
Caspase Inhibition Modulates Monocyte-Derived Macrophage Polarization in Damaged Tissues
 , ,
, ,  , , ,
, , ,  , ,
, , 
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Caspases Modulate CSF1-Induced Monocyte Differentiation
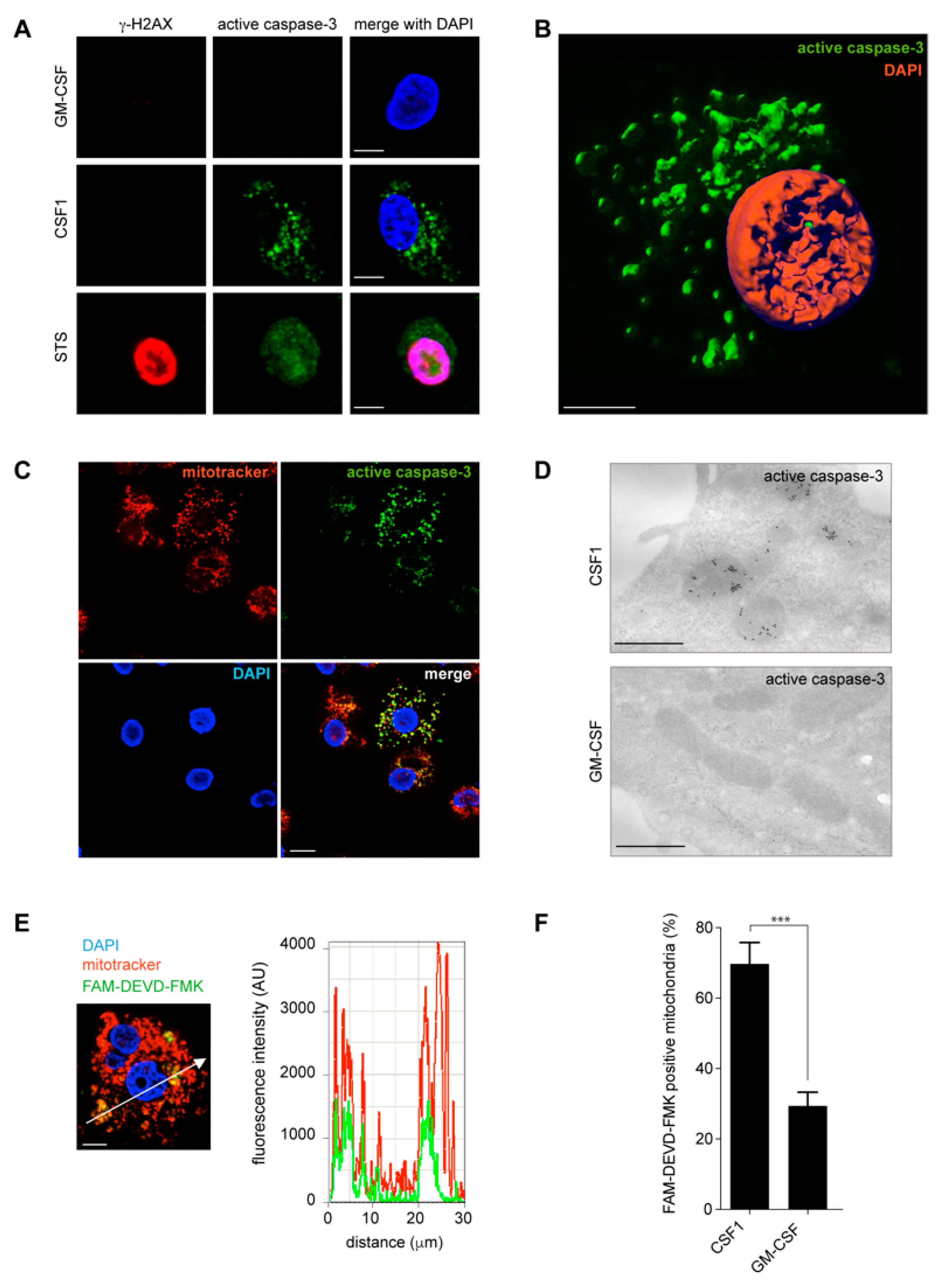
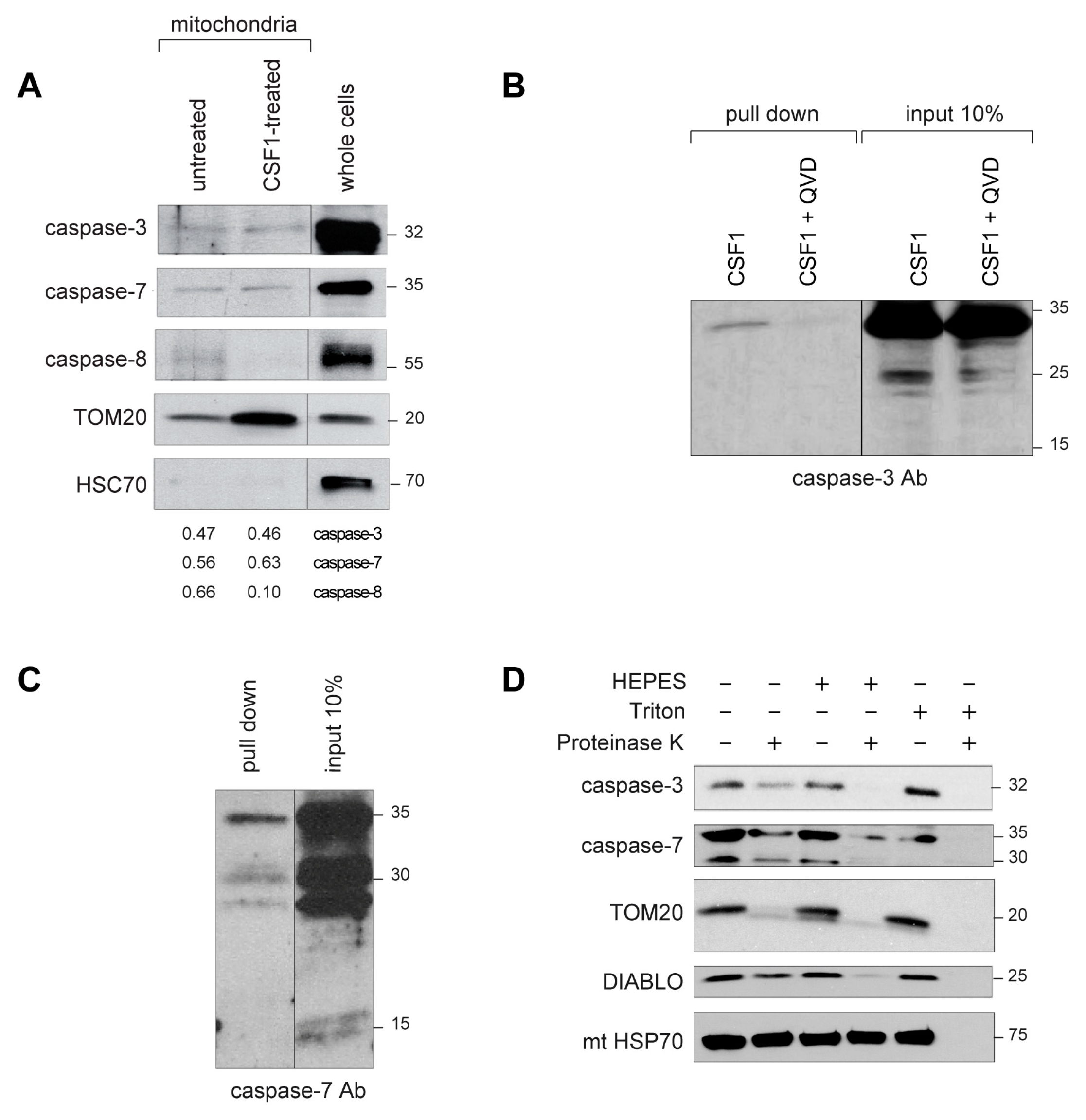
2.2. Active Caspase-3 and Caspase-7 Are Spatially Restricted to the Mitochondria Outer Membrane
2.3. Caspase Activation Promotes the Generation of Cytosolic Radical Oxygen Species
2.4. Active Caspase-7 Promotes NOX2 Activation through p47PHOX Cleavage
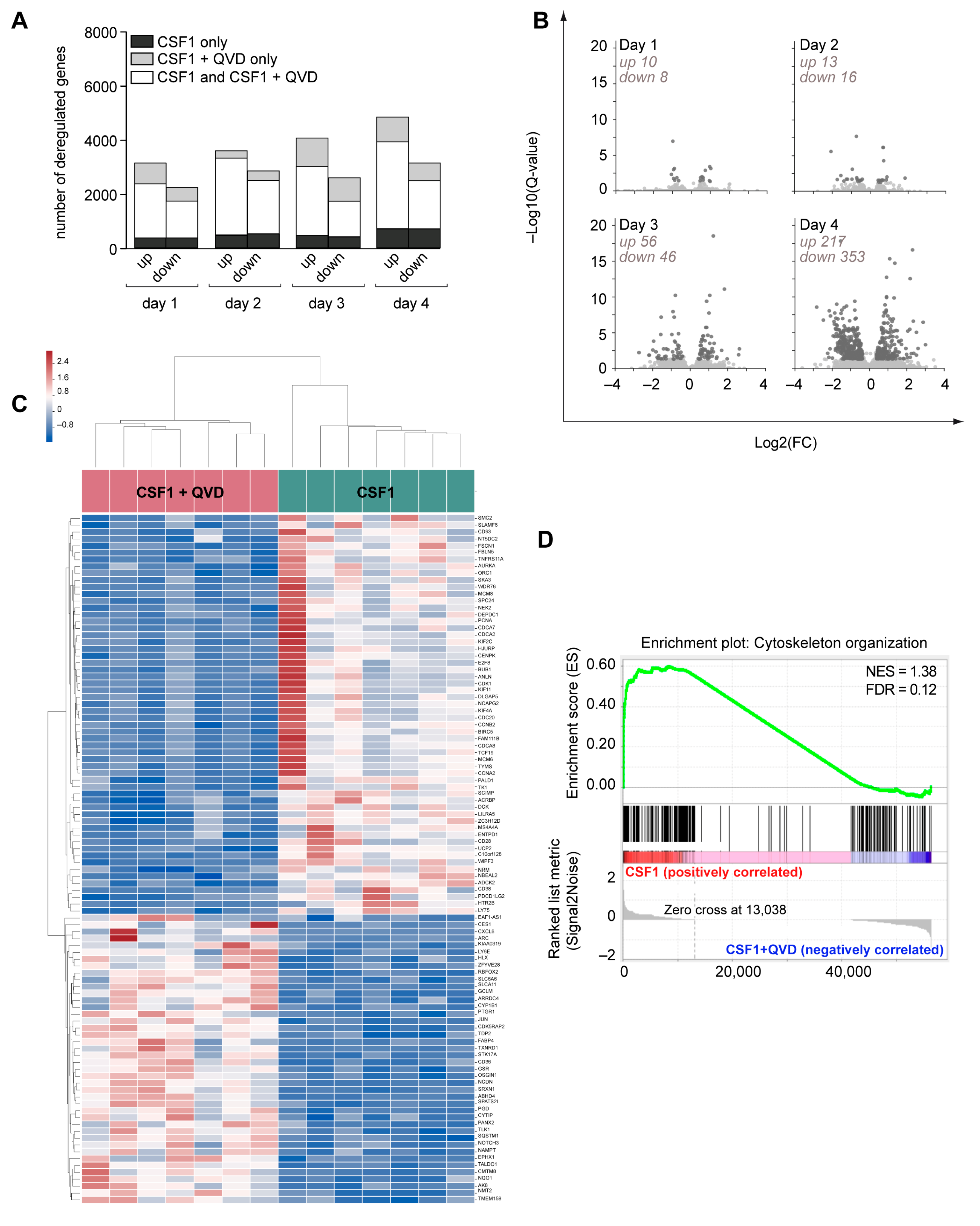
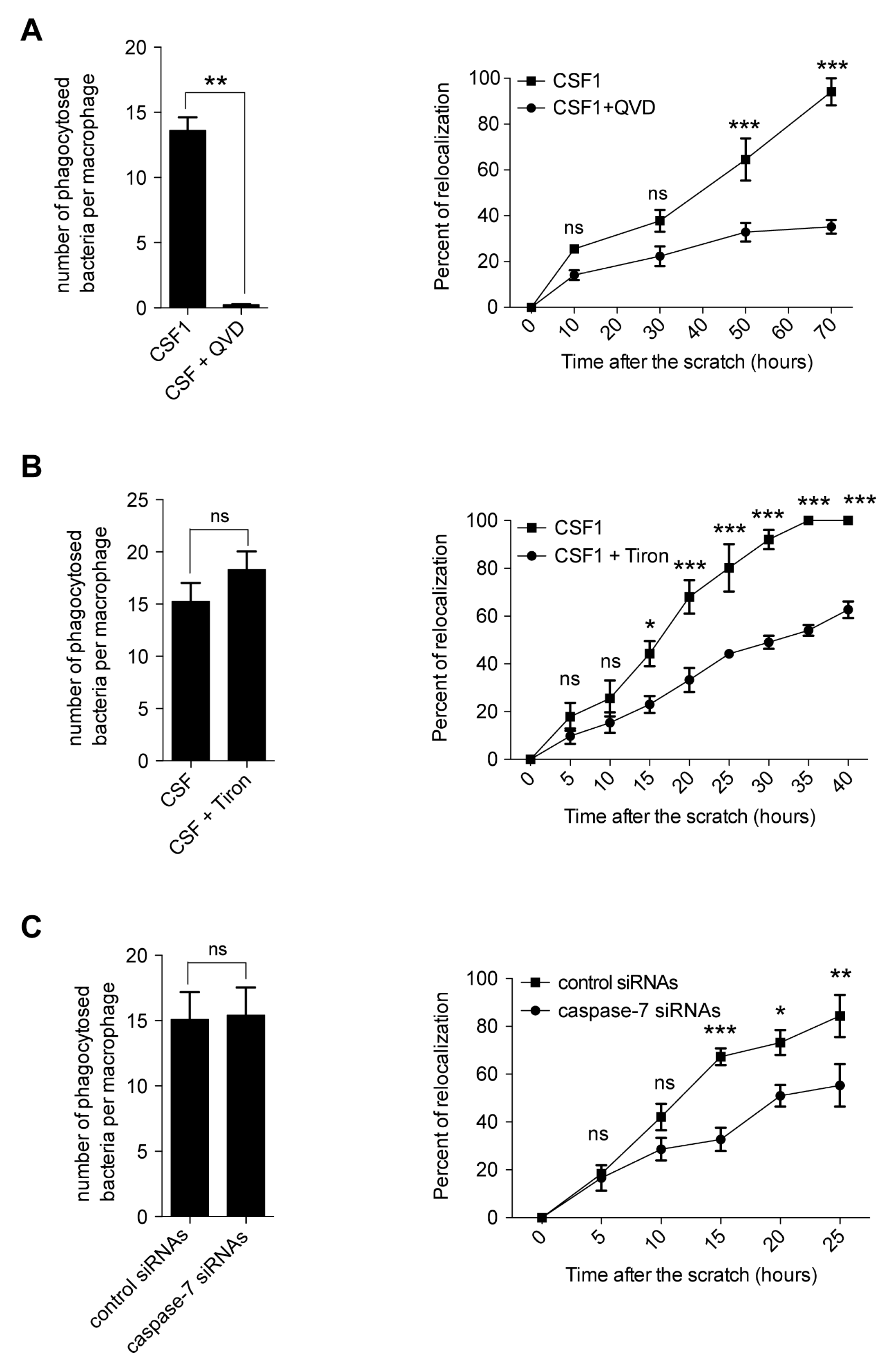
2.5. Caspase Inhibition Alters the Migration of CSF-1 Induced Macrophages
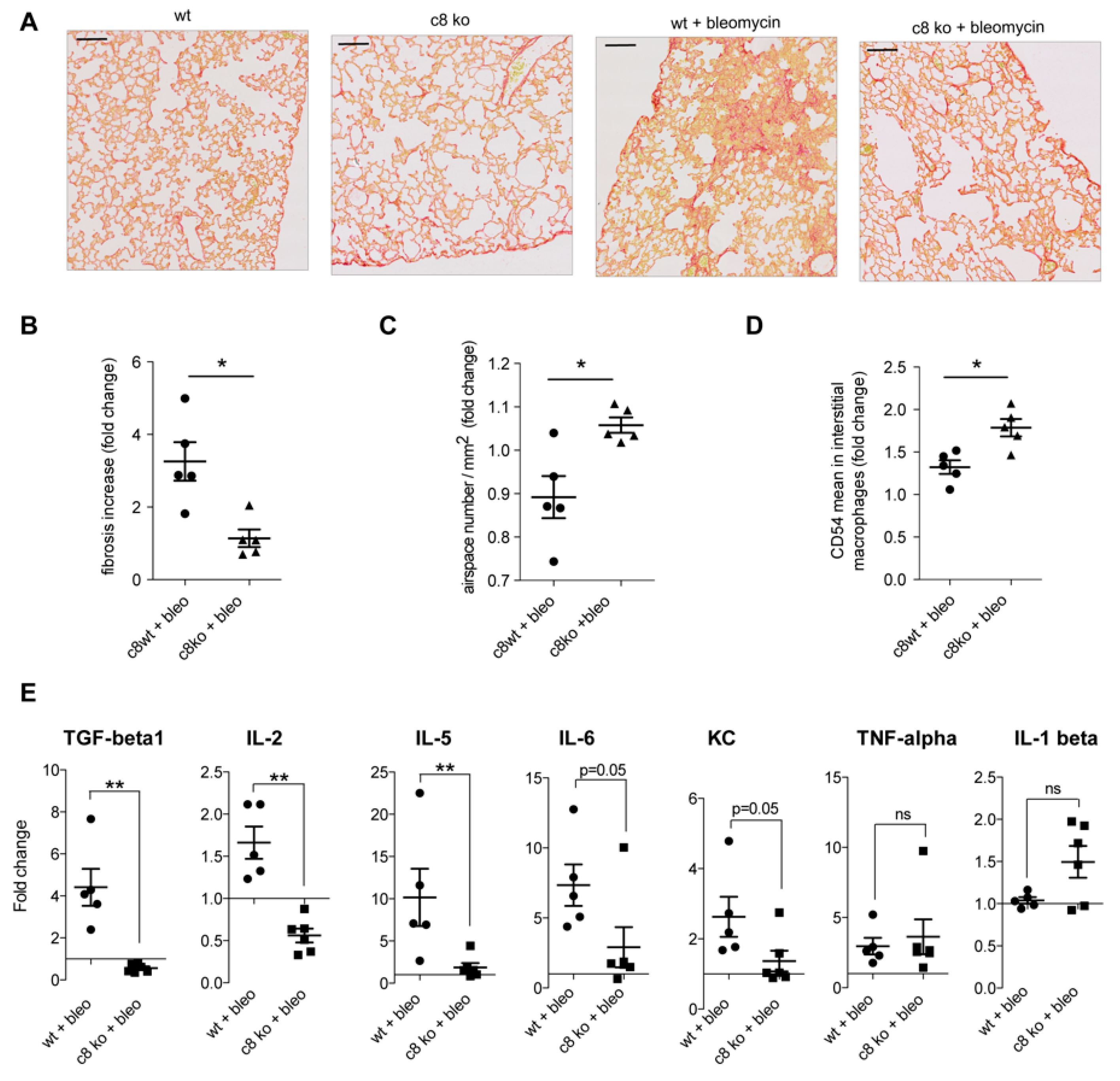
2.6. Caspase Inhibition in Monocytes/Macrophages Prevents Bleomycin-Induced Lung Fibrosis
3. Discussion
4. Materials and Methods
4.1. Study Design
4.2. Chemical Reagents and Antibodies
4.3. Cell Culture
4.4. Flow Cytometry Analysis of Cell Phenotype
4.5. Cell Morphology
4.6. Immunofluorescence Microscopy
4.7. In Situ Proximity Ligation Assay
4.8. Electron Microscopy
4.9. Caspase Activity
4.10. Immunoblotting
4.11. In Situ Caspase Trapping
4.12. Phagocytosis
4.13. Wound-Healing Assay
4.14. Cytokine Profile in Human Macrophages Supernatants
4.15. p47PHOX Proteolytic Cleavage
4.16. RNA Sequencing
4.17. Animal Models
4.18. Murine Macrophage Generation and Analysis
4.19. Lung Fibrosis Model
4.20. Fibrosis Quantification
4.21. Murine Interstitial Macrophage Collection and Analysis
4.22. Bronchoalveolar Lavage Fluid (BALF) Collection and Analysis
4.23. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef]
- Hoeffel, G.; Wang, Y.; Greter, M.; See, P.; Teo, P.; Malleret, B.; Leboeuf, M.; Low, D.; Oller, G.; Almeida, F.; et al. Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. J. Exp. Med. 2012, 209, 1167–1181. [Google Scholar] [CrossRef] [PubMed]
- Geissmann, F.; Manz, M.G.; Jung, S.; Sieweke, M.H.; Merad, M.; Ley, K. Development of monocytes, macrophages, and dendritic cells. Science 2010, 327, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu. Rev. Pathol. 2020, 15, 123–147. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014, 25, 846–859. [Google Scholar] [CrossRef]
