

Highly Efficient Cationic Polymerization of β-Pinene, a Bio-Based, Renewable Olefin, with TiCl4 Catalyst from Cryogenic to Energy-Saving Room Temperature Conditions



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
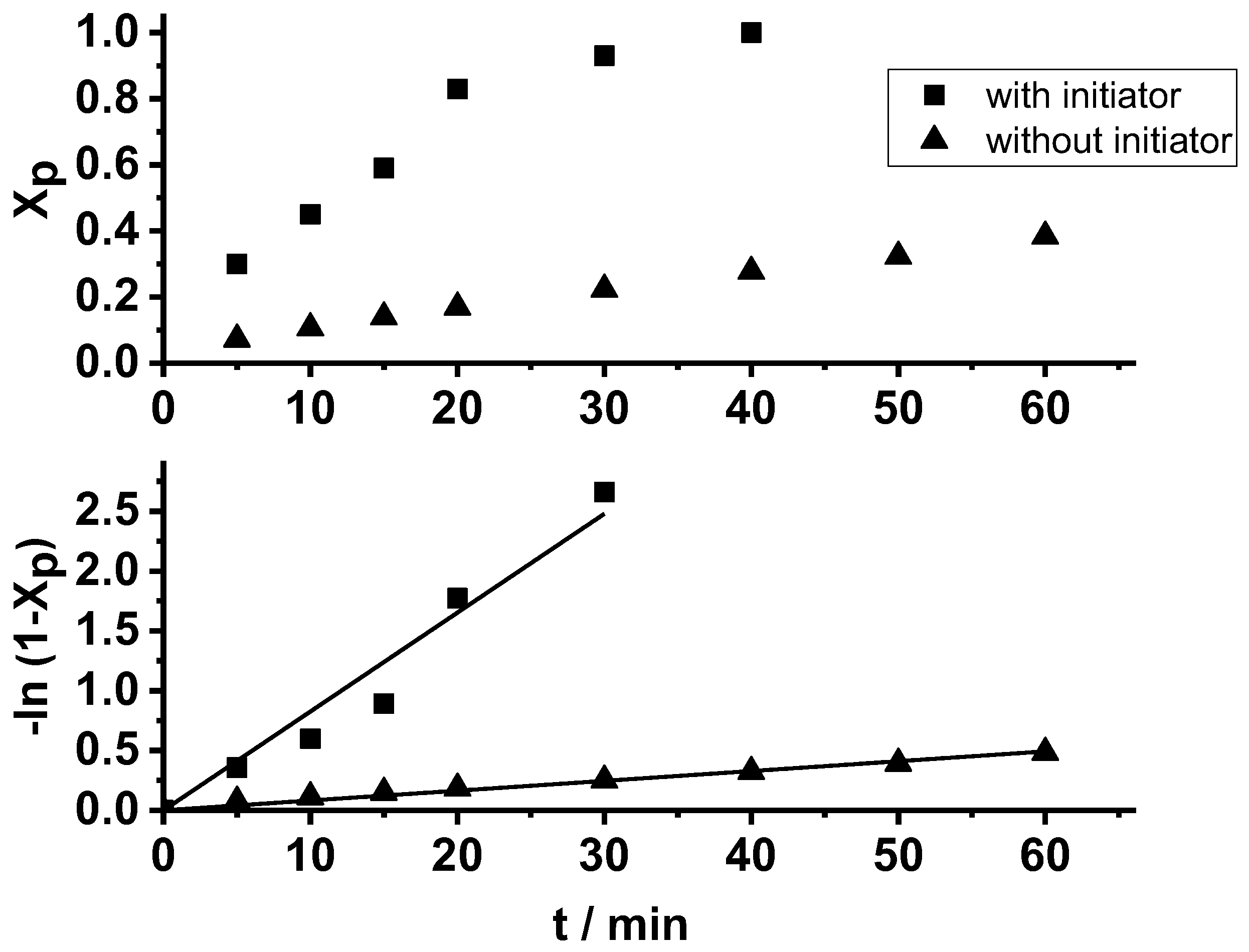
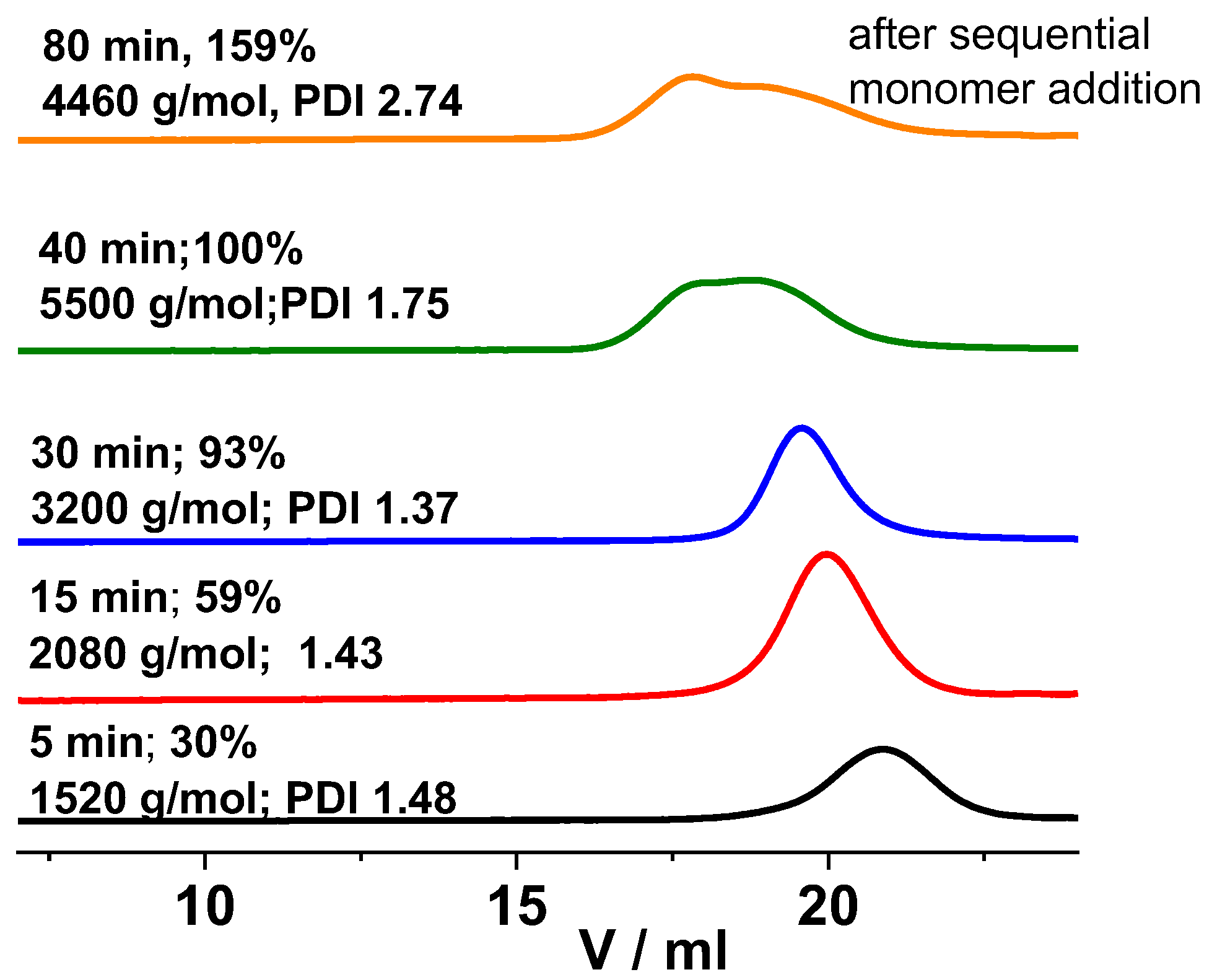
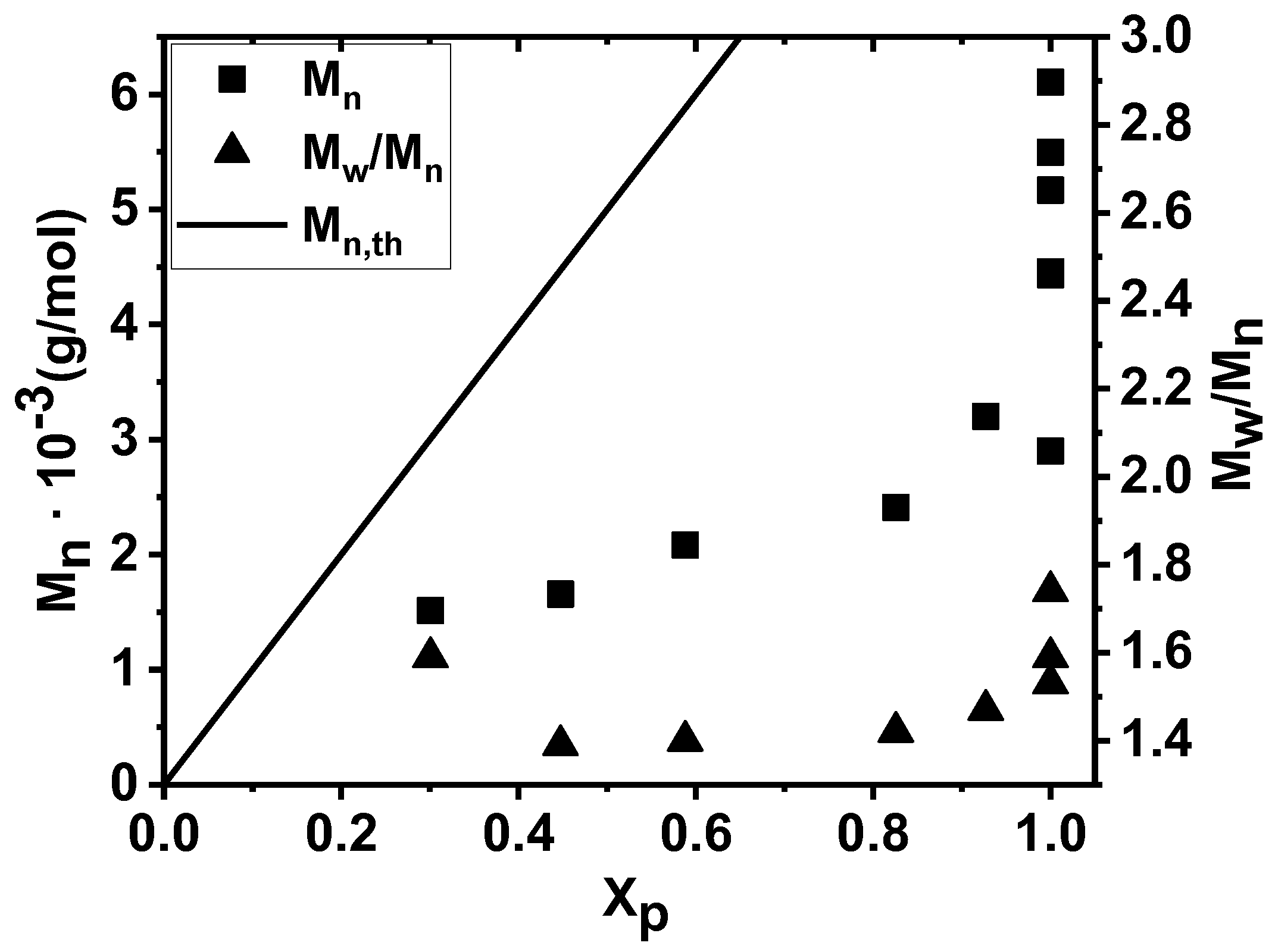
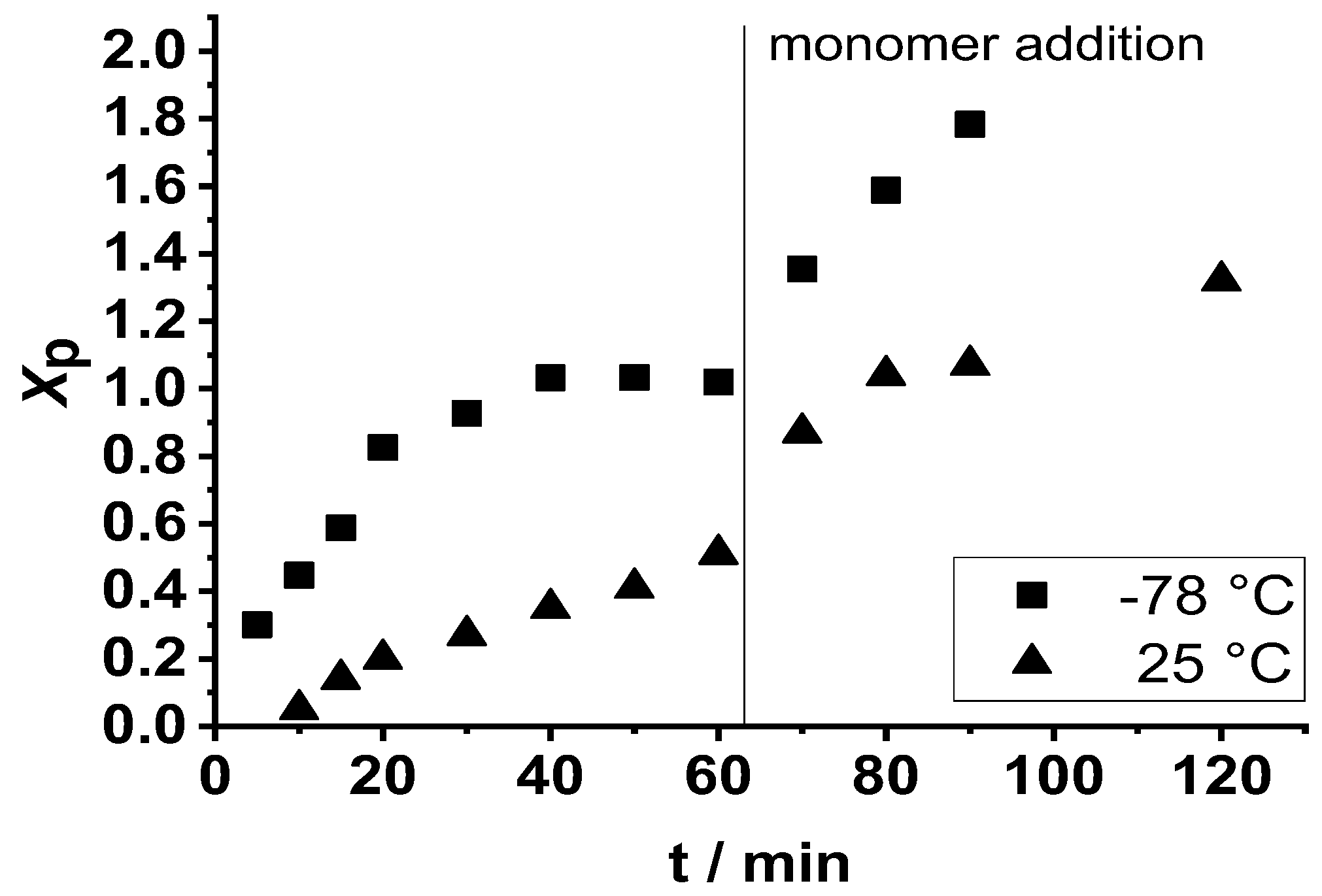
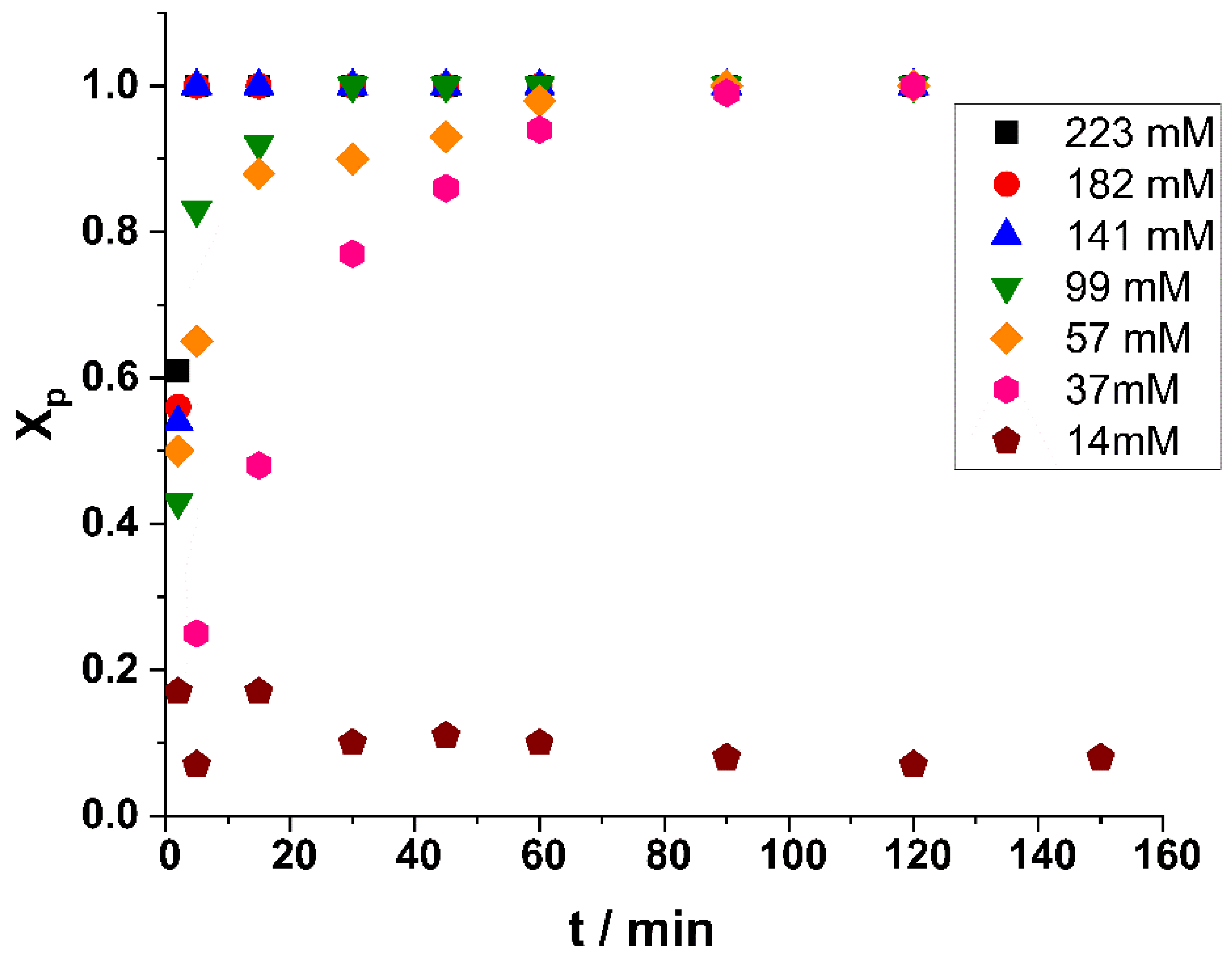
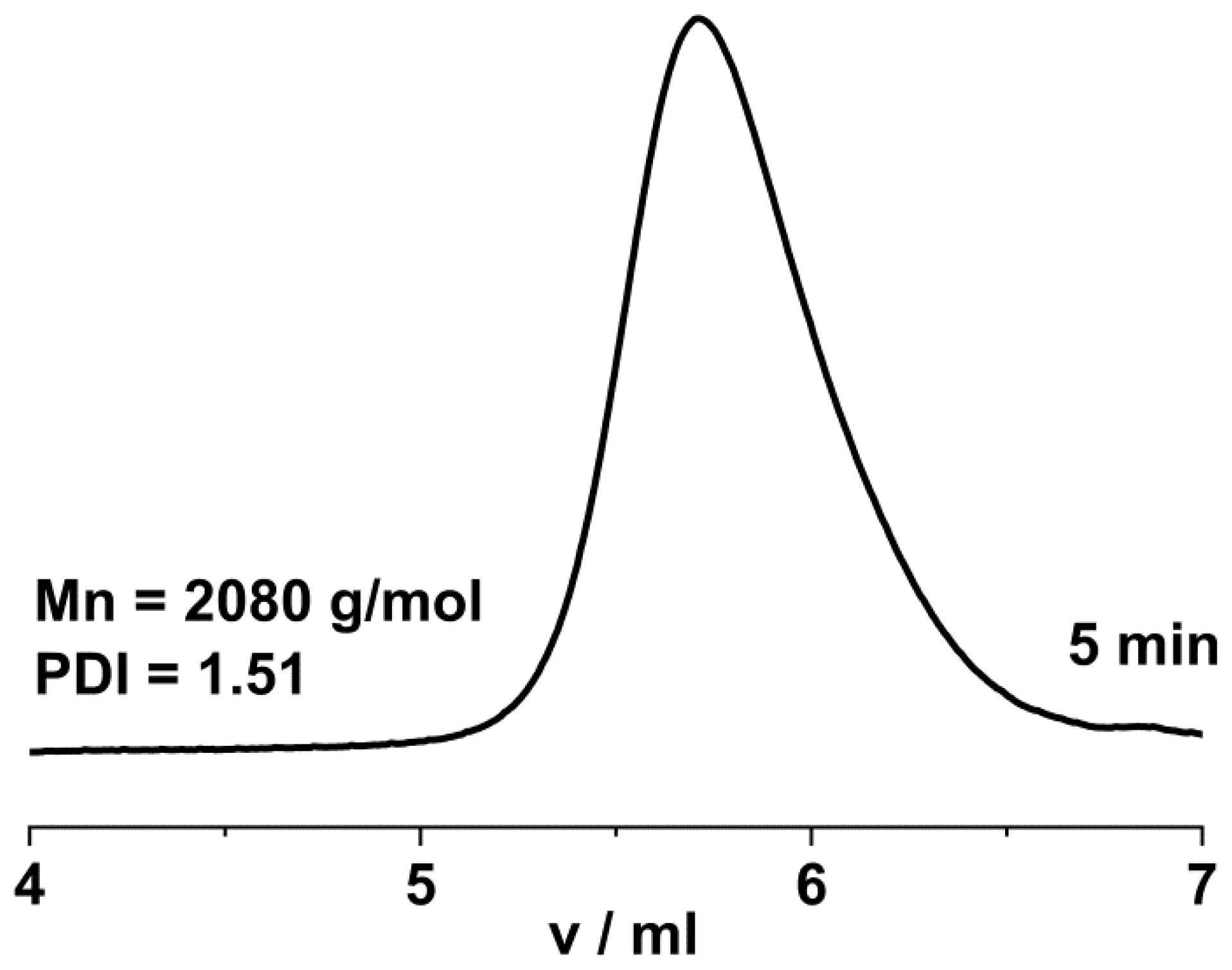
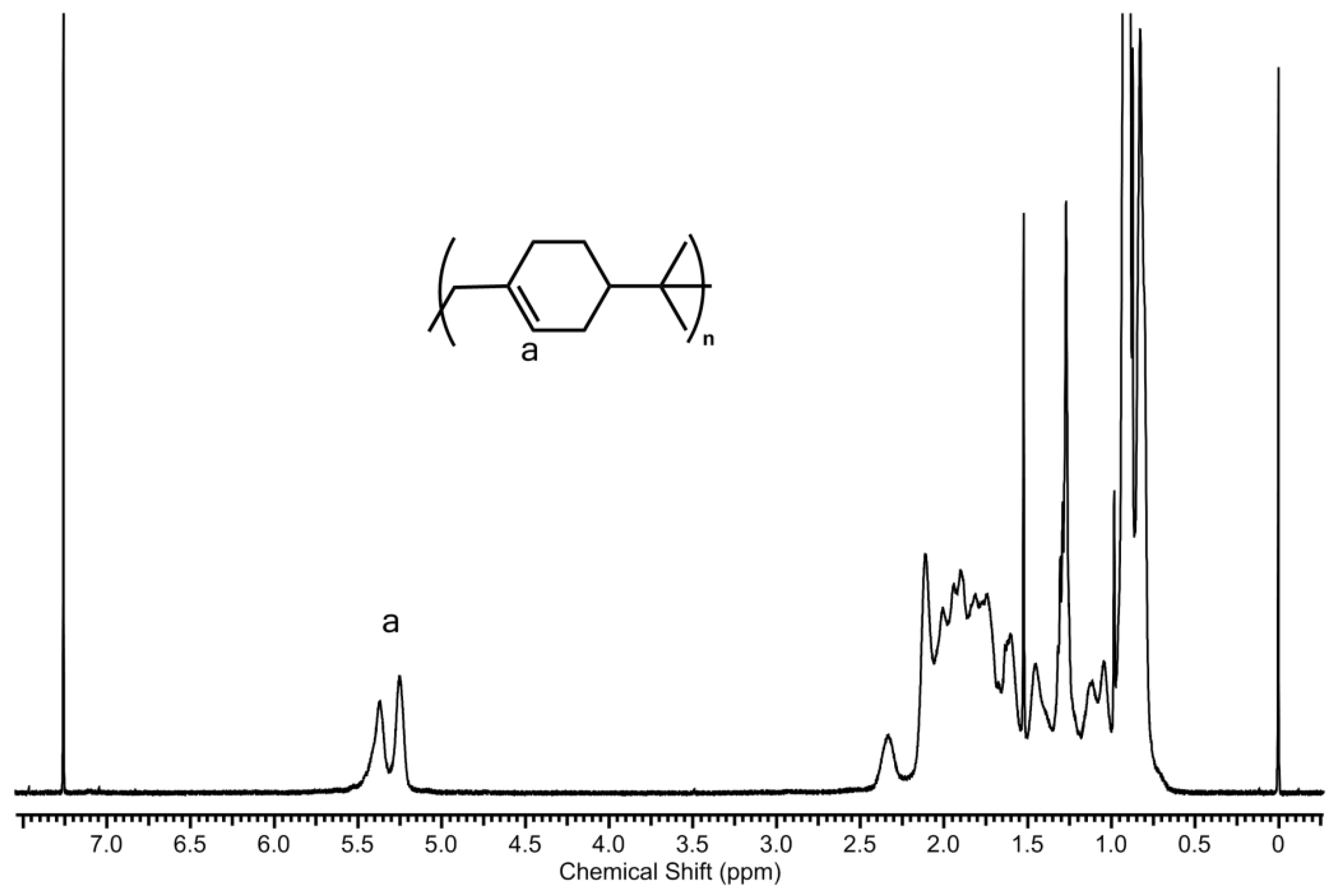
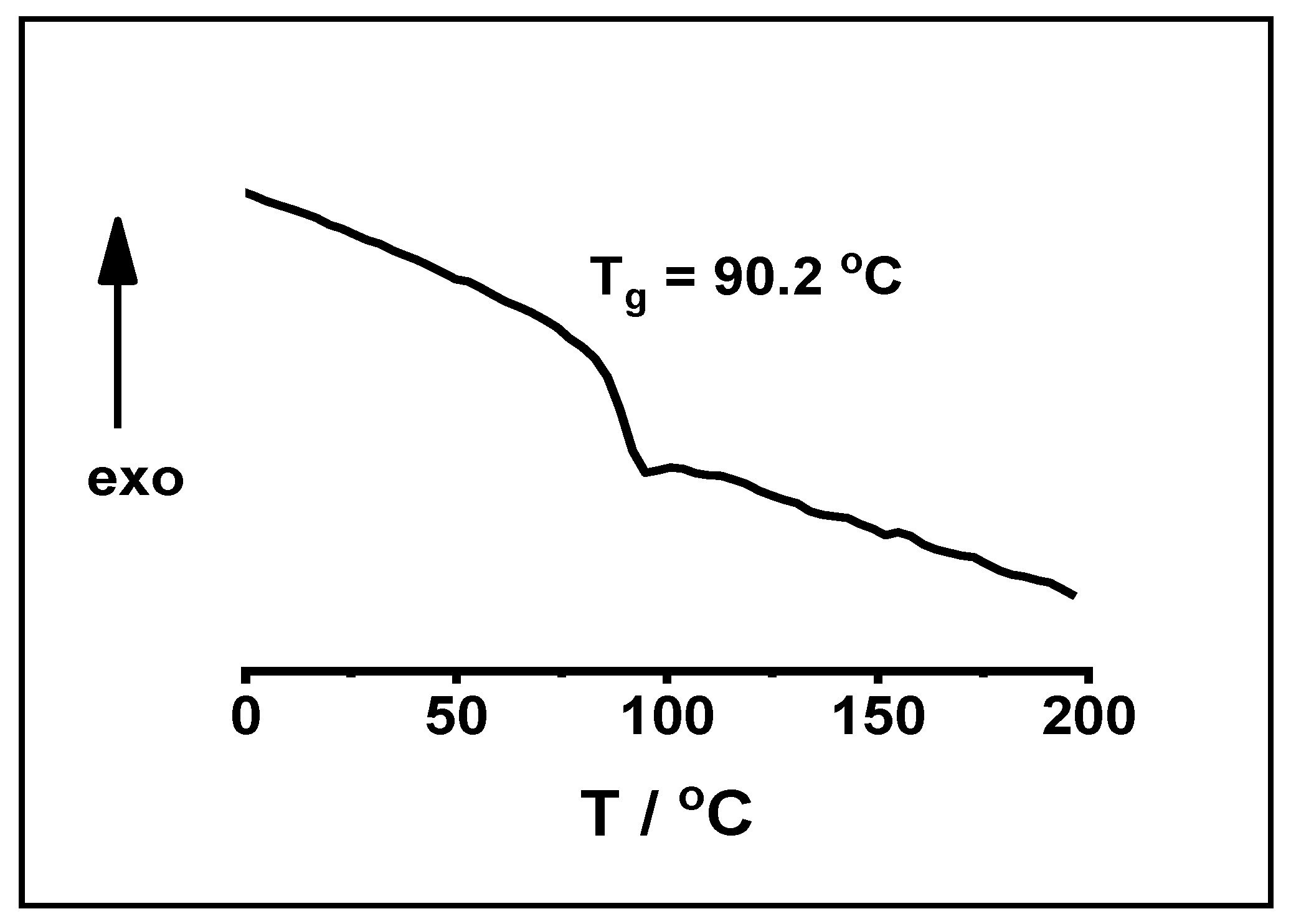
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- IEA. Oil 2019: Analysis and Forecasts to 2024; IEA: Paris, France, 2019. [Google Scholar] [CrossRef]
- Roddy, D.J. Biomass in a petrochemical world. Interface Focus 2013, 3, 20120038. [Google Scholar] [CrossRef]
- Cywar, R.M.; Rorrer, N.A.; Hoyt, C.B.; Beckham, G.T.; Chen, E.Y.X. Bio-based polymers with performance-advantaged properties. Nat. Rev. Mater. 2022, 7, 83–102. [Google Scholar] [CrossRef]
- Zhu, Y.; Romain, C.; Williams, C.K. Sustainable polymers from renewable resources. Nature 2016, 540, 354–362. [Google Scholar] [CrossRef]
- Maraveas, C.; Bayer, I.S.; Bartzanas, T. Recent advances in antioxidant polymers: From sustainable and natural monomers to synthesis and applications. Polymers 2021, 13, 2465. [Google Scholar] [CrossRef]
- Voet, V.S.; Guit, J.; Loos, K. Sustainable photopolymers in 3D printing: A review on biobased, biodegradable, and recyclable alternatives. Macromol. Rapid Commun. 2021, 42, 2000475. [Google Scholar] [CrossRef]
- Lacruz, A.; Salvador, M.; Blanco, M.; Vidal, K.; Goitandia, A.M.; Martinková, L.; Kyselka, M.; de Ilarduya, A.M. Biobased waterborne polyurethane-ureas modified with POSS-OH for fluorine-free hydrophobic textile coatings. Polymers 2021, 13, 3526. [Google Scholar] [CrossRef]
- Hulnik, M.I.; Kuharenko, O.V.; Vasilenko, I.V.; Timashev, P.; Kostjuk, S.V. Quasiliving cationic polymerization of anethole: Accessing high-performance plastic from the biomass-derived monomer. ACS Sust. Chem. Eng. 2021, 9, 6841–6854. [Google Scholar] [CrossRef]
- Rosenboom, J.G.; Langer, R.; Traverso, G. Bioplastics for a circular economy. Nat. Rev. Mater. 2022, 7, 117–137. [Google Scholar] [CrossRef]
- Niedner, L.; Kali, G. Green engineered polymers: Solvent free, room-temperature polymerization of monomer from a renewable resource, without utilizing initiator. Chem. Select 2019, 4, 3495–3499. [Google Scholar] [CrossRef]
- Mohanty, A.K.; Vivekanandhan, S.; Pin, J.M.; Misra, M. Composites from renewable and sustainable resources: Challenges and innovations. Science 2018, 362, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Gandini, A.; M. Lacerda, T. Monomers and Macromolecular Materials from Renewable Resources: State of the Art and Perspectives. Molecules 2022, 27, 159. [Google Scholar] [CrossRef] [PubMed]
- Nada, A.A.; Andicsová, A.E.; Mosnácek, J. Irreversible and Self-Healing Electrically Conductive Hydrogels Made of Bio-Based Polymers. Int. J. Mol. Sci. 2022, 23, 842. [Google Scholar] [CrossRef] [PubMed]
