] [CrossRef] [PubMed]
- Basak, S.; Vilasagaram, S.; Naidu, K.; Duttaroy, A.K. Insulin-Dependent, Glucose Transporter 1 Mediated Glucose Uptake and Tube Formation in the Human Placental First Trimester Trophoblast Cells. Mol. Cell. Biochem. 2019, 451, 91–106. [Google Scholar] [CrossRef]
- Zhang, L.; Yu, X.; Wu, Y.; Fu, H.; Xu, P.; Zheng, Y.; Wen, L.; Yang, X.; Zhang, F.; Hu, M.; et al. Gestational Diabetes Mellitus-Associated Hyperglycemia Impairs Glucose Transporter 3 Trafficking in Trophoblasts Through the Downregulation of AMP-Activated Protein Kinase. Front. Cell Dev. Biol. 2021, 9, 3032. [Google Scholar] [CrossRef] [PubMed]
- Gordon, M.C.; Zimmerman, P.D.; Landon, M.B.; Gabbe, S.G.; Kniss, D.A. Insulin and Glucose Modulate Glucose Transporter Messenger Ribonucleic Acid Expression and Glucose Uptake in Trophoblasts Isolated from First-Trimester Chorionic Villi. Am. J. Obs. Gynecol. 1995, 173, 1089–1097. [Google Scholar] [CrossRef]
- Cawyer, C.R.; Horvat, D.; Leonard, D.; Allen, S.R.; Jones, R.O.; Zawieja, D.C.; Kuehl, T.J.; Uddin, M.N. Hyperglycemia Impairs Cytotrophoblast Function via Stress Signaling. Am. J. Obstet. Gynecol. 2014, 211, 541-e1. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Diagnostic Criteria and Classification of Hyperglycaemia First Detected in Pregnancy; World Health Organization: Geneva, Switzerland, 2013. [Google Scholar]
- Miller, R.K.; Genbacev, O.; Turner, M.A.; Aplin, J.D.; Caniggia, I.; Huppertz, B. Human Placental Explants in Culture: Approaches and Assessments. Placenta 2005, 26, 439–448. [Google Scholar] [CrossRef]
- Chen, X.; Huang, J.; Peng, Y.; Han, Y.; Wang, X.; Tu, C. The Role of CircRNA Polyribonucleotide Nucleoside Transferase 1 on Gestational Diabetes Mellitus. Cell. Mol. Biol. 2022, 68, 148–154. [Google Scholar] [CrossRef]
- Lappas, M.; Yee, K.; Permezel, M.; Rice, G.E. Release and Regulation of Leptin, Resistin and Adiponectin from Human Placenta, Fetal Membranes, and Maternal Adipose Tissue and Skeletal Muscle from Normal and Gestational Diabetes Mellitus-Complicated Pregnancies. J. Endocrinol. 2005, 186, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, T.; Tani, K.; Maki, J.; Eguchi, T.; Tamada, S.; Eto, E.; Hayata, K.; Masuyama, H. Upregulation of Angiogenic Factors via Protein Kinase c and Hypoxia-Induced Factor-1a Pathways under High-Glucose Conditions in the Placenta. Acta Med. Okayama 2018, 72, 359–367. [Google Scholar] [CrossRef]
- Ujvari, D.; Jakson, I.; Babayeva, S.; Salamon, D.; Rethi, B.; Gidlöf, S.; Hirschberg, A.L. Dysregulation of in Vitro Decidualization of Human Endometrial Stromal Cells by Insulin via Transcriptional Inhibition of Forkhead Box Protein O1. PLoS ONE 2017, 12, e0171004. [Google Scholar] [CrossRef]
- Kaitu’u-Lino, T.J.; Brownfoot, F.C.; Beard, S.; Cannon, P.; Hastie, R.; Nguyen, T.V.; Binder, N.K.; Tong, S.; Hannan, N.J. Combining Metformin and Esomeprazole Is Additive in Reducing SFlt-1 Secretion and Decreasing Endothelial Dysfunction—Implications for Treating Preeclampsia. PLoS ONE 2018, 13, e0188845. [Google Scholar] [CrossRef] [PubMed]