- Tap, W.D.; Wainberg, Z.A.; Anthony, S.P.; Ibrahim, P.N.; Zhang, C.; Healey, J.H.; Chmielowski, B.; Staddon, A.P.; Cohn, A.L.; Shapiro, G.I.; et al. Structure-Guided Blockade of CSF1R Kinase in Tenosynovial Giant-Cell Tumor. N. Engl. J. Med. 2015, 373, 428–437. [Google Scholar] [CrossRef]
- Quail, D.F.; Bowman, R.L.; Akkari, L.; Quick, M.L.; Schuhmacher, A.J.; Huse, J.T.; Holland, E.C.; Sutton, J.C.; Joyce, J.A. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 2016, 352, aad3018. [Google Scholar] [CrossRef] [PubMed]
- Hume, D.A.; Irvine, K.M.; Pridans, C. The Mononuclear Phagocyte System: The Relationship between Monocytes and Macrophages. Trends Immunol. 2019, 40, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.B.; Ben-Moshe, T.; Varfolomeev, E.E.; Pewzner-Jung, Y.; Yogev, N.; Jurewicz, A.; Waisman, A.; Brenner, O.; Haffner, R.; Gustafsson, E.; et al. Caspase-8 serves both apoptotic and nonapoptotic roles. J. Immunol. 2004, 173, 2976–2984. [Google Scholar] [CrossRef] [PubMed]
- Sordet, O.; Rebe, C.; Plenchette, S.; Zermati, Y.; Hermine, O.; Vainchenker, W.; Garrido, C.; Solary, E.; Dubrez-Daloz, L. Specific involvement of caspases in the differentiation of monocytes into macrophages. Blood 2002, 100, 4446–4453. [Google Scholar] [CrossRef] [PubMed]
- Jacquel, A.; Benikhlef, N.; Paggetti, J.; Lalaoui, N.; Guery, L.; Dufour, E.K.; Ciudad, M.; Racoeur, C.; Micheau, O.; Delva, L.; et al. Colony-stimulating factor-1-induced oscillations in phosphatidylinositol-3 kinase/AKT are required for caspase activation in monocytes undergoing differentiation into macrophages. Blood 2009, 114, 3633–3641. [Google Scholar] [CrossRef]
- Rebe, C.; Cathelin, S.; Launay, S.; Filomenko, R.; Prevotat, L.; L’Ollivier, C.; Gyan, E.; Micheau, O.; Grant, S.; Dubart-Kupperschmitt, A.; et al. Caspase-8 prevents sustained activation of NF-kappaB in monocytes undergoing macrophagic differentiation. Blood 2007, 109, 1442–1450. [Google Scholar] [CrossRef]
- Cathelin, S.; Rebe, C.; Haddaoui, L.; Simioni, N.; Verdier, F.; Fontenay, M.; Launay, S.; Mayeux, P.; Solary, E. Identification of proteins cleaved downstream of caspase activation in monocytes undergoing macrophage differentiation. J. Biol. Chem. 2006, 281, 17779–17788. [Google Scholar] [CrossRef]