- Lakatos, C.; Kordován, M.Á.; Czifrák, K.; Nagy, L.; Vadkerti, B.; Daróczi, L.; Zsuga, M.; Kéki, S. Synthesis of Sucrose-HDI Cooligomers: New Polyols for Novel Polyurethane Networks. Int. J. Mol. Sci. 2022, 23, 1444. [Google Scholar] [CrossRef] [PubMed]
- Mahamat Ahmat, Y.; Madadi, S.; Charbonneau, L.; Kaliaguine, S. Epoxidation of terpenes. Catalysts 2021, 11, 847. [Google Scholar] [CrossRef]
- Nishida, T.; Satoh, K.; Tamura, M.; Li, Y.; Tomishige, K.; Caillol, S.; Ladmiral, V.; Vayer, M.; Mahut, F.; Sinturel, C.; et al. Terpenoid-derived conjugated dienes with exo-methylene and a 6-membered ring: High cationic reactivity, regioselective living cationic polymerization, and random and block copolymerization with vinyl ethers. Polym. Chem. 2021, 12, 1186–1198. [Google Scholar] [CrossRef]
- Palenzuela, M.; Sánchez-Roa, D.; Damián, J.; Sessini, V.; Mosquera, M.E. Polymerization of terpenes and terpenoids using metal catalysts. Adv. Organomet. Chem. 2021, 75, 55–93. [Google Scholar] [CrossRef]
- Lamparelli, D.H.; Kleybolte, M.M.; Winnacker, M.; Capacchione, C. Sustainable myrcene-based elastomers via a convenient anionic polymerization. Polymers 2021, 13, 838. [Google Scholar] [CrossRef]
- Mosquera, M.E.G.; Jiménez, G.; Tabernero, V.; Vinueza-Vaca, J.; García-Estrada, C.; Kosalková, K.; Sola-Landa, A.; Monje, B.; Acosta, C.; Alonso, R.; et al. Terpenes and terpenoids: Building blocks to produce biopolymers. Sustain. Chem. 2021, 2, 467–492. [Google Scholar] [CrossRef]
- Wahlen, C.; Frey, H. Anionic polymerization of terpene monomers: New options for bio-based thermoplastic elastomers. Macromolecules 2021, 54, 7323–7336. [Google Scholar] [CrossRef]
- Derdar, H.; Mitchell, G.R.; Mahendra, V.S.; Benachour, M.; Haoue, S.; Cherifi, Z.; Bachari, K.; Harrane, A.; Meghabar, R. Green nanocomposites from rosin-limonene copolymer and Algerian clay. Polymers 2020, 12, 1971. [Google Scholar] [CrossRef]
- Sahu, P.; Bhowmick, A.K.; Kali, G. Terpene based elastomers: Synthesis, properties, and applications. Processes 2020, 8, 553. [Google Scholar] [CrossRef]
- Della Monica, F.; Kleij, A.W. From terpenes to sustainable and functional polymers. Polym. Chem. 2020, 11, 5109–5127. [Google Scholar] [CrossRef]
- Wilbon, P.A.; Chu, F.; Tang, C. Progress in renewable polymers from natural terpenes, terpenoids, and rosin. Macromol. Rapid Commun. 2013, 34, 8–37. [Google Scholar] [CrossRef] [PubMed]
- Winnacker, M. Pinenes: Abundant and renewable building blocks for a variety of sustainable polymers. Angew. Chem. Int. Ed. 2018, 57, 14362–14371. [Google Scholar] [CrossRef] [PubMed]
- Nyamwihura, R.J.; Ogungbe, I.V. The pinene scaffold: Its occurrence, chemistry, synthetic utility, and pharmacological importance. RSC Adv. 2022, 12, 11346–11375. [Google Scholar] [CrossRef]
- Kleybolte, M.M.; Winnacker, M. β-pinene-derived polyesteramides and their blends: Advances in their upscaling, processing, and characterization. Macromol. Rapid Commun. 2021, 42, 2100065. [Google Scholar] [CrossRef]