- Flores-Espinosa, P.; Preciado-Martínez, E.; Mejía-Salvador, A.; Sedano-González, G.; Bermejo-Martínez, L.; Parra-Covarruvias, A.; Estrada-Gutiérrez, G.; Vega-Sánchez, R.; Méndez, I.; Quesada-Reyna, B.; et al. Selective Immuno-Modulatory Effect of Prolactin upon pro-Inflammatory Response in Human Fetal Membranes. J. Reprod. Immunol. 2017, 123, 58–64. [Google Scholar] [CrossRef]
- Olmos-Ortiz, A.; Déciga-García, M.; Preciado-Martínez, E.; Bermejo-Martínez, L.; Flores-Espinosa, P.; Mancilla-Herrera, I.; Irles, C.; Helguera-Repetto, A.C.; Quesada-Reyna, B.; Goffin, V.; et al. Prolactin Decreases LPS-Induced Inflammatory Cytokines by Inhibiting TLR-4/NFκB Signaling in the Human Placenta. Mol. Hum. Reprod. 2019, 25, 660–667. [Google Scholar] [CrossRef]
- Zavan, B.; de Almeida, E.M.; Salles, É.D.S.L.; do Amarante-Paffaro, A.M.; Paffaro, V.A. COX-2 Plays a Role in Angiogenic DBA+ UNK Cell Subsets Activation and Pregnancy Protection in LPS-Exposed Mice. Placenta 2016, 44, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Miao, H.; Li, X.; Hu, Y.; Sun, H.; Hou, Y. Curcumin Inhibits Placental Inflammation to Ameliorate LPS-Induced Adverse Pregnancy Outcomes in Mice via Upregulation of Phosphorylated Akt. Inflamm. Res. 2017, 66, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Tchirikov, M.; Schlabritz-Loutsevitch, N.; Maher, J.; Buchmann, J.; Naberezhnev, Y.; Winarno, A.S.; Seliger, G. Mid-Trimester Preterm Premature Rupture of Membranes (PPROM): Etiology, Diagnosis, Classification, International Recommendations of Treatment Options and Outcome. J. Perinat. Med. 2018, 46, 465–488. [Google Scholar] [CrossRef]
- Jialal, I.; Rajamani, U. Endotoxemia of Metabolic Syndrome: A Pivotal Mediator of Meta-Inflammation. Metab. Syndr. Relat. Disord. 2014, 12, 454–456. [Google Scholar] [CrossRef]
- Ferriere, A.; Santa, P.; Garreau, A.; Bandopadhyay, P.; Blanco, P.; Ganguly, D.; Sisirak, V. Self-Nucleic Acid Sensing: A Novel Crucial Pathway Involved in Obesity-Mediated Metaflammation and Metabolic Syndrome. Front. Immunol. 2021, 11, 624256. [Google Scholar] [CrossRef]
- Mikamo, H.; Yamagishi, Y.; Sugiyama, H.; Sadakata, H.; Miyazaki, S.; Sano, T.; Tomita, T. High Glucose-Mediated Overexpression of ICAM-1 in Human Vaginal Epithelial Cells Increases Adhesion of Candida Albicans. J. Obs. Gynaecol. 2018, 38, 226–230. [Google Scholar] [CrossRef]
- Vitko, N.P.; Grosser, M.R.; Khatri, D.; Lance, T.R.; Richardson, A.R. Expanded Glucose Import Capability Affords Staphylococcus Aureus Optimized Glycolytic Flux during Infection. mBio 2016, 7, e00296-16. [Google Scholar] [CrossRef]
- Sakowicz-Burkiewicz, M.; Kocbuch, K.; Grden, M.; Maciejewska, I.; Szutowicz, A.; Pawelczyk, T. High Glucose Concentration Impairs ATP Outflow and Immunoglobulin Production by Human Peripheral B Lymphocytes: Involvement of P2X7 Receptor. Immunobiology 2013, 218, 591–601. [Google Scholar] [CrossRef]
- Luk, A.O.Y.; Lau, E.S.H.; Cheung, K.K.T.; Kong, A.P.S.; Ma, R.C.W.; Ozaki, R.; Chow, F.C.C.; So, W.Y.; Chan, J.C.N. Glycaemia Control and the Risk of Hospitalisation for Infection in Patients with Type 2 Diabetes: Hong Kong Diabetes Registry. Diabetes Metab. Res. Rev. 2017, 33, e2923. [Google Scholar] [CrossRef]
- Kai-Larsen, Y.; Gudmundsson, G.H.; Agerberth, B. A Review of the Innate Immune Defence of the Human Foetus and Newborn, with the Emphasis on Antimicrobial Peptides. Acta Paediatr. Int. J. Paediatr. 2014, 103, 1000–1008. [Google Scholar] [CrossRef]