- Guery, L.; Benikhlef, N.; Gautier, T.; Paul, C.; Jego, G.; Dufour, E.; Jacquel, A.; Cally, R.; Manoury, B.; Vanden Berghe, T.; et al. Fine-tuning nucleophosmin in macrophage differentiation and activation. Blood 2011, 118, 4694–4704. [Google Scholar] [CrossRef]
- Boorsma, C.E.; Draijer, C.; Melgert, B.N. Macrophage heterogeneity in respiratory diseases. Mediat. Inflamm. 2013, 2013, 769214. [Google Scholar] [CrossRef]
- Duffield, J.S.; Lupher, M.; Thannickal, V.J.; Wynn, T.A. Host responses in tissue repair and fibrosis. Annu. Rev. Pathol. 2013, 8, 241–276. [Google Scholar] [CrossRef]
- Gibbons, M.A.; MacKinnon, A.C.; Ramachandran, P.; Dhaliwal, K.; Duffin, R.; Phythian-Adams, A.T.; van Rooijen, N.; Haslett, C.; Howie, S.E.; Simpson, A.J.; et al. Ly6Chi monocytes direct alternatively activated profibrotic macrophage regulation of lung fibrosis. Am. J. Respir. Crit. Care Med. 2011, 184, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Okuma, T.; Terasaki, Y.; Kaikita, K.; Kobayashi, H.; Kuziel, W.A.; Kawasuji, M.; Takeya, M. C-C chemokine receptor 2 (CCR2) deficiency improves bleomycin-induced pulmonary fibrosis by attenuation of both macrophage infiltration and production of macrophage-derived matrix metalloproteinases. J. Pathol. 2004, 204, 594–604. [Google Scholar] [CrossRef]
- Redente, E.F.; Keith, R.C.; Janssen, W.; Henson, P.M.; Ortiz, L.A.; Downey, G.P.; Bratton, D.L.; Riches, D.W. Tumor necrosis factor-alpha accelerates the resolution of established pulmonary fibrosis in mice by targeting profibrotic lung macrophages. Am. J. Respir. Cell Mol. Biol. 2014, 50, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Lacey, D.C.; Achuthan, A.; Fleetwood, A.J.; Dinh, H.; Roiniotis, J.; Scholz, G.M.; Chang, M.W.; Beckman, S.K.; Cook, A.D.; Hamilton, J.A. Defining GM-CSF- and macrophage-CSF-dependent macrophage responses by in vitro models. J. Immunol. 2012, 188, 5752–5765. [Google Scholar] [CrossRef] [PubMed]
- Solier, S.; Pommier, Y. The apoptotic ring: A novel entity with phosphorylated histones H2AX and H2B and activated DNA damage response kinases. Cell Cycle 2009, 8, 1853–1859. [Google Scholar] [CrossRef]