- Stamm, A.; Öhlin, J.; Mosbech, C.; Olsén, P.; Guo, B.; Söderberg, E.; Biundo, A.; Fogelström, L.; Bhattacharyya, S.; Bornscheuer, U.T.; et al. Pinene-based oxidative synthetic toolbox for scalable polyester synthesis. JACS Au 2021, 1, 1949–1960. [Google Scholar] [CrossRef]
- Atkinson, R.L.; Monaghan, O.R.; Elsmore, M.T.; Topham, P.D.; Toolan, D.T.; Derry, M.J.; Taresco, V.; Stockman, R.A.; De Focatiis, D.S.A.; Irvine, D.J.; et al. RAFT polymerisation of renewable terpene (meth)acrylates and the convergent synthesis of methacrylate–acrylate–methacrylate triblock copolymers. Polym. Chem. 2021, 12, 3177–3189. [Google Scholar] [CrossRef]
- Fried, A.D.; Brantley, J.N. Controlled polymerization of β-pinadiene: Accessing unusual polymer architectures with biomass-derived monomers. ACS Macro Lett. 2020, 9, 595–599. [Google Scholar] [CrossRef]
- Feng, X.; Xiao, Z.; Yang, Y.; Chen, S.; Liao, S.; Luo, H.; Lu, H.; Wang, Z.; Fan, G. β-pinene derived products with enhanced in vitro antimicrobial activity. Nat. Prod. Commun. 2021, 16, 1–8. [Google Scholar] [CrossRef]
- Xu, J.; Zhu, P.; Liu, X.; Hou, Y.; Yang, X.; Shan, S.; Ma, Y.; Pan, D.; Dong, B.; Guo, Z. Preparation of high-density fuel through dimerization of β-pinene. Chem. Eng. Technol. 2020, 43, 2259–2265. [Google Scholar] [CrossRef]
- Salehi, B.; Upadhyay, S.; Erdogan Orhan, I.; Kumar Jugran, A.; LD Jayaweera, S.; ADias, D.; Sharopov, F.; Taheri, Y.; Martins, N.; Baghalpour, N.; et al. Therapeutic potential of α- and β-pinene: A miracle gift of nature. Biomolecules 2019, 9, 738. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Feng, X.Z.; Xiao, Z.Q.; Fan, G.R.; Chen, S.X.; Liao, S.L.; Luo, H.; Wang, Z.D. Design, synthesis, antibacterial, antifungal and anticancer evaluations of novel β-pinene quaternary ammonium salts. Int. J. Mol. Sci. 2021, 22, 11299. [Google Scholar] [CrossRef]
- Adamski, A.; Adamska, J. The use of essential oils–alpha and β-pinene in the treatment of COVID-19. Ann. Clin. Med. Case Rep. 2021, 7, 1–4. [Google Scholar]
- Roberts, W.J.; Day, A.R. A study of the polymerization of α- and β-pinene with Friedel—Crafts type catalysts. J. Am. Chem. Soc. 1950, 72, 1226–1230. [Google Scholar] [CrossRef]
- Pietila, H.; Sivola, A.; Sheffer, H. Cationic polymerization of β-pinene, styrene and α-methylstyrene. J. Polym. Sci. Part A Polym. Chem. 1970, 8, 727–737. [Google Scholar] [CrossRef]
- Huet, J.M.; Marechal, E. Study of cationic polymerization of beta-pinene. C. R. Acad. Sci. Paris 1970, 271, 1058–1061. [Google Scholar]
- Snyder, C.; McIver, W.; Sheffer, H. Cationic polymerization of β-pinene and styrene. J. Appl. Polym. Sci. 1977, 21, 131–139. [Google Scholar] [CrossRef]
- Kennedy, J.P.; Liao, T.P.; Guhaniyogi, S.; Chang, V.S. Poly (β-pinenes) carrying one, two, or three functional end groups. I. The effect of reaction conditions on the polymerization of β-pinene. J. Polym. Sci. Polym. Chem. Ed. 1982, 20, 3219–3227. [Google Scholar] [CrossRef]