- King, A.E.; Paltoo, A.; Kelly, R.W.; Sallenave, J.M.; Bocking, A.D.; Challis, J.R.G. Expression of Natural Antimicrobials by Human Placenta and Fetal Membranes. Placenta 2007, 28, 161–169. [Google Scholar] [CrossRef]
- Olmos-Ortiz, A.; García-Quiroz, J.; Avila, E.; Caldiño-Soto, F.; Halhali, A.; Larrea, F.; Díaz, L. Lipopolysaccharide and CAMP Modify Placental Calcitriol Biosynthesis Reducing Antimicrobial Peptides Gene Expression. Am. J. Reprod. Immunol. 2018, 79, e12841. [Google Scholar] [CrossRef]
- Olmos-Ortiz, A.; Hernández-Pérez, M.; Flores-Espinosa, P.; Sedano, G.; Helguera-Repetto, A.C.; Villavicencio-Carrisoza, Ó.; Valdespino-Vazquez, M.Y.; Flores-Pliego, A.; Irles, C.; Rivas-Santiago, B.; et al. Compartmentalized Innate Immune Response of Human Fetal Membranes against Escherichia Coli Choriodecidual Infection. Int. J. Mol. Sci. 2022, 23, 2994. [Google Scholar] [CrossRef]
- Zaga-Clavellina, V.; Garcia-Lopez, G.; Flores-Espinosa, P. Evidence of in Vitro Differential Secretion of Human Beta-Defensins-1, -2, and -3 after Selective Exposure to Streptococcus Agalactiae in Human Fetal Membranes. J. Matern.-Fetal Neonatal Med. 2012, 25, 358–363. [Google Scholar] [CrossRef]
- Froy, O.; Hananel, A.; Chapnik, N.; Madar, Z. Differential Effect of Insulin Treatment on Decreased Levels of Beta-Defensins and Toll-like Receptors in Diabetic Rats. Mol. Immunol. 2007, 44, 796–802. [Google Scholar] [CrossRef]
- Szukiewicz, D.; Alkhalayla, H.; Pyzlak, M.; Szewczyk, G. High Glucose Culture Medium Downregulates Production of Human β-Defensin-2 (HBD-2) in Human Amniotic Epithelial Cells (HAEC). In Proceedings of the Experimental Biology 2016 Meeting, San Diego, CA, USA, 2–6 April 2016. [Google Scholar]
- Montoya-Rosales, A.; Castro-Garcia, P.; Torres-Juarez, F.; Enciso-Moreno, J.A.; Rivas-Santiago, B. Glucose Levels Affect LL-37 Expression in Monocyte-Derived Macrophages Altering the Mycobacterium Tuberculosis Intracellular Growth Control. Microb. Pathog. 2016, 97, 148–153. [Google Scholar] [CrossRef]
- Xia, S.L.; Li, X.F.; Abasubong, K.P.; Xu, C.; Shi, H.J.; Liu, W.B.; Zhang, D.D. Effects of Dietary Glucose and Starch Levels on the Growth, Apparent Digestibility, and Skin-Associated Mucosal Non-Specific Immune Parameters in Juvenile Blunt Snout Bream (Megalobrama Amblycephala). Fish Shellfish Immunol. 2018, 79, 193–201. [Google Scholar] [CrossRef]
- King, A.E.; Kelly, R.W.; Sallenave, J.M.; Bocking, A.D.; Challis, J.R.G. Innate Immune Defences in the Human Uterus during Pregnancy. Placenta 2007, 28, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Duits, L.A.; Ravensbergen, B.; Rademaker, M.; Hiemstra, P.S.; Nibbering, P.H. Expression of β-Defensin 1 and 2 MRNA by Human Monocytes, Macrophages and Dendritic Cells. Immunology 2002, 106, 517–525. [Google Scholar] [CrossRef]
- King, A.E.; Critchley, H.O.D.; Sallenave, J.M.; Kelly, R.W. Elafin in Human Endometrium: An Antiprotease and Antimicrobial Molecule Expressed during Menstruation. J. Clin. Endocrinol. Metab. 2003, 88, 4426–4431. [Google Scholar] [CrossRef]