- Xu, Q.; Choksi, S.; Qu, J.; Jang, J.; Choe, M.; Banfi, B.; Engelhardt, J.F.; Liu, Z.G. NADPH Oxidases Are Essential for Macrophage Differentiation. J. Biol. Chem. 2016, 291, 20030–20041. [Google Scholar] [CrossRef]
- Cachat, J.; Deffert, C.; Hugues, S.; Krause, K.H. Phagocyte NADPH oxidase and specific immunity. Clin. Sci. (Lond.) 2015, 128, 635–648. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Lejal, N.; Truchet, S.; Bechor, E.; Bouguyon, E.; Khedkar, V.; Bertho, N.; Vidic, J.; Adenot, P.; Solier, S.; Pick, E.; et al. Turning off NADPH oxidase-2 by impeding p67(phox) activation in infected mouse macrophages reduced viral entry and inflammation. Biochim. Et Biophys. Acta (BBA)-Gen. Subj. 2018, 1862, 1263–1275. [Google Scholar] [CrossRef]
- Gharib, S.A.; Johnston, L.K.; Huizar, I.; Birkland, T.P.; Hanson, J.; Wang, Y.; Parks, W.C.; Manicone, A.M. MMP28 promotes macrophage polarization toward M2 cells and augments pulmonary fibrosis. J. Leukoc. Biol. 2014, 95, 9–18. [Google Scholar] [CrossRef]
- Manoury, B.; Nenan, S.; Leclerc, O.; Guenon, I.; Boichot, E.; Planquois, J.M.; Bertrand, C.P.; Lagente, V. The absence of reactive oxygen species production protects mice against bleomycin-induced pulmonary fibrosis. Respir. Res. 2005, 6, 11. [Google Scholar] [CrossRef]
- Nakajima, Y.I.; Kuranaga, E. Caspase-dependent non-apoptotic processes in development. Cell Death Differ. 2017, 24, 1422–1430. [Google Scholar] [CrossRef] [PubMed]
- Solier, S.; Fontenay, M.; Vainchenker, W.; Droin, N.; Solary, E. Non-apoptotic functions of caspases in myeloid cell differentiation. Cell Death Differ. 2017, 24, 1337–1347. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Mei, Y.; Schipma, M.J.; Roth, E.W.; Bleher, R.; Rappoport, J.Z.; Wickrema, A.; Yang, J.; Ji, P. Nuclear Condensation during Mouse Erythropoiesis Requires Caspase-3-Mediated Nuclear Opening. Dev. Cell 2016, 36, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, S.; Nagasaki, M.; Kunishima, S.; Sawaguchi, A.; Sakata, A.; Sakaguchi, H.; Ohmori, T.; Manabe, I.; Italiano, J.E., Jr.; Ryu, T.; et al. IL-1alpha induces thrombopoiesis through megakaryocyte rupture in response to acute platelet needs. J. Cell Biol. 2015, 209, 453–466. [Google Scholar] [CrossRef]