- Kennedy, J.P.; Liao, T.P.; Guhaniyogi, S.; Chang, V.S. Poly (β-pinenes) carrying one, two, and three functional end groups. II. Syntheses and characterization. J. Polym. Sci. Polym. Chem. Ed. 1982, 20, 3229–3240. [Google Scholar] [CrossRef]
- Martinez, F. Cationic polymerization of β-pinene. J. Polym. Sci. Polym. Chem. Ed. 1984, 22, 673–677. [Google Scholar] [CrossRef]
- Keszler, B.; Kennedy, J.P. Synthesis of high molecular weight poly(β-pinene). Adv. Polym. Sci. 1992, 100, 1–9. [Google Scholar] [CrossRef]
- Satoh, K.; Sugiyama, H.; Kamigaito, M. Biomass-derived heat-resistant alicyclic hydrocarbonpolymers: Poly(terpenes) and their hydrogenated derivatives. Green Chem. 2006, 8, 878–882. [Google Scholar] [CrossRef]
- Guiné, R.P.F.; Castro, J.A.A.M. Polymerization of β-pinene with ethylaluminum dichloride (C2H5AlCl2). J. Appl. Polym. Sci. 2001, 82, 2558–2565. [Google Scholar] [CrossRef]
- Lu, J.; Kamigaito, M.; Sawamoto, M.; Higashimura, T.; Deng, Y.X. Living cationic isomerization polymerization of β-pinene. 1. Initiation with HCl−2-chloroethyl vinyl ether adduct/TiCl3(OiPr) in conjunction with nBu4NCl. Macromolecules 1997, 30, 22–26. [Google Scholar] [CrossRef]
- Lu, J.; Kamigaito, M.; Sawamoto, M.; Higashimura, T.; Deng, Y.X. Living cationic isomerization polymerization of β-pinene. 2. Synthesis of block and random copolymers with styrene or p-methylstyrene. Macromolecules 1997, 30, 27–31. [Google Scholar] [CrossRef]
- Lu, J.; Kamigaito, M.; Sawamoto, M.; Higashimura, T.; Deng, Y.X. Living cationic isomerization polymerization of β-pinene. III. Synthesis of end-functionalized polymers and graft copolymers. J. Polym. Sci. Part A Polym. Chem. 1997, 35, 1423–1430. [Google Scholar] [CrossRef]
- Kukhta, N.A.; Vasilenko, I.V.; Kostjuk, S.V. Room temperature cationic polymerization of β-pinene using modified AlCl3 catalyst: Toward sustainable plastics from renewable biomass resources. Green Chem. 2011, 13, 2362–2364. [Google Scholar] [CrossRef]
- Satoh, K.; Nakahara, A.; Mukunoki, K.; Sugiyama, H.; Saito, H.; Kamigaito, M. Sustainable cycloolefin polymer from pine tree oil for optoelectronics material: Living cationic polymerization of β-pinene and catalytic hydrogenation of high-molecular-weight hydrogenated poly(β-pinene). Polym. Chem. 2014, 5, 3222–3230. [Google Scholar] [CrossRef]
- Destephen, A.; González de San Román, E.; Martínez-Tong, D.E.; Ballard, N. Cationic polymerization of β-pinene using B(C6F5)3 as a Lewis acid for the synthesis of tackifiers in pressure sensitive adhesives. Macromol. Mater. Eng. 2021, 306, 2100194. [Google Scholar] [CrossRef]
- Lu, J.; Kamigaito, M.; Sawamoto, M.; Higashimura, T.; Deng, Y.-X. Cationic polymerization of β-pinene with the AlCl3/SbCl3 binary catalyst: Comparison with α-pinene polymerization. J. Appl. Polym. Sci. 1996, 61, 1011–1016. [Google Scholar] [CrossRef]