- Huang, C.T.; Lue, J.H.; Cheng, T.H.; Tsai, Y.J. Glycemic Control with Insulin Attenuates Sepsis-Associated Encephalopathy by Inhibiting Glial Activation via the Suppression of the Nuclear Factor Kappa B and Mitogen-Activated Protein Kinase Signaling Pathways in Septic Rats. Brain Res. 2020, 1738, 146822. [Google Scholar] [CrossRef]
- Martins, J.O.; Ferracini, M.; Ravanelli, N.; Landgraf, R.G.; Jancar, S. Insulin Suppresses LPS-Induced INOS and COX-2 Expression and NF-ΚB Activation in Alveolar Macrophages. Cell. Physiol. Biochem. 2008, 22, 279–286. [Google Scholar] [CrossRef]
- Lin, Y.; Ye, S.; He, Y.; Li, S.; Chen, Y.; Zhai, Z. Short-Term Insulin Intensive Therapy Decreases MCP-1 and NF-ΚB Expression of Peripheral Blood Monocyte and the Serum MCP-1 Concentration in Newly-Diagnosed Type 2 Diabetics. Arch. Endocrinol. Metab. 2018, 62, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Liu, Y.; Huan, Z.; Wang, Y.; Xu, J. Metformin Protects Chondrocytes against IL-1β Induced Injury by Regulation of the AMPK/NF-ΚB Signaling Pathway. Pharmazie 2020, 75, 632–636. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Peng, B.; Qin, Z.; Zhu, W.; Guo, C. Metformin Attenuates LPS-Induced Neuronal Injury and Cognitive Impairments by Blocking NF-ΚB Pathway. BMC Neurosci. 2021, 22, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Carlos, A.; Valdez-Miramontes, C.; Marin-Luevano, P.; González-Curiel, I.; Enciso-Moreno, J.A.; Rivas-Santiago, B. Metformin Promotes Mycobacterium Tuberculosis Killing and Increases the Production of Human β-Defensins in Lung Epithelial Cells and Macrophages. Microbes Infect. 2020, 22, 111–118. [Google Scholar] [CrossRef]
- Xiao, Y.; Liu, F.; Li, S.; Jiang, N.; Yu, C.; Zhu, X.; Qin, Y.; Hui, J.; Meng, L.; Song, C.; et al. Metformin Promotes Innate Immunity through a Conserved PMK-1/P38 MAPK Pathway. Virulence 2020, 11, 39–48. [Google Scholar] [CrossRef]
- Hulme, K.D.; Yan, L.; Marshall, R.J.; Bloxham, C.J.; Upton, K.R.; Hasnain, S.Z.; Bielefeldt-Ohmann, H.; Loh, Z.; Ronacher, K.; Chew, K.Y.; et al. High Glucose Levels Increase Influenzaassociated Damage to the Pulmonary Epithelial-Endothelial Barrier. eLife 2020, 9, e56907. [Google Scholar] [CrossRef] [PubMed]
- Giri, B.; Dey, S.; Das, T.; Sarkar, M.; Banerjee, J.; Dash, S.K. Chronic Hyperglycemia Mediated Physiological Alteration and Metabolic Distortion Leads to Organ Dysfunction, Infection, Cancer Progression and Other Pathophysiological Consequences: An Update on Glucose Toxicity. Biomed. Pharmacother. 2018, 107, 306–328. [Google Scholar] [CrossRef] [PubMed]
- Molina, S.A.; Moriarty, H.K.; Infield, D.T.; Imhoff, B.R.; Vance, R.J.; Kim, A.H.; Hansen, J.M.; Hunt, W.R.; Koval, M.; McCarty, N.A. Insulin Signaling via the PI3-Kinase/Akt Pathway Regulates Airway Glucose Uptake and Barrier Function in a CFTR-Dependent Manner. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L688–L702. [Google Scholar] [CrossRef] [PubMed]
- Rong, X.; Ji, Y.; Zhu, X.; Yang, J.; Qian, D.; Mo, X.; Lu, Y. Neuroprotective Effect of Insulin-Loaded Chitosan Nanoparticles/PLGA-PEG-PLGA Hydrogel on Diabetic Retinopathy in Rats. Int. J. Nanomed. 2019, 14, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Han, J.T.; Zhang, W.F.; Wang, Y.C.; Cai, W.X.; Lv, G.F.; Hu, D.H. Insulin Protects against Damage to Pulmonary Endothelial Tight Junctions after Thermal Injury: Relationship with Zonula Occludens-1, F-Actin, and AKT Activity. Wound Repair Regen. 2014, 22, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Jiang, P.; Huang, C.; Li, J.; Chen, J.; Wang, L.; Lin, Y.; Wang, F.; Liu, J. Metformin Alters the Chemotaxis and Flagellar Motility of Escherichia Coli. Front. Microbiol. 2022, 12, 792406. [Google Scholar] [CrossRef]