- Frisan, E.; Vandekerckhove, J.; de Thonel, A.; Pierre-Eugene, C.; Sternberg, A.; Arlet, J.B.; Floquet, C.; Gyan, E.; Kosmider, O.; Dreyfus, F.; et al. Defective nuclear localization of Hsp70 is associated with dyserythropoiesis and GATA-1 cleavage in myelodysplastic syndromes. Blood 2012, 119, 1532–1542. [Google Scholar] [CrossRef]
- Ribeil, J.A.; Zermati, Y.; Vandekerckhove, J.; Cathelin, S.; Kersual, J.; Dussiot, M.; Coulon, S.; Moura, I.C.; Zeuner, A.; Kirkegaard-Sorensen, T.; et al. Hsp70 regulates erythropoiesis by preventing caspase-3-mediated cleavage of GATA-1. Nature 2007, 445, 102–105. [Google Scholar] [CrossRef]
- Amcheslavsky, A.; Wang, S.; Fogarty, C.E.; Lindblad, J.L.; Fan, Y.; Bergmann, A. Plasma Membrane Localization of Apoptotic Caspases for Non-apoptotic Functions. Dev. Cell 2018, 45, 450–464.e453. [Google Scholar] [CrossRef]
- Aram, L.; Braun, T.; Braverman, C.; Kaplan, Y.; Ravid, L.; Levin-Zaidman, S.; Arama, E. A Krebs Cycle Component Limits Caspase Activation Rate through Mitochondrial Surface Restriction of CRL Activation. Dev. Cell 2016, 37, 15–33. [Google Scholar] [CrossRef]
- Minis, A.; Steller, H. Krebs Cycle Moonlights in Caspase Regulation. Dev. Cell 2016, 37, 1–2. [Google Scholar] [CrossRef]
- Park, H.H. Structural features of caspase-activating complexes. Int. J. Mol. Sci. 2012, 13, 4807–4818. [Google Scholar] [CrossRef] [PubMed]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656. [Google Scholar] [CrossRef] [PubMed]
- Anson, F.; Thayumanavan, S.; Hardy, J.A. Exogenous Introduction of Initiator and Executioner Caspases Results in Different Apoptotic Outcomes. JACS Au 2021, 1, 1240–1256. [Google Scholar] [CrossRef] [PubMed]
- Allan, L.A.; Clarke, P.R. Phosphorylation of caspase-9 by CDK1/cyclin B1 protects mitotic cells against apoptosis. Mol. Cell 2007, 26, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Cardone, M.H.; Roy, N.; Stennicke, H.R.; Salvesen, G.S.; Franke, T.F.; Stanbridge, E.; Frisch, S.; Reed, J.C. Regulation of cell death protease caspase-9 by phosphorylation. Science 1998, 282, 1318–1321. [Google Scholar] [CrossRef] [PubMed]