- Cataldo, F.; Gobbino, M.; Ursini, O.; Angelini, G. A study on the optically active polymer poly-β-pinene. J. Macromol. Sci. Part A Pure Appl. Chem. 2007, 44, 1225–1234. [Google Scholar] [CrossRef]
- Hayatifar, M.; Marchetti, F.; Pampaloni, G.; Patil, Y.; Galletti, A.M.R. Room-temperature polymerization of β-pinene by niobium and tantalum halides. Catal. Today 2012, 192, 177–182. [Google Scholar] [CrossRef]
- Karasawa, Y.; Kimura, M.; Kanazawa, A.; Kanaoka, S.; Aoshima, S. New initiating systems for cationic polymerization of plant-derived monomers: GaCl3/alkylbenzene-induced controlled cationic polymerization of β-pinene. Polym. J. 2015, 47, 152–157. [Google Scholar] [CrossRef]
- Zhu, H.; Liu, Z.; Zhang, T.; Zeng, W.; An, X.; Lei, F. β-Pinene cationic polymerization using Keggin heteropolyacid catalysts. React. Kinet. Mech. Catal. 2010, 99, 463–470. [Google Scholar] [CrossRef]
- Zhu, H.; Liu, Z.; An, X.; Lei, F. Keggin heteropolyacids as catalyst for the polymerization of β-pinene. React. Kinet. Mech. Catal. 2010, 100, 355–361. [Google Scholar] [CrossRef]
- Liu, Z.G.; Zhu, H.L.; Wang, S.; Zhao, L.L.; Lei, F.H. Synthesis of substituted phosphotungstic acids and their catalytic performance on cationic polymerization of β-pinene. Adv. Mater. Res. 2013, 634, 616–619. [Google Scholar] [CrossRef]
- Liu, Z.G.; Zeng, W.; Zhang, T.S.; Wang, S.; Zhu, H.L.; Lei, F.H. Cationic polymerization of β-pinene using Keggin phosphotungstates as catalysts. Adv. Mater. Res. 2012, 550, 296–300. [Google Scholar] [CrossRef]
- Liu, Z.; Cao, S.; Wang, S.; Zeng, W.; Zhang, T.; Li, P.; Lei, F. Activated-carbon-supported phosphotungstic acid as novel heterogeneous catalysts for cationic polymerization of β-pinene. J. Chem. Eng. Jpn. 2015, 48, 29–34. [Google Scholar] [CrossRef]
- Liu, Z.; Cao, S.; Wang, S.; Zeng, W.; Zhang, T. Silica-supported phosphotungstic acid as a novel heterogeneous catalyst for β-pinene polymerization. React. Kinet. Mech. Catal. 2014, 111, 577–590. [Google Scholar] [CrossRef]
- Akeb, M.; Harrane, A.; Belbachir, M. Polymerization of β-pinene by using natural montmorillonite clay as a green catalyst. Green Mater. 2018, 6, 58–64. [Google Scholar] [CrossRef]
- Poco, J.G.R.; Danese, M.; Giudici, R. Investigation of Cationic Polymerization of β-Pinene Using Calorimetric Measurements. Macromol. React. Eng. 2010, 4, 145–154. [Google Scholar] [CrossRef]
- Destephen, A.; Ballard, N. On the limitations of cationic polymerization of vinyl monomers in aqueous dispersed media. Polym. Chem. 2021, 12, 6444–6455. [Google Scholar] [CrossRef]
- Gaspar, A.S.; Cordeiro, J.B.; Simoes, P.A.; Gameiro, D.; Rocha, F.A.; Serra, A.C.; Coelho, J.F.J.; Fonseca, A.C. Poly(β-pinene) as an efficient biobased tackifier for metallocene poly(ethylene) based hotmelt adhesives. Int. J. Adh. Adh. 2022, 114, 103111. [Google Scholar] [CrossRef]
- Zhang, X.; Li, N.; Han, S.; Wei, Z.; Dai, B. Terpene resin prepared from renewable turpentine oil as a new type of cold flow improver for soybean biodiesel-diesel blends. Fuel 2022, 320, 123844. [Google Scholar] [CrossRef]