- Zhang, M.; Sun, W.; Du, J.; Gou, Y.; Liu, L.; Wang, R.; Xu, X. Protective Effect of Metformin on Sepsis Myocarditis in Zebrafish. Dose-Response 2020, 18, 1559325820938543. [Google Scholar] [CrossRef]
- Lang, C.H.; Spitzer, J.A. Glucose Kinetics and Development of Endotoxin Tolerance during Long-Term Continuous Endotoxin Infusion. Metabolism 1987, 36, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, A.E.; Kopydlowski, K.M.; Vogel, S.N. Inhibition of Lipopolysaccharide-Induced Signal Transduction in Endotoxin-Tolerized Mouse Macrophages: Dysregulation of Cytokine, Chemokine, and Toll-Like Receptor 2 and 4 Gene Expression. J. Immunol. 2000, 164, 5564–5574. [Google Scholar] [CrossRef]
- Seeley, J.J.; Ghosh, S. Molecular Mechanisms of Innate Memory and Tolerance to LPS. J. Leukoc. Biol. 2017, 101, 107–119. [Google Scholar] [CrossRef]
- Foster, S.L.; Hargreaves, D.C.; Medzhitov, R. Gene-Specific Control of Inflammation by TLR-Induced Chromatin Modifications. Nature 2007, 447, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Zubair, K.; You, C.; Kwon, G.; Kang, K. Two Faces of Macrophages: Training and Tolerance. Biomedicines 2021, 9, 1596. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhang, J.; Yu, S.; Li, Y.; Zhu, J.; Zhang, K.; Zhang, R. Cell Pyroptosis in Health and Inflammatory Diseases. Cell Death Discov. 2022, 8, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and Functions of Inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef]
- Humberg, A.; Fortmann, I.; Siller, B.; Kopp, M.V.; Herting, E.; Göpel, W.; Härtel, C. Preterm Birth and Sustained Inflammation: Consequences for the Neonate. Semin. Immunopathol. 2020, 42, 451–468. [Google Scholar] [CrossRef]
- Green, E.S.; Arck, P.C. Pathogenesis of Preterm Birth: Bidirectional Inflammation in Mother and Fetus. Semin. Immunopathol. 2020, 42, 413–429. [Google Scholar] [CrossRef]
- Li, Y.; Luo, W.; Zhang, J.; Luo, Y.; Han, W.; Wang, H.; Xia, H.; Chen, Z.; Yang, Y.; Chen, Q.; et al. Maternal Inflammation Exaggerates Offspring Susceptibility to Cerebral Ischemia–Reperfusion Injury via the COX-2/PGD2/DP2 Pathway Activation. Oxid. Med. Cell. Longev. 2022, 2022, 1571705. [Google Scholar] [CrossRef]
- Song, H.; Hu, K.; Du, X.; Zhang, J.; Zhao, S. Risk Factors, Changes in Serum Inflammatory Factors, and Clinical Prevention and Control Measures for Puerperal Infection. J. Clin. Lab. Anal. 2020, 34, e23047. [Google Scholar] [CrossRef]
- Kielkopf, C.L.; Bauer, W.; Urbatsch, I.L. Bradford Assay for Determining Protein Concentration. Cold Spring Harb. Protoc. 2020, 2020, 102269. [Google Scholar] [CrossRef]
- Scottish Intercollegiate Guidelines Network (SIGN) Management of Suspected Bacterial Lower Urinary Tract Infection in Adult Women. (SIGN publication no. 160) 2020. Available online: https://www.sign.ac.uk/our-guidelines/management-of-suspected-bacterial-lower-urinary-tract-infection-in-adult-women/ (accessed on 25 October 2022).