- Sohn, D.; Schulze-Osthoff, K.; Janicke, R.U. Caspase-8 can be activated by interchain proteolysis without receptor-triggered dimerization during drug-induced apoptosis. J. Biol. Chem. 2005, 280, 5267–5273. [Google Scholar] [CrossRef]
- Thomsen, N.D.; Koerber, J.T.; Wells, J.A. Structural snapshots reveal distinct mechanisms of procaspase-3 and -7 activation. Proc. Natl. Acad. Sci. USA 2013, 110, 8477–8482. [Google Scholar] [CrossRef]
- Kamber Kaya, H.E.; Ditzel, M.; Meier, P.; Bergmann, A. An inhibitory mono-ubiquitylation of the Drosophila initiator caspase Dronc functions in both apoptotic and non-apoptotic pathways. PLoS Genet. 2017, 13, e1006438. [Google Scholar] [CrossRef]
- Murray, J.; Renslo, A.R. Modulating caspase activity: Beyond the active site. Curr. Opin. Struct. Biol. 2013, 23, 812–819. [Google Scholar] [CrossRef]
- Salvesen, G.S.; Walsh, C.M. Functions of caspase 8: The identified and the mysterious. Semin. Immunol. 2014, 26, 246–252. [Google Scholar] [CrossRef]
- Orning, P.; Lien, E. Multiple roles of caspase-8 in cell death, inflammation, and innate immunity. J. Leucoc. Biol. 2021, 109, 121–141. [Google Scholar] [CrossRef] [PubMed]
- Leverrier, S.; Salvesen, G.S.; Walsh, C.M. Enzymatically active single chain caspase-8 maintains T-cell survival during clonal expansion. Cell Death Differ. 2011, 18, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Ch’en, I.L.; Beisner, D.R.; Degterev, A.; Lynch, C.; Yuan, J.; Hoffmann, A.; Hedrick, S.M. Antigen-mediated T cell expansion regulated by parallel pathways of death. Proc. Natl. Acad. Sci. USA 2008, 105, 17463–17468. [Google Scholar] [CrossRef]
- Vercammen, D.; Brouckaert, G.; Denecker, G.; Van de Craen, M.; Declercq, W.; Fiers, W.; Vandenabeele, P. Dual signaling of the Fas receptor: Initiation of both apoptotic and necrotic cell death pathways. J. Exp. Med. 1998, 188, 919–930. [Google Scholar] [CrossRef]
- Henry, C.M.; Martin, S.J. Caspase-8 Acts in a Non-enzymatic Role as a Scaffold for Assembly of a Pro-inflammatory "FADDosome" Complex upon TRAIL Stimulation. Mol. Cell 2017, 65, 715–729.e5. [Google Scholar] [CrossRef] [PubMed]