- Rodrigues, P.R.; Goncalves, S.A.; Vieira, R.P. Organocatalyzed β-pinene polymerization in UV light: Assessment of reaction conditions and material characterization. Eur. Polym. J. 2021, 147, 110303. [Google Scholar] [CrossRef]
- Rodrigues, P.R.; Junior, L.M.; de Souza, W.F.C.; Sato, H.H.; Alves, R.M.V.; Vieira, R.P. O-ATRP synthesized poly(β-pinene) blended with chitosan for antimicrobial and antioxidant bio-based films production. Int. J. Biol. Macromol. 2021, 193, 425–432. [Google Scholar] [CrossRef]
- Rodrigues, P.R.; Nascimento, L.E.S.; Godoy, H.T.; Vieira, R.P. Improving chitosan performance in the simultaneous adsorption of multiple polycyclic aromatic hydrocarbons by oligo(β-pinene) incorporation. Carbohydrate Polym. 2023, 302, 120379. [Google Scholar] [CrossRef]
- Rodrigues, P.R.; Wang, X.; Li, Z.; Lyu, J.; Wang, W.; Vieira, R.P. A new nano hyperbranched β-pinene polymer: Controlled synthesis and nonviral gene delivery. Colloids Surf. B Biointerfaces 2023, 222, 113032. [Google Scholar] [CrossRef]
- Yu, P.; Li, A.L.; Liang, H.; Lu, J. Polymerization of β-pinene with Schiff-base nickel complexes catalyst: Synthesis of relatively high molecular weight poly (β-pinene) at high temperature with high productivity. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 3739–3746. [Google Scholar] [CrossRef]
- Kawamura, K.; Nomura, K. Ethylene copolymerization with limonene and β-pinene: New bio-based polyolefins prepared by coordination polymerization. Macromolecules 2021, 54, 4693–4703. [Google Scholar] [CrossRef]
- Sheffer, H.; Greco, G.; Paik, G. The characterization of styrene–β-pinene polymers. J. Appl. Polym. Sci. 1983, 28, 1701–1705. [Google Scholar] [CrossRef]
- Li, A.; Zhang, W.; Liang, H.; Lu, J. Living cationic random copolymerization of β-pinene and isobutylene with 1-phenylethyl chloride/TiCl4/Ti(OiPr)4/nBu4NCl. Polymer 2004, 45, 6533–6537. [Google Scholar] [CrossRef]
- Dey, A.; Haldar, U.; De, P. Block copolymer synthesis by the combination of living cationic polymerization and other polymerization methods. Front. Chem. 2021, 9, 644547. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Lu, J. Synthesis of poly (β-pinene)-g-polystyrene from allylic brominated poly (β-pinene). J. Appl. Polym. Sci. 2000, 75, 599–603. [Google Scholar] [CrossRef]
- Lu, J.; Liang, H.; Li, A.; Cheng, Q. Synthesis of block and graft copolymers of β-pinene and styrene by transformation of living cationic polymerization to atom transfer radical polymerization. Eur. Polym. J. 2004, 40, 397–402. [Google Scholar] [CrossRef]
- Lu, J.; Liang, H.; Zhang, W.; Cheng, Q. Synthesis of poly(β-pinene)-g-poly(meth)acrylate by the combination of living cationic polymerization and atom transfer radical polymerization. J. Polym. Sci. Part A Polym. Chem. 2003, 41, 1237–1242. [Google Scholar] [CrossRef]
- Lu, J.; Liang, H.; Zhang, R.; Li, B. Synthesis of poly(β-pinene)-b-polytetrahydrofuran from β-pinene-based macroinitiator. Polymer 2004, 42, 4549–4553. [Google Scholar] [CrossRef]