- Kass, E.H. Bacteriuria and the Diagnosis of Infections of the Urinary Tract: With Observations on the Use of Methionine as a Urinary Antiseptic. AMA Arch. Intern. Med. 1957, 100, 709–714. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Parameter | Mean ± SD (n = 35) | Range (Min–Max) |
|---|---|---|
| Maternal age (years) | 30.6 ± 5.1 | 19–40 |
| Pre-gestational BMI (kg/m2) | 25.4 ± 3.3 | 18.6–30.5 |
| Number of pregnancies | 2 ± 1 | 1–5 |
| Parity (Primiparous/Multiparous) (%/%) | 9/26 | 25.7%/74.3% |
| Gestational age (weeks) | 38.4 ± 0.6 | 37.2–40 |
| SBP (mm Hg) | 112 ± 10 | 99–140 |
| DBP (mm Hg) | 75 ± 9 | 60–107 |
| Maternal weight gain (kg) | 11.1 ± 4.1 | 3.5–20 |
| NB weight (kg) | 3.1 ± 0.3 | 2.5–3.9 |
| NB weight centiles | 49 ± 25 | 15–97 |
| NB length (cm) | 49 ± 1 | 47.5–53 |
| NB length centiles | 61 ± 25 | 15–98 |
| NB head circumference (cm) | 35 ± 1 | 32.5–37.5 |
| NB head circumference centiles | 77 ± 24 | 17–99 |
| APGAR 1 min | 8.4 ± 0.5 | 7–9 |
| APGAR 5 min | 9.2 ± 0.4 | 9–10 |
| Placenta weight (g) | 536 ± 122 | 312.4–728.8 |
| NB sex (male/female) (%/%) | 17/18 | 48.5%/51.5% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiménez-Escutia, R.; Vargas-Alcantar, D.; Flores-Espinosa, P.; Helguera-Repetto, A.C.; Villavicencio-Carrisoza, O.; Mancilla-Herrera, I.; Irles, C.; Torres-Ramos, Y.D.; Valdespino-Vazquez, M.Y.; Velázquez-Sánchez, P.; et al. High Glucose Promotes Inflammation and Weakens Placental Defenses against E. coli and S. agalactiae Infection: Protective Role of Insulin and Metformin. Int. J. Mol. Sci. 2023, 24, 5243. https://doi.org/10.3390/ijms24065243
Jiménez-Escutia R, Vargas-Alcantar D, Flores-Espinosa P, Helguera-Repetto AC, Villavicencio-Carrisoza O, Mancilla-Herrera I, Irles C, Torres-Ramos YD, Valdespino-Vazquez MY, Velázquez-Sánchez P, et al. High Glucose Promotes Inflammation and Weakens Placental Defenses against E. coli and S. agalactiae Infection: Protective Role of Insulin and Metformin. International Journal of Molecular Sciences. 2023; 24(6):5243. https://doi.org/10.3390/ijms24065243
Chicago/Turabian StyleJiménez-Escutia, Rodrigo, Donovan Vargas-Alcantar, Pilar Flores-Espinosa, Addy Cecilia Helguera-Repetto, Oscar Villavicencio-Carrisoza, Ismael Mancilla-Herrera, Claudine Irles, Yessica Dorin Torres-Ramos, María Yolotzin Valdespino-Vazquez, Pilar Velázquez-Sánchez, and et al. 2023. "High Glucose Promotes Inflammation and Weakens Placental Defenses against E. coli and S. agalactiae Infection: Protective Role of Insulin and Metformin" International Journal of Molecular Sciences 24, no. 6: 5243. https://doi.org/10.3390/ijms24065243
APA StyleJiménez-Escutia, R., Vargas-Alcantar, D., Flores-Espinosa, P., Helguera-Repetto, A. C., Villavicencio-Carrisoza, O., Mancilla-Herrera, I., Irles, C., Torres-Ramos, Y. D., Valdespino-Vazquez, M. Y., Velázquez-Sánchez, P., Zamora-Escudero, R., Islas-López, M., Carranco-Salinas, C., Díaz, L., Zaga-Clavellina, V., & Olmos-Ortiz, A. (2023). High Glucose Promotes Inflammation and Weakens Placental Defenses against E. coli and S. agalactiae Infection: Protective Role of Insulin and Metformin. International Journal of Molecular Sciences, 24(6), 5243. https://doi.org/10.3390/ijms24065243