- Weaver, B.P.; Weaver, Y.M.; Mitani, S.; Han, M. Coupled Caspase and N-End Rule Ligase Activities Allow Recognition and Degradation of Pluripotency Factor LIN-28 during Non-Apoptotic Development. Dev. Cell 2017, 41, 665–673.e666. [Google Scholar] [CrossRef]
- Singel, K.L.; Segal, B.H. NOX2-dependent regulation of inflammation. Clin. Sci. (Lond.) 2016, 130, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Groemping, Y.; Lapouge, K.; Smerdon, S.J.; Rittinger, K. Molecular basis of phosphorylation-induced activation of the NADPH oxidase. Cell 2003, 113, 343–355. [Google Scholar] [CrossRef]
- El-Benna, J.; Dang, P.M.; Gougerot-Pocidalo, M.A.; Marie, J.C.; Braut-Boucher, F. p47phox, the phagocyte NADPH oxidase/NOX2 organizer: Structure, phosphorylation and implication in diseases. Exp. Mol. Med. 2009, 41, 217–225. [Google Scholar] [CrossRef]
- Chaubey, S.; Jones, G.E.; Shah, A.M.; Cave, A.C.; Wells, C.M. Nox2 is required for macrophage chemotaxis towards CSF-1. PLoS ONE 2013, 8, e54869. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, S.A.; Jadhavar, P.S.; Miglani, S.K.; Singh, M.P.; Kalane, D.P.; Agarwal, A.K.; Sathe, B.D.; Mukherjee, K.; Gupta, A.; Haldar, S.; et al. Design, synthesis and optimization of bis-amide derivatives as CSF1R inhibitors. Bioorganic Med. Chem. Lett. 2017, 27, 2153–2160. [Google Scholar] [CrossRef] [PubMed]
- Ye, T.H.; Yang, F.F.; Zhu, Y.X.; Li, Y.L.; Lei, Q.; Song, X.J.; Xia, Y.; Xiong, Y.; Zhang, L.D.; Wang, N.Y.; et al. Inhibition of Stat3 signaling pathway by nifuroxazide improves antitumor immunity and impairs colorectal carcinoma metastasis. Cell Death Dis. 2017, 8, e2534. [Google Scholar] [CrossRef]
- Luo, Y.; Zhou, H.; Krueger, J.; Kaplan, C.; Lee, S.H.; Dolman, C.; Markowitz, D.; Wu, W.; Liu, C.; Reisfeld, R.A.; et al. Targeting tumor-associated macrophages as a novel strategy against breast cancer. J. Clin. Investig. 2006, 116, 2132–2141. [Google Scholar] [CrossRef]
- Guerriero, J.L.; Sotayo, A.; Ponichtera, H.E.; Castrillon, J.A.; Pourzia, A.L.; Schad, S.; Johnson, S.F.; Carrasco, R.D.; Lazo, S.; Bronson, R.T.; et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature 2017, 543, 428–432. [Google Scholar] [CrossRef]
- McWhorter, F.Y.; Wang, T.; Nguyen, P.; Chung, T.; Liu, W.F. Modulation of macrophage phenotype by cell shape. Proc. Natl. Acad. Sci. USA 2013, 110, 17253–17258. [Google Scholar] [CrossRef] [PubMed]