- Derakhshan, A.A.; Pirsaheb, M.; Zinadini, S. Synthesis of sustainable poly(S-Abietic-co-pinene) through inverse vulcanization of Kurdica gum and used to fabricate durable and recyclable super-hydrophobic cotton wool filter: Oil-water separation application. Prog. Org. Coat. 2022, 168, 106862. [Google Scholar] [CrossRef]
- Wang, S.; Zhou, Y.; Wang, Y.; Dong, F.; Liu, H.; Xu, X. A natural polymer with desirable self-healing and recyclable, antibacterial, and adhesive properties based on turpentine monomer. Mater. Chem. Front. 2023, 7, 333–344. [Google Scholar] [CrossRef]
- Kennedy, J.P.; Iván, B. Designed Polymers by Carbocationic Macromolecular Engineering: Theory and Practice; Hanser Publishers: Munich, NY, USA, 1992. [Google Scholar] [CrossRef]
- Iván, B.; Szanka, I.; Szabó, Á.; Pásztor, S.; Pásztói, B.; Stumphauser, T.; Kasza, G.; Szarka, G.; Kalocsai, D.; Bajcsi, Á.; et al. Endfunctional polyisobutylenes by quasiliving carbocationic polymerization and bi-and multicomponent macromolecular architectures therefrom. In Macromolecular Engineering: Design, Synthesis and Applications of Polymers; Lubnin, A., Erdodi, G., Eds.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 23–49. [Google Scholar] [CrossRef]
- Everland, H.; Kops, J.; Nielsen, A.; Iván, B. Living carbocationic polymerization of isobutylene and synthesis of ABA block copolymers by conventional laboratory techniques. Polym. Bull. 1993, 31, 159–166. [Google Scholar] [CrossRef]
- Tawada, M.; Faust, R. Living cationic polymerization of isobutylene with mixtures of titanium tetrachloride/titanium tetrabromide. Macromolecules 2005, 38, 4989–4995. [Google Scholar] [CrossRef]
- Fodor, Z.; Faust, R. Polyisobutylene-based thermoplastic elastomers. IV. Synthesis of poly (styrene-block-isobutylene-block-styrene) triblock copolymers using n-butyl chloride as solvent. J. Macromol. Sci. Part A Pure Appl. Chem. 1996, 33, 305–324. [Google Scholar] [CrossRef]
- Storey, R.F.; Thomas, Q.A. Quasi-living cationic polymerization of styrene and isobutylene: Measurement of run number and calculation of apparent rate constant of ionization by TiCl4. Macromolecules 2003, 36, 5065–5071. [Google Scholar] [CrossRef]
- Kaszas, G.; Puskas, J.E.; Kennedy, J.P.; Hager, W.G. Polyisobutylene-containing block polymers by sequential monomer addition. II. Polystyrene–polyisobutylene–polystyrene triblock polymers: Synthesis, characterization, and physical properties. J. Polym. Sci. Part A Polym. Chem. 1991, 29, 427–435. [Google Scholar] [CrossRef]
- Makarevich, M.I.; Nikishau, P.A.; Berezianko, I.A.; Glushkova, T.V.; Rezvova, M.A.; Ovcharenko, E.A.; Bekmukhamedov, G.E.; Yakhvarov, D.G.; Kostjuk, S.V. Aspects of the Synthesis of Poly(styrene-block-isobutylene-block-styrene) by TiCl4-Co-initiated Cationic Polymerization in Open Conditions. Macromol 2021, 1, 243–255. [Google Scholar] [CrossRef]
- Kennedy, J.P.; Maréchal, E. Carbocationic Polymerization; John Wiley & Sons Ltd.: New York, NY, USA, 1982. [Google Scholar] [CrossRef]
- Iván, B. Open mechanistic problems of quasiliving carbocationic polymerization of olefins mediated by nucleophilic additives. Macromol. Symp. 1988, 132, 65–74. [Google Scholar] [CrossRef]
- Held, D.; Iván, B.; Müller, A.H.; de Jong, F.; Graafland, T. Stability of propagating species in living cationic polymerization of isobutylene. ACS Symp. Ser. 1997, 665, 63–74. [Google Scholar]
- Storey, R.F.; Chisholm, B.J.; Masse, M.A. Morphology and physical properties of poly (styrene-b-isobutylene-b-styrene) block copolymers. Polymer 1996, 37, 2925–2938. [Google Scholar] [CrossRef]
- Nugay, N.; Nugay, T.; Deodhar, T.; Keszler, B.L.; Kennedy, J.P. Low cost bifunctional initiators for bidirectional living cationic polymerization of olefins. II. Hyperbranched styrene–isobutylene–styrene triblocks with superior combination of properties. J. Polym. Sci. Part A Polym. Chem. 2018, 56, 705–713. [Google Scholar] [CrossRef]
- Kali, G.; Szesztay, M.; Bodor, A.; Iván, B. A New Synthetic Method for the Preparation of Star-Shaped Polyisobutylene with Hyperbranched Polystyrene Core. Macromol. Chem. Phys. 2007, 208, 1388–1393. [Google Scholar] [CrossRef]
- Iván, B. Terminology and classification of quasiliving polymerizations and ideal living polymerizations on the basis of the logic of elementary polymerization reactions, and comments on using the term “controlled”. Macromol. Chem. Phys. 2000, 201, 2621–2628. [Google Scholar]
- Soares, A.; Pestana, M. Polyterpene Resins: Part I—A Brief Historical Review. Silva Lusit. 2020, 28, 181–195. [Google Scholar] [CrossRef]













Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verebélyi, K.; Szabó, Á.; Réti, Z.; Szarka, G.; Villányi, Á.; Iván, B. Highly Efficient Cationic Polymerization of β-Pinene, a Bio-Based, Renewable Olefin, with TiCl4 Catalyst from Cryogenic to Energy-Saving Room Temperature Conditions. Int. J. Mol. Sci. 2023, 24, 5170. https://doi.org/10.3390/ijms24065170
Verebélyi K, Szabó Á, Réti Z, Szarka G, Villányi Á, Iván B. Highly Efficient Cationic Polymerization of β-Pinene, a Bio-Based, Renewable Olefin, with TiCl4 Catalyst from Cryogenic to Energy-Saving Room Temperature Conditions. International Journal of Molecular Sciences. 2023; 24(6):5170. https://doi.org/10.3390/ijms24065170
Chicago/Turabian StyleVerebélyi, Klára, Ákos Szabó, Zsombor Réti, Györgyi Szarka, Ákos Villányi, and Béla Iván. 2023. "Highly Efficient Cationic Polymerization of β-Pinene, a Bio-Based, Renewable Olefin, with TiCl4 Catalyst from Cryogenic to Energy-Saving Room Temperature Conditions" International Journal of Molecular Sciences 24, no. 6: 5170. https://doi.org/10.3390/ijms24065170
APA StyleVerebélyi, K., Szabó, Á., Réti, Z., Szarka, G., Villányi, Á., & Iván, B. (2023). Highly Efficient Cationic Polymerization of β-Pinene, a Bio-Based, Renewable Olefin, with TiCl4 Catalyst from Cryogenic to Energy-Saving Room Temperature Conditions. International Journal of Molecular Sciences, 24(6), 5170. https://doi.org/10.3390/ijms24065170