- Boussetta, T.; Gougerot-Pocidalo, M.A.; Hayem, G.; Ciappelloni, S.; Raad, H.; Arabi Derkawi, R.; Bournier, O.; Kroviarski, Y.; Zhou, X.Z.; Malter, J.S.; et al. The prolyl isomerase Pin1 acts as a novel molecular switch for TNF-alpha-induced priming of the NADPH oxidase in human neutrophils. Blood 2010, 116, 5795–5802. [Google Scholar] [CrossRef]
- Taleb, K.; Auffray, C.; Villefroy, P.; Pereira, A.; Hosmalin, A.; Gaudry, M.; Le Bon, A. Chronic Type I IFN Is Sufficient To Promote Immunosuppression through Accumulation of Myeloid-Derived Suppressor Cells. J. Immunol. 2017, 198, 1156–1163. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solier, S.; Mondini, M.; Meziani, L.; Jacquel, A.; Lacout, C.; Berghe, T.V.; Julé, Y.; Martinou, J.-C.; Pierron, G.; Rivière, J.; et al. Caspase Inhibition Modulates Monocyte-Derived Macrophage Polarization in Damaged Tissues. Int. J. Mol. Sci. 2023, 24, 4151. https://doi.org/10.3390/ijms24044151
Solier S, Mondini M, Meziani L, Jacquel A, Lacout C, Berghe TV, Julé Y, Martinou J-C, Pierron G, Rivière J, et al. Caspase Inhibition Modulates Monocyte-Derived Macrophage Polarization in Damaged Tissues. International Journal of Molecular Sciences. 2023; 24(4):4151. https://doi.org/10.3390/ijms24044151
Chicago/Turabian StyleSolier, Stéphanie, Michele Mondini, Lydia Meziani, Arnaud Jacquel, Catherine Lacout, Tom Vanden Berghe, Yvon Julé, Jean-Claude Martinou, Gérard Pierron, Julie Rivière, and et al. 2023. "Caspase Inhibition Modulates Monocyte-Derived Macrophage Polarization in Damaged Tissues" International Journal of Molecular Sciences 24, no. 4: 4151. https://doi.org/10.3390/ijms24044151
APA StyleSolier, S., Mondini, M., Meziani, L., Jacquel, A., Lacout, C., Berghe, T. V., Julé, Y., Martinou, J.-C., Pierron, G., Rivière, J., Deloger, M., Dupuy, C., Slama-Schwok, A., Droin, N., Vandenabeele, P., Auberger, P., Deutsch, E., El-Benna, J., Dang, P. M.-C., & Solary, E. (2023). Caspase Inhibition Modulates Monocyte-Derived Macrophage Polarization in Damaged Tissues. International Journal of Molecular Sciences, 24(4), 4151. https://doi.org/10.3390/ijms24044151